

Abstract
In this study the regulation of cardiac sympathetic outflow by presynaptic P2X receptor-gated ion channels was examined.
ATP (30 μM–1 mM) and other P2-receptor agonists elicited [3H]-noradrenaline ([3H]-NA) outflow from the isolated guinea-pig right atrium with the potency order of ATP>2-methyl-thioATP>α,β-methylene-ATP=ADP, whereas β,γ-methylene-L-ATP was inactive.
Ca2+-free conditions abolished both electrical field stimulation (EFS)- and ATP-evoked release of tritium. Unlike from EFS-induced outflow, ATP-induced [3H]-NA outflow was not reduced by ω-Conotoxin-GVIA (100 nM), Cd2+ (100 μM) and tetrodotoxin (1 μM).
The rapid extracellular decomposition of ATP was revealed by HPLC analysis. However, the effect of ATP to promote [3H]-NA release was not prevented by 8-cyclopentyl-1,3-dipropylxanthine (DPCPX, 250 nM), 3,7-dimethyl-1-propargylxanthine (DMPX, 250 nM), or by reactive blue 2 (RB2, 10 μM), antagonists of A1-, A2- and inhibitory P2 receptors.
Zn2+ (50 μM), the P2X-receptor modulator potentiated, and P2X receptor antagonists, i.e. suramin (300 μM), pyridoxal-phosphate-6-azophenyl-2′,4′-disulphonic acid (PPADS, 30 μM) and 2′-o-(trinitrophenyl)-adenosine 5′-triphosphate (TNP-ATP, 30 μM) antagonized the ATP (1 mM)-evoked response.
RT–PCR study revealed the expression of P2X2 and P2X3 receptor mRNAs in guinea-pig superior cervical ganglion.
PPADS (30 μM) significantly reduced the EFS-induced [3H]-NA outflow in the presence DPCPX (250 nM) and RB2 (10 μM).
In summary a P2X-type purinoceptor regulates noradrenaline release from the isolated right atrium of the guinea-pig. The pharmacological profile of the receptor resemble to homo-oligomeric P2X3 or hetero-oligomeric P2X2/P2X3 complexes, and provide a new target to intervene on sympathetic neuroeffector transmission at the presynaptic site.
Keywords: Noradrenaline, release, P2X-purinoceptors, atrium, guinea-pig, RT–PCR
Introduction
Presynaptic ligand-gated ion channels provide an important target to amplify neuroeffector transmission (MacDermott et al., 1999). ATP gated ion channels, also known as P2X receptors, form a unique family of ligand-gated ion channels, characterized by two membrane spanning regions and a large extracellular loop (Brake et al., 1994). Until now seven different members of this family has been identified which display distinct but overlapping distribution and pharmacological features (North & Surprenant, 2000; Ralevic & Burnstock, 1998). These seven P2X proteins, numbered by P2X1 to P2X7, however, do not function as individual receptors, but they form homo- or hetero-oligomeric complexes in expression systems, corresponding to functional ATP-gated ion channels in native tissues. Recent biochemical evidence suggest that as much as 17 different homo- or hetero-oligomeric combinations of P2X receptor subtypes result in functional receptor (Torres et al., 1999). The coassembly of different individual P2X subtypes may result in a completely novel pharmacological phenotype (Haines et al., 1999; Le et al., 1998, 1999; Lewis et al., 1995), but this is not necessarily the case; in other combinations one subtype dominates over the other in terms of pharmacological profile (King et al., 2000). Given this considerable diversity, a major challenge has recently been shown to characterize P2X receptor assemblies in expression systems and to identify physiological responses by their native counterparts. In the nervous system P2X receptors mediate the well-known fast transmitter action of extracellular ATP in neuro-neuronal (Edwards et al., 1992) and neuro-effector synapses (Evans et al., 1992). As P2X receptors are also known for their high Ca2+ permeability (Rogers et al., 1997), whereby Ca2+ signal and neurotransmitter release could be directly initiated, another possible function of neuronal P2X receptors is that they modulate neurotransmitter release. In our earlier study we demonstrated presynaptic P2 receptor on cholinergic nerve terminals (Sperlágh & Vizi, 1991), which was confirmed by electrophysiological studies (Sun & Stanley, 1996), and followed by reports on P2X receptors regulating glutamate (Gu & MacDermott, 1997) dopamine (Zhang et al., 1996) and GABA release (Hugel & Schlichter, 2000). Apart from our previous study, where the presence of P2 receptors on sympathetic nerve terminals has been first suggested (Sperlágh & Vizi, 1991), recent reports identified stimulatory P2X receptors on noradrenergic axon terminals of cultured rat sympathetic neurons (Boehm, 1999, von Kugelgen et al., 1999). However, the organization and receptor distribution of cultured neurons might not reflect that of normal tissue and no such data is available concerning the noradrenaline release in whole tissue. Furthermore a wealth of data suggest that there is considerable inter-species and inter-ganglionic variation in the expression of P2X receptors in sympathetic ganglia of different species (Evans & Surprenant, 1996; Xiang et al., 1998; Zhong et al., 2000a,2000b), therefore one could not extrapolate from one species to another, or from one ganglion to another.
The main question addressed in this study was, therefore, to show whether the release of noradrenaline could be modulated by presynaptic P2X receptors in the isolated right atrium of the guinea-pig, and to characterize pharmacologically the receptor-subtype involved.
Methods
[3H]-Noradrenaline release from the isolated right atrium of the guinea-pig
[3H]-Noradrenaline release experiments were carried out according to the method described in our earlier papers (e.g. Oe et al., 1999). Male albino guinea-pigs (Richter Gedeon Co., Budapest, Hungary) of 300–500 g body weights were anaesthetized with saturating concentrations of diethyl-ether, and then stunned and exsanguinated. The right atria were dissected in ice-cold Krebs' solution saturated with 95% O2 and 5% CO2 and incubated in 1 ml of modified Krebs solution (mM: NaCl 113, KCl 4.7, CaCl2 2.5, KH2PO4 1.2, MgSO4 1.2, NaHCO3 25.0, glucose 11.5, pH 7.8) containing 370 kBq ml−1 [3H]-NA (0.27 μM, specific activity 1.35 TBq mmol−1; Amersham), ascorbic acid (30 μM) and Na2EDTA (100 μM) for 60 min. The medium was bubbled continuously with 95% O2 and 5% CO2 and maintained at 37°C. After incubation the tissues were rinsed and transferred to tissue chambers of 4-ml volume and perfused continuously with modified Krebs' solution at a rate of 1 ml min−1. In order to wash out the excess radioactivity and to allow tissue equilibration, the preparations were perfused for 60-min. Subsequently, perfusate samples were collected over a 3-min period and assayed for [3H]-NA. During the sample collection period, electrical field stimulation was applied twice 30 min apart (S1, S2), using a Grass S88 stimulator (Quincy, MA, U.S.A.), with the following parameters: alternate square-wave pulses (25 V cm−1, 2.5-ms duration) applied at 2 Hz; a total of 240 shocks were delivered during both S1 and S2. In some experiments DPCPX and RB2 were perfused during the entire collection period and PPADS was applied 18 min before S2 and thereafter. In other experiments only one electrical stimulation (S1) was applied, and 12 min after stimulation P2 receptor agonists (ATP, 2-methyl-thioATP, ADP, α,β-methylene-ATP, β,γ-methylene-L-ATP) were applied for a 10 min perfusion period. Purinoceptor antagonists (PPADS, DPCPX, DMPX, suramin, TNP-ATP, RB2 and NF 279) and Zn2+ were administered 10 min before ATP, tetrodotoxin, Cd2+, and ω-conotoxin GVIA were perfused from 12 min before S1, Ca2+ free solution was applied 60 min before S1 and thereafter.
The radioactivity released from the preparations was measured with a Packard 1900 Tricarb (Packard, Canberra, Australia) liquid scintillation spectrometer. A 0.5-ml aliquot of the perfusate sample was added to 4 ml of liquid scintillation fluid (Packard Ultima Gold) and counts were determined. For determining the residual radioactivity, the tissues were weighed and homogenized, and the radioactivity was extracted with 10% trichloroacetic acid. The counts were converted to absolute activity by the external standard method. Release of [3H]-NA was expressed in Bq g−1 and as a percentage of the amount of radioactivity in the tissue at the sample collection time (fractional release). It was reported previously that the majority of tritium outflow under identical conditions is derived from [3H]-NA (Nakatsuka et al., 1995). Basal outflow was calculated as the fractional release measured in a 3-min sample in the absence and presence of drugs, respectively. Drug-induced NA outflow was expressed by calculating the net release in response to drug application by the area-under-the-curve method, i.e. by subtracting the release before the drug application from the values measured after drug application. Similarly, electrical-stimulation evoked NA release was calculated by subtracting the release before the stimulation from the values measured after the respective stimulation (S1,S2). The effect of drugs on the electrical stimulation-evoked release of [3H]-NA was expressed as S2/S1 ratios, measured in the absence and presence of the drug.
ATP metabolism studies
Right atria were dissected from guinea-pigs as described above, subdivided into three pieces and incubated in 3 ml of Krebs solution at 37°C, bubbled with 95% O2+5% CO2. Subsequently 60 nmol of ATP was added to the 3-ml bath and aliquots of 70 μl were taken out 2.5, 5, 10, 15, 20 and 30 min after the addition of ATP. The concentrations of ATP, ADP, AMP, adenosine and inosine in the aliquots were measured by high-performance liquid chromatography combined with ultraviolet detection (HPLC-UV) according to the method described earlier (Sperlágh et al., 1995). The actual concentrations of ATP, ADP, and AMP were expressed in μM. For the determination of the kinetic parameters of ectoATPase, a linear regression for ATP or AMP concentrations as a function of time was calculated from the concentrations of the first five samples (0, 2.5, 5, 10 and 15) after three different initial concentrations of ATP or AMP (20, 100 and 500 μM), and the slope was used as initial velocity (vi). These initial velocities were used for calculation of the parameters from the Lineweaver–Burk plot using linear line regression:
where vi is the velocity measured when very little substrate has reacted, [S] is the concentration of the substrate, Vmax (maximal velocity) is a point where the enzyme is saturated with the substrate and KM (Michaelis constant) is the concentration that produces half-maximal velocity.
RT–PCR study
Dissected organs (guinea-pig superior cervical ganglion, or hippocampus) were collected into liquid nitrogen. Total RNA from tissue samples was isolated by a modified guanidine isothiocyanate method with TRIZOL Reagent (Life Technologies, Rockville, MD, U.S.A.). DNA contamination of RNA preparations was eliminated by treatment with RNase free DNase (Promega, Madison, WI, U.S.A.) as described Ausubel et al. (1988). First-strand cDNA templates were synthesized from 5 μg of DNase treated RNA samples with SuperScript Preamplification System (Life Technologies) using random nonamer primers in 20 μl total volume and 5 μl of the reactions were applied for PCR amplification using 0.4 μM forward and reverse primers. Amplifications were carried out as follows: 5 min initial denaturation at 96°C then 2 u of Taq DNA polymerase (Promega) was added to the reactions and 35 cycles at 94°C for 1 min, 59°C for 1 min, 72°C for 1 min followed by 5 min final extension at 72°C.
Primers for amplication of guinea-pig P2X2 cDNA were designed to the common region of the known three splice variants (AF053327, AF053328, AF053329) (Parker et al., 1998) between positions 406 (AGCATCATCACCAGGATTGAG) and 897 (GATGATGACGCCAATAACACC). Oligonucleotides were synthesized by MWG Biotech AG (Ebersberg, Germany). Primers for amplification of the P2X3 cDNA were designed to rat P2X3 sequence (X91167) between positions 742 (CTCCTGCCTAACCTCACCGACAAGGACATAAAGAGGTGCCGCTTC) and 1321 (CTAGTGACCAATAGAATAGGCCCCTGAGTCTGTAGACTGCTTCTC) and were kindly provided by J. Simon. Since the guinea-pig sequence is not available, mouse β-actin primers were used for control amplification (AGCTGAGAGGGAAATCGTGC and GATGGAGGGCCGGCTCAT, positions 569 and 1048 on cDNA) (Tokunaga et al., 1986). Amplification products were analysed by agarose gelelectrophoresis.
Materials
The following chemicals were used: [3H]-noradrenaline, ([3H]-NA, Amersham, Little Chalfont, U.K.) adenosine 5′-triphosphate (ATP), adenosine 5′-diphosphate (ADP), adenosine 5′-monophosphate (AMP), adenosine, inosine, tetrodotoxin (TTX), α,β-methylene-adenosine 5′-triphosphate (α,β-methylene-ATP) (all from Sigma, St. Louis, MO, U.S.A.), 8-cyclopentyl-1,3-dipropylxanthine (DPCPX), 3.7-dimethyl-1-propargylxanthine (DMPX), 2-methylthioadenosine triphosphate (2-methyl-thioATP), pyridoxal-phosphate-6-azophenyl-2′,4′-disulphonic acid tetrasodium (PPADS) (RBI, Natick, MA, U.S.A.), ZnCl2 CdCl2 (Reanal, Budapest, Hungary), ω-conotoxin GVIA (Alomone Labs, Jerusalem, Israel), suramin (Bayer, Leverkusen, Germany), 2′-o-(trinitrophenyl) adenosine 5′-triphosphate (TNP-ATP, Molecular Probes, Eugene, OR, U.S.A.), β,γ-methylene-L-adenosine triphosphate (β,γ-methylene-L-ATP), NF 279 (Tocris Cookson, Ballwin, MO, U.S.A.), reactive blue 2 (RB2) (Aldrich, Steinheim, Germany). All solutions were freshly prepared on the day of use.
Statistics
All data were expressed as means±s.e.mean of n observations. The statistical analysis were made by one-way analysis of variance (ANOVA) followed by Dunnett test (multiple comparisons), or Student's t-test (pairwise comparisons). P values of less than 0.05 were considered statistically significant. Concentration-response curves were constructed using the sigmoidal logistic equation of the GraphPad Prism (San Diego, CA, U.S.A.) software.
Results
[3H]-noradrenaline release experiments on the isolated right atrium of the guinea-pig
After 60 min preperfusion the basal tritium efflux measured in a 3 min sample was 0.32±0.02% (n=8) of tissue tritium content, which remained relatively constant during the subsequent sample collections. Electrical field stimulation of low frequency (2 Hz, 240 shocks) increased the outflow of [3H]-NA: the electrical stimulation-evoked release was 1.22±0.13%, (n=8, P<0.001) in control experiments (Figure 1a). The second stimulation (S2) elicited a similar amount of tritium, resulting in an S2/S1 ratio of 0.96±0.02 (n=8). In the presence of the sodium channel inhibitor tetrodotoxin (TTX, 1 μM) the basal outflow of [3H]-NA did not change significantly (0.34±0.01, n=6, P>0.05); in contrast there was no detectable increase in the outflow, in response to stimulation (see Figure 2).
Figure 1.
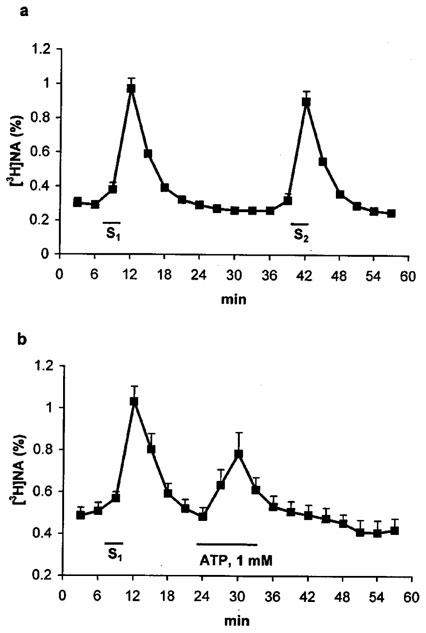
Electrical field stimulation- and ATP-induced release of [3H]-NA from the isolated guinea-pig right atrium. Tissues were superfused with Krebs' solution for 60 min and then subjected to electrical field stimulations (a,b) S1, S2, 25 V cm−1, 2 Hz, 2.5 ms, 240 shocks) or to ATP (1 mM) application (b) as indicated. The tritium content in the perfusate samples was measured by liquid scintillation spectrometry and was expressed in fractional release (%, for calculation, see Methods), as a function of time. Data represent the means±s.e.mean of 6–8 identical experiments.
Figure 2.
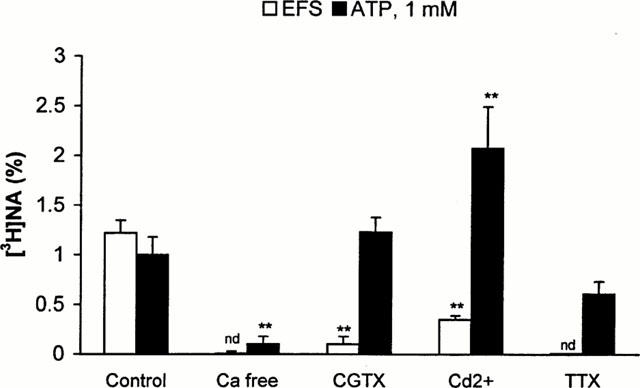
Effect of Ca2+ free medium, voltage dependent Ca2+ channel antagonists and tetrodotoxin on the electrical field stimulation-evoked and ATP-induced outflow of [3H]-NA in the isolated guinea-pig right atrium. The preparations were superfused and subjected to electrical field stimulation (S1, 25 V cm−1, 2 Hz, 2.5 ms, 240 shocks) or ATP (1 mM) application, according to the experimental protocol shown in Figure 1b. Ca2+ free solution (Ca2+ free), supplemented with 1 mM EGTA was applied from 60 min before the start of sample collection, and thereafter, ω-conotoxin GVIA (CGTX, 0.1 μM), Cd2+ (Cd2+, 100 μM) and tetrodotoxin (TTX, 1 μM) was perfused from 10 min before the start of the sample collection period, and thereafter. The net tritium release evoked by electrical field stimulation (EFS, open bars) or ATP (ATP, solid bars) were calculated by the area-under-the-curve method and expressed in fractional release (%). Data show the means±s.e.mean of 6–8 identical experiments. Asterisks indicate significant differences from respective controls, calculated by ANOVA followed by Dunnett test (**P>0.01). n.d. not detectable.
A 10-min perfusion of the preparations with ATP (1 mM) caused a transient increase in the efflux of [3H]-NA (Figure 1b) which peaked 6 min after ATP administration and then gradually declined and returned to the baseline level in the following 18 min. The net release evoked by ATP (1 mM) (1.23±0.2%), was comparable to that evoked by field stimulation, and was concentration-dependent between 0.03 and 1 mM (see Figure 5). As it is known that high concentration of ATP might have non-specific depolarizing effect, higher concentration than 1 mM was not tested.
Figure 5.
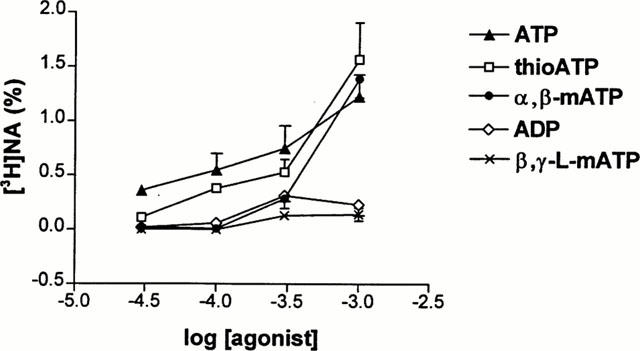
Concentration-response relationship of [3H]-NA outflow evoked by different P2X receptor agonists. Isolated guinea-pig right atrium preparations were superfused and subjected to ATP (triangles), ADP (diamonds), 2-methyl-thioATP (thioATP, squares), α,β-methylene-ATP (α,β-mATP, circles) or β,γ-methylene-L-ATP (β,γ-L-mATP, crosses) application in different concentrations indicated on the abscissa, ranging from 30 μM to 1 mM according to the experimental protocol shown in Figure 1b. The net tritium release evoked by different agonists were calculated by the area-under-the-curve method and expressed in fractional release (%). Data show the means±s.e.mean of 6–8 identical experiments.
In order to explore the potential mechanism whereby ATP promotes the outflow of noradrenaline, in subsequent experiments electrical field stimulation (EFS)-induced and ATP-induced [3H]-NA outflow were compared under different experimental conditions. When tissues were exposed to Ca2+ free solution supplemented with 1 mM EGTA both EFS- and ATP induced [3H]-NA outflow was abolished (Figure 2). ω-Conotoxin-GVIA (100 nM) the selective blocker of the N-type voltage dependent Ca2+ channels inhibited EFS-evoked tritium outflow, whereas it did not affect significantly basal (0.33±0.02, n=6, P>0.05) and ATP-induced (Figure 2) tritium outflow. Similarly, Cd2+ (100 μM) which is known to inhibit all types of voltage dependent Ca2+ channels at this concentration (Miller, 1987) almost totally inhibited EFS-evoked outflow, while it did not decrease basal (0.31±0.03, n=6, P>0.05) and ATP-evoked outflow, moreover, the latter was potentiated (Figure 2). Blockade of sodium-dependent action potential propagation by TTX (1 μM), which prevented electrical field stimulation-evoked [3H]-NA outflow, did not significantly affect ATP-induced [3H]-NA outflow (Figure 2).
Since a relatively high concentration of ATP was necessary to obtain this effect, the possibility that ATP might have been broken down rapidly in the extracellular space was envisaged in the following experiments. The decomposition of exogenous ATP was determined by HPLC-UV technique (Figure 3). Sixty nmol ATP, added to the preparations was readily hydrolyzed to ADP, AMP and adenosine by the ectoATPase, ectoATPdiphosphohydrolase and 5′nucleotidase enzymes, and as an end-product of inactivation, inosine also appeared in the extracellular fluid, showing the activity of the adenosine deaminase enzyme. The vmax and KM values of ectoATPase enzyme, obtained in initial rate measurements were 14±4.18 nmol min−1 prep. and 445±92 μM respectively (n=3). Hence, ATP itself, but its breakdown products acting on adenosine receptors might be also responsible for increased outflow of tritium in response to ATP challenge. In the presence of DPCPX (250 nM), the A1-selective adenosine receptor antagonist, and DMPX (250 nM), the relatively selective A2 receptor antagonist the basal efflux of [3H]-NA did not change significantly (0.49±0.05, n=6, P>0.05 and 0.41±0.02, n=6, P>0.05 in the presence of DPCPX and DMPX, respectively). Neither DPCPX (250 nM), nor DMPX (250 nM) affected [3H]-NA outflow induced by 1 mM ATP (Figure 4). As it has been demonstrated that noradrenaline release from sympathetic neurons is under the control of inhibitory P2Y-like purinoceptors (von Kugelgen et al., 1993; 1995) the effect of reactive blue 2 (RB2, 10 μM), a P2 receptor antagonist known to antagonize this inhibitory neuromodulation was also tested: nevertheless, RB2 was ineffective to modify either basal (0.49±0.24, n=6, P>0.05), or ATP (1 mM)-induced outflow (Figure 4). In contrast, PPADS (30 μM) another P2 receptor-antagonist completely inhibited, while Zn2+ (50 μM) the allosteric modulator of ATP gated ion channels significantly potentiated ATP-induced response (Figure 4), while no change in basal efflux of tritium was observed (0.39±0.03, n=6, P>0.05 and 0.31±0.01, n=8, P>0.05, in the presence of PPADS and Zn2+, respectively).
Figure 3.
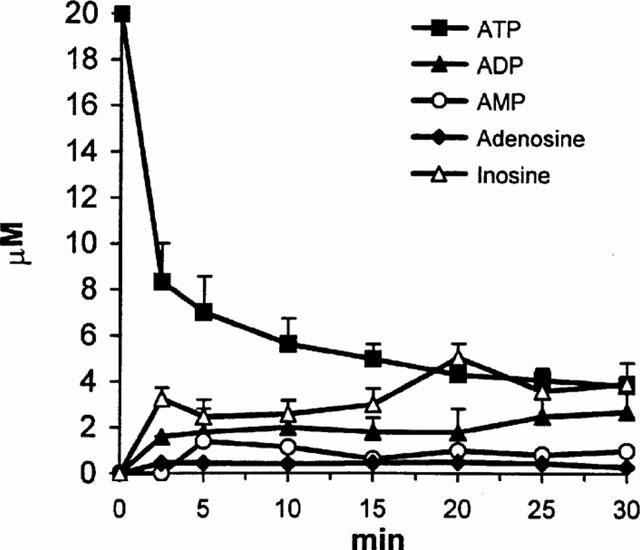
Extracellular decomposition of ATP in the isolated guinea-pig right atrium. Preparations were incubated in 3 ml Krebs' solution in the presence of 60 nmol ATP. Aliquots (70 μl) were taken 2.5, 5, 10, 20, 25, and 30 min after the addition of ATP. The amount of nucleotides (ATP, ADP, AMP) and nucleosides (ADO, adenosine, INO, inosine) in the aliquots were determined by HPLC-UV method and expressed in μM. Data show the means±s.e.mean of three identical experiments.
Figure 4.
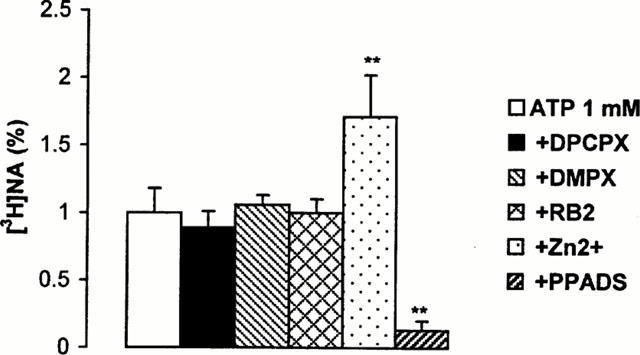
Effect of different antagonists and Zn2+ on ATP-induced outflow of [3H]-NA. Isolated guinea-pig right atrium preparations were superfused and subjected to ATP (1 mM) application, according to the experimental protocol shown in Figure 1b. DPCPX (250 nM), DMPX (250 nM), reactive blue 2 (RB2, 10 μM), Zn2+ (50 μM) and PPADS (30 μM) were perfused 10 min before ATP application and thereafter. The net tritium release evoked by ATP was calculated by the area-under-the-curve method and expressed in fractional release (%). Data show the means±s.e.mean of 6–8 identical experiments. **P<0.01 indicates significant differences between ATP-evoked tritium outflow in the absence and presence of drugs or Zn2+, calculated by ANOVA followed by Dunnett test.
Apart from ATP, other agonists, known to act on P2 receptors were also active to promote tritium release. Figure 5 shows the concentration-response curves of different P2 agonists to elicit [3H]-NA outflow. ATP appeared to be the most potent agonist, whereas 2-methyl-thioATP and α,β-methylene-ATP also behaved as less potent, but full agonists (Figure 5). ADP exhibited similar potency than α,β-methylene-ATP, however its effect declined at 1 mM and it released significantly less tritium than ATP at this concentration (0.23±0.01% of tissue tritium, n=6, P<0.05), indicating that it acts as a partial agonist. β,γ-methylene-L-ATP, a relatively selective antagonist of P2X1 receptors (Trezise et al., 1995) appeared as an even weaker agonist, being ineffective at releasing substantial amounts of tritrium under our experimental conditions (Figure 5).
The effect of ATP was also tested in the presence of a variety of P2 receptor antagonists. PPADS (30 μM), suramin (300 μM) and TNP-ATP (30 μM) all decreased or abolished the response obtained by ATP at different concentrations (Figure 6). NF279 (2 μM), an antagonist of P2X1 receptor did not affect significantly [3H]-NA outflow evoked by 1 mM ATP (1.85±0.22, n=6, P>0.05).
Figure 6.
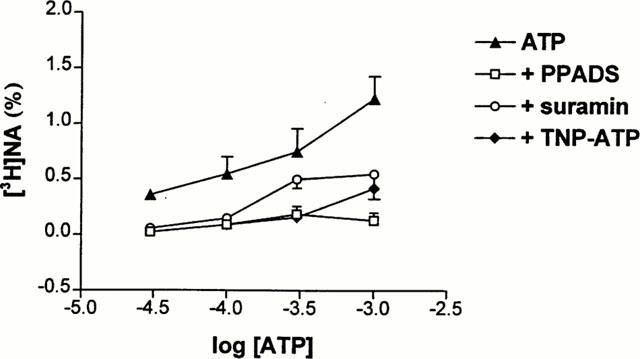
Effect of P2X receptor antagonists on [3H]-NA outflow evoked by ATP. Concentration-response curves for ATP were made in the absence (triangles) or presence of PPADS (30 μM, squares), suramin (300 μM, circles) and TNP-ATP (30 μM, diamonds). The net tritium release evoked by different concentrations of ATP ranging from 30 μM to 1 mM were calculated by the area-under-the-curve method and expressed in fractional release (%). Data show the means±s.e.mean of 4–8 identical experiments.
Since the above findings outlined a pharmacological profile resembling to P2X3 and P2X2/P2X3 receptor, a RT–PCR study was performed to confirm that P2X2 and P2X3 receptor mRNA could be coexpressed in guinea-pig superior cervical ganglion (SCG) which is known to supply the sympathetic innervation of the heart.
RT–PCR study
Specific primers were designed to the common regions of the known three splice variants of guinea-pig P2X2 receptor cDNA. Since the sequence of the guinea-pig P2X3 receptor cDNA has not been reported so far, in case of P2X3 receptor, primers designed to the rat P2X3 receptor cDNA were used. As shown in Figure 7, mRNAs encoding both P2X2 and P2X3 receptors were present in the guinea-pig SCG and hippocampus, the latter was used as a positive control. The length of both P2X2 and P2X3 specific PCR products were as expected. When PCR was performed on RNA samples without reverse transcription, there were no amplification products, indicating that the bands on the gel generated by RT–PCR couldn't be derived from direct amplification of genomic DNA.
Figure 7.
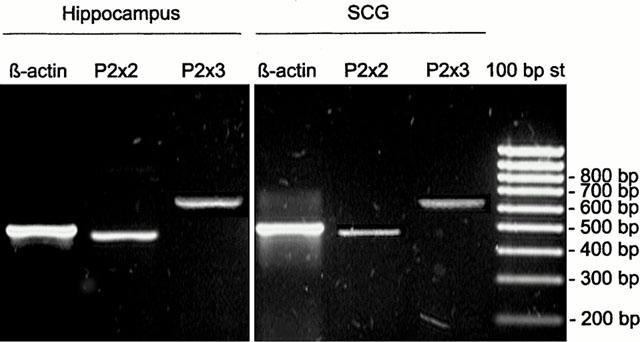
RT–PCR analysis of P2X2 and P2X3 receptor subtype expression in the guinea-pig SCG and hippocampus. Total RNA samples from the SCG and hippocampus were reverse transcribed and amplified by PCR using primers specific to P2X2 and P2X3 transcripts. Amplification of β-actin was used as internal control. A 100 bp DNA ladder (Fermentas, Vilnius, Lithuania) was used to identify PCR fragment sizes. The gels shown are representative of at least three independent experiments.
Regulation of EFS-induced [3H]-NA outflow by P2 receptors activated by endogenous ATP
In order to exclude interaction of endogenous ATP with inhibitory modulation via A1 and P2Y-like receptors, the following experiments were carried out in the presence of DPCPX (250 nM) and RB2 (10 μM), the antagonists of these receptors, which were perfused from the beginning of the sample collection period. Under these conditions the S2/S1 ratio was 1.14±0.05 (n=6). When PPADS (30 μM) was perfused before the second stimulation, the resting outflow did not change significantly (data not shown). In contrast a slight but significant decrease in the EFS-evoked [3H]-NA outflow was observed in the presence of PPADS: the S2/S1 ratio changed to 0.87±0.06 (n=6, P<0.01).
Discussion
ATP has been known for a long time to act as an extracellular messenger in the cardiovascular system (Drury & Szent-Györgyi, 1929). It is co-released with noradrenaline from sympathetic nerve terminal in response to neuronal activity (Fredholm et al., 1982; Sperlágh & Vizi, 2000), but it could also be released from postsynaptic target cells (Vizi & Sperlágh, 1999; Vizi et al., 1992), i.e. from endothelial cells (Milner et al., 1990; Yang et al., 1994), and from cardiac muscle cells upon energy deprivation (Borst & Schrader, 1991). ATP activates multiple subtypes of ionotropic (P2X) and metabotropic (P2Y) receptors, regulating myocardial function, haemostasis and proliferation of the endothelium (Boarder & Hourani, 1998; Rongen et al., 1997). A further important site of action of extracellular ATP could be neuronal P2 receptors, whereby neuroregulation of cardiac function could be locally and effectively controlled. Neuronal P2X receptor subtypes have been extensively characterized in cellular systems (North & Surprenant, 2000; Ralevic & Burnstock, 1998), however much less is known about their identity and functional properties in whole tissues; in particular how they control neurotransmitter release. Here we provide pharmacological evidence that the release of NA is under the control of P2X receptors, located on cardiac sympathetic nerve terminals.
In our experiments ATP and other P2 receptor agonists elicited concentration-dependent [3H]-NA outflow, which was comparable to that obtained after axonal stimulation of sympathetic nerves. ATP-induced [3H]-NA outflow proved to be entirely dependent on extracellular Ca2+, but apart from EFS-evoked outflow it was not abolished in the presence of voltage-dependent calcium channel blockers. Knowing that P2X receptors have relatively high Ca2+ permeability (Rogers et al., 1997), the potential mechanism whereby ATP promotes the release of NA could be the direct influx of Ca2+ through the receptor ion channel complex, rather than the subsequent activation of voltage dependent calcium channels. On the other hand, blockade of voltage dependent sodium channels by TTX did not significantly affect ATP induced [3H]-NA outflow, indicating that sodium channels do not play any part in the initiation of this kind of release. This result also suggests that the underlying receptor is located downstream from action potential initiation site and is supposedly at the varicose axon terminals of sympathetic neurons.
The concentration range of ATP to elicit this response was between 30 μM and 1 mM, which is relatively higher than values obtained in cellular systems where ectoATPase activity is inhibited or tightly controlled (Crack et al., 1995; Khakh & Kennedy, 1998). The most plausible explanation of this discrepancy is that ATP has broken down in the extracellular space to inactive metabolites by the ectonucleotidase cascade located on endothelial surface (Pearson et al., 1980). Therefore it was important to determine the degradation rate of ATP in this tissue. Indeed, exogenous ATP has been readily hydrolyzed to ADP, AMP and adenosine, due to the activity of ectoATPase, ectoATPdiphosphohydrolase and 5′nucleotidase enzymes, and as an end product of this inactivation pathway, inosine also appeared in the extracellular fluid due to the activity of adenosine deaminase enzyme. Nevertheless, neither DPCPX, the A1-selective adenosine receptor antagonist, nor DMPX, the relatively selective A2 receptor antagonist, affected significantly [3H]-NA outflow induced by 1 mM ATP, suggesting that metabolites of ATP acting on adenosine receptors, did not contribute to this effect.
As several studies have shown, the release of NA from sympathetic nerve terminals is under the control of inhibitory P2Y-like receptors (von Kugelgen et al., 1993; 1995), and this inhibitory neuromodulation is sensitive to RB2, an antagonist of P2 receptors, the effect of this drug was also tested against ATP induced [3H]-NA outflow. However, RB2 was ineffective to modify ATP-evoked [3H]-NA outflow, showing that distinct receptors are responsible for inhibition and stimulation of NA release by extracellular ATP. In contrast other P2 receptor antagonists, such as PPADS and suramin, inhibited, whereas Zn2+, the allosteric modulator of certain ATP gated ion channels, potentiated ATP-induced [3H]-NA outflow consistently with the idea that it is mediated by P2X receptors. Although at present there is no ligand which is able to differentiate between P2X and P2Y receptor families, the notion, we favour, that P2X rather than P2Y receptors are responsible for this effect, relies on the following arguments: (1) The response was sensitive to modulation by Zn2+ and Cd2+, and (2) was entirely [Ca2+]o-dependent, features characteristic for ATP gated ion channels (Li et al., 1993; Nakazawa & Ohno, 1997) but not to metabotropic P2Y receptors; (3) ADP exhibited a weak, partial agonist effect which excluded the involvement of P2Y4 and P2Y6 receptors, where ADP is equipotent or more potent than ATP, whereas the agonist activity of α,β-methylene-ATP ruled out the involvement of P2Y1 and P2Y2 (Ralevic & Burnstock, 1998); (4) to date almost all P2Y-like receptors which have been involved in the modulation of neurotransmitter release have been proved to be inhibitory receptor (e.g. von Kugelgen et al., 1993; 1995), while positive modulation of neurotransmitter release is generally mediated by P2X-like receptors (e.g. Boehm, 1999; Gu & MacDermott, 1997; Hugel & Schlichter, 2000).
Our findings support previous observation of Boehm (1999) who found that cultured rat sympathetic neurons are equipped with presynaptic P2X receptors, and confirm that these receptors are functional in a whole tissue, where synaptic organization and receptor distribution is retained. However, the pharmacological profile of the receptor identified in our experiments is dissimilar from that described in the above study, indicating that a different subtype of P2X receptor is involved. Furthermore, the pharmacological pattern outlined by this study allows identification of the P2X receptor subunit composition of the underlying receptor, at least from the known pharmacological phenotypes of P2X receptor assemblies: these are six functional homomeric (P2X1, P2X2, P2X3, P2X4, P2X5, P2X7) and four heteromeric P2X, (P2X2/P2X3, P2X1/P2X5, P2X2/P2X6, P2X4/P2X6) combinations (Haines et al., 1999; King et al., 2000; Le et al., 1998; 1999; North & Surprenant, 2000). As the most important feature, α,β-methylene-ATP acted as full agonist in our study, which rules out the involvement of all homomeric combinations, except P2X1 and P2X3, and the heteromeric assemblies, except P2X2/P2X3 and P2X4/P2X6. This finding is also indicative for the phenotypic difference between P2X receptors regulating NA release in the rat and guinea-pig sympathetic nerves, i.e. at the rat receptor which was identified as a P2X2 receptor, α,β-methylene-ATP was inactive (Boehm, 1999). Since β,γ-methylene-L-ATP, which has been claimed as a selective P2X1 receptor agonist (Trezise et al., 1995) was virtually inactive in our experiments, the involvement of P2X1 receptors seems also unlikely. Furthermore, reactive blue 2, which has been shown to antagonize responses mediated by P2X1 (Brake et al., 1994) and heterologously expressed P2X4/P2X6 receptors (Le et al., 1998), did not affect the response evoked by ATP in our study, which limits the likely candidates to P2X3 or P2X2/P2X3 combinations. Supporting this assumption, TNP-ATP that is reported to be active at these two (but not at all) P2X receptors (Virginio et al., 1998) antagonized the response evoked by ATP. Although TNP-ATP has been reported as nanomolar affinity ligand at these receptor assemblies (Lewis et al., 1998; Virginio et al., 1998) it exhibited lower affinity in whole tissues, most likely because it is subject to dephosphorylation by ectonucleotidases (Lewis et al., 1998). The most important phenotypic difference between P2X3 and P2X2/3 receptor is their different desensitization properties (Lewis et al., 1995); however, the temporal resolution of the release technique does not allow direct investigation of receptor desensitization. Instead, a RT–PCR study was carried out to prove whether these P2X receptor subunits could be coexpressed in guinea-pig SCG, the sympathetic ganglion providing the sympathetic innervation of the cardiac tissue. In the case of P2X2 receptors, specific primers were designed to those regions of guinea-pig P2X2 cDNA which are present in all splice variants of this receptor subtype (Parker et al., 1998), and our data showed that this receptor is expressed in the hippocampus and in the SCG. We have used rat-specific P2X3 primers to detect the P2X3 receptor mRNA in a guinea-pig, since to date P2X2 receptor is the only P2X subtype which has been molecularly identified in this species (Parker et al., 1998). Nevertheless mRNA for this receptor was also clearly detected both in the hippocampus and atrium, indicating that the rat primers could amplify the guinea-pig P2X3 receptor cDNA, and there should be high sequence homology between rat and guinea-pig P2X3 genes. This is not surprising, taking into account the relatively high homology between rat and guinea-pig P2X2 (Parker et al., 1998). Our data are also in agreement with the very recent immunohistochemical study of Zhong et al., who found that P2X2 and P2X3 receptors are the only P2X subtypes which are expressed in guinea-pig SCG (Zhong et al., 2000a) as opposed to rat SCG, where P2X1, P2X2, P2X4 and P2X6 are expressed (Xiang et al., 1998). Therefore our data adds further support to the growing notion that the composition of P2X receptor subunits expressed in sympathetic ganglia of rat and guinea-pig is different (Zhong et al., 2000a,2000b), and this species heterogeneity is also manifested at those receptors which are expressed at the nerve terminal level. Nevertheless, our data does not rule out the possibility that the expression pattern of P2X receptors in cardiac sympathetic nerve terminals is inhomogeneous and individual nerve terminals might express different subunit assemblies i.e. either P2X3 or P2X2/3 or even P2X2.
As it is well known that endogenous ATP is co-released with NA from the sympathetic nerves (Sperlágh & Vizi, 2000; Vizi et al., 1992) and from the postsynaptic target cell upon neuronal stimulation (Vizi & Sperlágh, 1999; Vizi et al., 1992), and early studies showed stimulation-dependent release of ATP from sympathetic nerves supplying the heart (Fredholm et al., 1982), it seemed worthwhile to challenge the hypothesis, whether the release of noradrenaline evoked by axonal stimulation is subject to modulation by endogenous ATP through presynaptic P2X receptors described in the previous experiments. Since NA release from sympathetic nerves is under the tonic control of inhibitory P2 receptors (von Kugelgen et al., 1995) and A1 adenosine receptors (Hedqvist & Fredholm, 1979), the possible interference of endogenous ATP with these receptors was prevented by the application of RB2 and DPCPX. Under these conditions PPADS (the P2 receptor antagonist) slightly but significantly reduced the electrical field stimulation-induced NA outflow, raising the possibility that the release of NA upon neuronal activity is under the autofacilitatory control of P2 receptors located on varicose terminals of sympathetic nerves. However, as PPADS has also inhibitory activity on ectoATPase enyzme (Bultmann et al., 1996) and on intracellular Ca2+ mobilization (Vigne et al., 1996) this assumption needs further investigation.
In summary, we show here that the release of NA from cardiac sympatheic nerves is regulated by presynaptic P2X receptors. The pharmacological phenotype of this receptor is similar to homo-oligomeric P2X3 and to hetero-oligomeric P2X2/P2X3 receptors, consistent with the expression of the receptor subunit mRNA in the SCG. These receptors may serve as a target site for endogenous ATP released from various sources (Sperlágh & Vizi, 2000) or drugs acting in a non-synaptic fashion (Vizi, 2000) in the cardiovascular system under physiological and pathological conditions.
Acknowledgments
This study was supported by grants from the Hungarian Research Foundation (OTKA T025614, T029859) and Hungarian Medical Research Council (ETT27/2000). The authors are grateful to Dr Joseph Simon for providing RT–PCR primers, for Ms Zsuzzsanna Körössy for excellent technical assistance, and for Ms Éva Szénássy for HPLC measurements.
Abbreviations
- ADP
adenosine 5′-diphosphate
- AMP
adenosine 5′-monophosphate
- ATP
adenosine 5′-triphosphate
- DMPX
3,7-dimethyl-1-propargylxanthine
- DPCPX
8-cyclopentyl-1,3-dipropylxanthine
- EFS
electrical field stimulation
- [3H]-NA
[3H]-noradrenaline
- α,β-methylene-ATP
α,β-methylene-adenosine 5′-triphosphate
- β,γ-methylene-L-ATP
β,γ-methylene-L-adenosine triphosphate
- 2-methyl-thioATP
2-methylthioadenosine triphosphate
- NA
noradrenaline
- PPADS
pyridoxal-phosphate-6-azophenyl-2′,4′-disulphonic acid
- RB2
reactive blue 2
- RT–PCR
reverse-transcription-coupled-polymerase-chain-reaction
- SCG
superior cervical ganglion
- TNP-ATP
2′-o-(trinitrophenyl) adenosine 5′-triphosphate
- TTX
tetrodotoxin
References
- AUSUBEL F.M., BRENT R., KINGTON R.E., MOORE D.D., SEIDMAN J.G., SMITH J.A., STRUHL K., CHANDA V.B.Removal of contaminating DNA Current Protocols in Molecular Biology 1988New York: John Wiley & Sons; edsCh. 4.1.4 [Google Scholar]
- BOARDER M.R., HOURANI S.M. The regulation of vascular function by P2 receptors: multiple sites and multiple receptors. Trends Pharmacol. Sci. 1988;19:99–107. doi: 10.1016/s0165-6147(98)01170-5. [DOI] [PubMed] [Google Scholar]
- BOEHM S. ATP stimulates sympathetic transmitter release via presynaptic P2X purinoceptors. J. Neurosci. 1999;19:737–746. doi: 10.1523/JNEUROSCI.19-02-00737.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BORST M.M., SCHRADER J. Adenine nucleotide release from isolated perfused guinea-pig hearts and extracellular formation of adenosine. Circ. Res. 1991;68:797–806. doi: 10.1161/01.res.68.3.797. [DOI] [PubMed] [Google Scholar]
- BRAKE A.J., WAGENBACH M.J., JULIUS D. New structural motif for ligand-gated ion channels defined by an ionotropic ATP receptor. Nature. 1994;371:519–523. doi: 10.1038/371519a0. [DOI] [PubMed] [Google Scholar]
- BULTMANN R., PAUSE B., WITTENBURG H., KURZ G., STARKE K. P2-purinoceptor antagonists: I. Blockade of P2-purinoceptor subtypes and ecto-nucleotidases by small aromatic isothiocyanato-sulphonates. Naunyn Schmiedeberg's Arch. Pharmacol. 1996;354:481–490. doi: 10.1007/BF00168440. [DOI] [PubMed] [Google Scholar]
- CRACK B.E., POLLARD C.E., BEUKERS M.W., ROBERTS S.M., HUNT S.F., INGALL A.H., MCKECHNIE K.C., AP I.J., LEFF P. Pharmacological and biochemical analysis of FPL 67156, a novel, selective inhibitor of ecto-ATPase. Br. J. Pharmacol. 1995;114:475–481. doi: 10.1111/j.1476-5381.1995.tb13251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DRURY A.N., SZENT-GYÖRGYI A. The physiological action of adenine compounds with especial reference to their action on the mammalian heart. J. Physiol. (Lond). 1929;68:214–237. doi: 10.1113/jphysiol.1929.sp002608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDWARDS F.A., GIBB A.J., COLQUHOUN D. ATP receptor-mediated synaptic currents in the central nervous system. Nature. 1992;359:144–147. doi: 10.1038/359144a0. [DOI] [PubMed] [Google Scholar]
- EVANS R.J., DERKACH V., SURPRENANT A. ATP mediates fast synaptic transmission in mammalian neurons. Nature. 1992;357:503–505. doi: 10.1038/357503a0. [DOI] [PubMed] [Google Scholar]
- EVANS R.J., SURPRENANT A. P2x receptors in autonomic and sensory neurons. Semin. Neurosci. 1996;8:217–223. [Google Scholar]
- FREDHOLM B.B., HEDQVIST P., LINDSTROM K., WENNMALM M. Release of nucleosides and nucleotides from the rabbit heart by sympathetic nerve stimulation. Acta Physiol. Scand. 1982;116:285–295. doi: 10.1111/j.1748-1716.1982.tb07142.x. [DOI] [PubMed] [Google Scholar]
- GU J.G., MACDERMOTT A.B. Activation of ATP P2X receptors elicits glutamate release from sensory neuron synapses. Nature. 1997;389:749–753. doi: 10.1038/39639. [DOI] [PubMed] [Google Scholar]
- HAINES W.R., TORRES G.E., VOIGT M.M., EGAN T.M. Properties of the novel ATP-gated ionotropic receptor composed of the P2X (1) and P2X(5) isoforms. Mol. Pharmacol. 1999;56:720–727. [PubMed] [Google Scholar]
- HEDQVIST P., FREDHOLM B.B. Inhibitory effect of adenosine on adrenergic neuroeffector transmission in the rabbit heart. Acta Physiol. Scand. 1979;105:120–122. doi: 10.1111/j.1748-1716.1979.tb06321.x. [DOI] [PubMed] [Google Scholar]
- HUGEL S., SCHLICHTER R. Presynaptic P2X receptors facilitate inhibitory GABAergic transmission between cultured rat spinal cord dorsal horn neurons. J. Neurosci. 2000;20:2121–2130. doi: 10.1523/JNEUROSCI.20-06-02121.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KHAKH B.S., KENNEDY C. Adenosine and ATP: progress in their receptors' structures and functions. Trends Pharmacol. Sci. 1998;19:39–41. doi: 10.1016/s0165-6147(97)01158-9. [DOI] [PubMed] [Google Scholar]
- KING B.F., TOWNSEND-NICHOLSON A., WILDMAN S.S., THOMAS T., SPYER K.M., BURNSTOCK G. Coexpression of Rat P2X2 and P2X6 Subunits in Xenopus Oocytes. J. Neurosci. 2000;20:4871–4877. doi: 10.1523/JNEUROSCI.20-13-04871.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LE K.T., BABINSKI K., SEGUELA P. Central P2X4 and P2X6 channel subunits coassemble into a novel heteromeric ATP receptor. J. Neurosci. 1998;18:7152–7159. doi: 10.1523/JNEUROSCI.18-18-07152.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LE K.T., BOUE-GRABOT E., ARCHAMBAULT V., SEGUELA P. Functional and biochemical evidence for heteromeric ATP-gated channels composed of P2X1 and P2X5 subunits. J. Biol. Chem. 1999;274:15415–15419. doi: 10.1074/jbc.274.22.15415. [DOI] [PubMed] [Google Scholar]
- LEWIS C., NEIDHART S., HOLY C., NORTH R.A., BUELL G., SURPRENANT A. Coexpression of P2X2 and P2X3 receptor subunits can account for ATP-gated currents in sensory neurons. Nature. 1995;377:432–435. doi: 10.1038/377432a0. [DOI] [PubMed] [Google Scholar]
- LEWIS C.J., SURPRENANT A., EVANS R.J. 2′,3′-O-(2,4,6-trinitrophenyl) adenosine 5′-triphosphate (TNP-ATP) – a nanomolar affinity antagonist at rat mesenteric artery P2X receptor ion channels. Br. J. Pharmacol. 1998;124:1463–1466. doi: 10.1038/sj.bjp.0702001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI C., PEOPLES R.W., LI Z., WEIGHT F.F. Zn2+ potentiates excitatory action of ATP on mammalian neurons. Proc. Natl. Acad. Sci. U.S.A. 1993;90:8264–8267. doi: 10.1073/pnas.90.17.8264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MACDERMOTT A.B., ROLE L.W., SIEGELBAUM S.A. Presynaptic ionotropic receptors and the control of transmitter release. Annu. Rev. Neurosci. 1999;22:443–485. doi: 10.1146/annurev.neuro.22.1.443. [DOI] [PubMed] [Google Scholar]
- MILLER R.J. Multiple calcium channels and neuronal function. Science. 1987;235:46–52. doi: 10.1126/science.2432656. [DOI] [PubMed] [Google Scholar]
- MILNER P., BODIN P., LOESCH A., BURNSTOCK G. Rapid release of endothelin and ATP from isolated aortic endothelial cells exposed to increased flow. Biochem. Biophys. Res. Commun. 1990;170:649–656. doi: 10.1016/0006-291x(90)92141-l. [DOI] [PubMed] [Google Scholar]
- NAKATSUKA H., NAGANO O., FOLDES F.F., NAGASHIMA H., VIZI E.S. Effects of adenosine on norepinephrine and acetylcholine release from guinea-pig right atrium: role of A1 receptors. Neurochem. Int. 1995;27:345–353. doi: 10.1016/0197-0186(95)00016-2. [DOI] [PubMed] [Google Scholar]
- NAKAZAWA K., OHNO Y. Effects of neuroamines and divalent cations on cloned and mutated ATP-gated channels. Eur. J. Pharmacol. 1997;325:101–108. doi: 10.1016/s0014-2999(97)00107-6. [DOI] [PubMed] [Google Scholar]
- NORTH R.A., SURPRENANT A. Pharmacology of cloned P2X receptors. Annu. Rev. Pharmacol. Toxicol. 2000;40:563–580. doi: 10.1146/annurev.pharmtox.40.1.563. [DOI] [PubMed] [Google Scholar]
- OE K., SPERLAGH B., SANTHA E., MATKO I., NAGASHIMA H., FOLDES F.F., VIZI E.S. Modulation of norepinephrine release by ATP-dependent K(+)-channel activators and inhibitors in guinea-pig and human isolated right atrium. Cardiovasc Res. 1999;43:125–134. doi: 10.1016/s0008-6363(99)00052-8. [DOI] [PubMed] [Google Scholar]
- PARKER M.S., LARROQUE M.L., CAMPBELL J.M., BOBBIN R.P., DEININGER P.L. Novel variant of the P2X2 ATP receptor from the guinea-pig organ of Corti. Hear. Res. 1998;121:62–70. doi: 10.1016/s0378-5955(98)00065-3. [DOI] [PubMed] [Google Scholar]
- PEARSON J.D., CARLETON J.S., GORDON J.L. Metabolism of adenine nucleotides by ectoenzymes of vascular endothelial and smooth-muscle cells in culture. Biochem. J. 1980;190:421–429. doi: 10.1042/bj1900421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RALEVIC V., BURNSTOCK G. Receptors for purines and pyrimidines. Pharmacol. Rev. 1998;50:413–492. [PubMed] [Google Scholar]
- ROGERS M., COLQUHOUN L.M., PATRICK J.W., DANI J.A. Calcium flux through predominantly independent purinergic ATP and nicotinic acetylcholine receptors. J. Neurophysiol. 1997;77:1407–1417. doi: 10.1152/jn.1997.77.3.1407. [DOI] [PubMed] [Google Scholar]
- RONGEN G.A., FLORAS J.S., LENDERS J.W., THIEN T., SMITS P. Cardiovascular pharmacology of purines. Clin. Sci. (Colch) 1997;92:13–24. doi: 10.1042/cs0920013. [DOI] [PubMed] [Google Scholar]
- SPERLAGH B., KITTEL A., LAJTHA A., VIZI E.S. ATP acts as fast neurotransmitter in rat habenula: neurochemical and enzymecytochemical evidence. Neurosci. 1995;66:915–920. doi: 10.1016/0306-4522(94)00588-v. [DOI] [PubMed] [Google Scholar]
- SPERLAGH B., VIZI E.S. Effect of presynaptic P2 receptor stimulation on transmitter release. J. Neurochem. 1991;56:1466–1470. doi: 10.1111/j.1471-4159.1991.tb02039.x. [DOI] [PubMed] [Google Scholar]
- SPERLAGH B., VIZI E.S.Regulation of purine release Handbook of Experimental Pharmacology. Purinergic and pyrimidinergic signalling 2000Heidelberg. Springer; (ed.) Abbracchio, M.P., Williams, M(in press) [Google Scholar]
- SUN X.P., STANLEY E.F. An ATP-activated, ligand-gated ion channel on a cholinergic presynaptic nerve terminal. Proc. Natl. Acad. Sci. U.S.A. 1996;93:1859–1863. doi: 10.1073/pnas.93.5.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TOKUNAGA K., TANIGUCHI H., YODA K., SHIMIZU M., SAKIYAMA S. Nucleotide sequence of a full-length cDNA for mouse cytoskeletal beta-actin mRNA. Nucleic Acids Res. 1986;14:28–29. doi: 10.1093/nar/14.6.2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TORRES G.E., EGAN T.M., VOIGT M.M. Hetero-oligomeric assembly of P2X receptor subunits. Specificities exist with regard to possible partners. J. Biol. Chem.. 1999;274:6653–6659. doi: 10.1074/jbc.274.10.6653. [DOI] [PubMed] [Google Scholar]
- TREZISE D.J., MICHEL A.D., GRAHAMES C.B., KHAKH B.S., SURPRENANT A., HUMPHREY P.P. The selective P2X purinoceptor agonist, beta, gamma-methylene-L-adenosine 5′-triphosphate, discriminates between smooth muscle and neuronal P2X purinoceptors. Naunyn Schmiedeberg's Arch. Pharmacol. 1995;351:603–609. doi: 10.1007/BF00170159. [DOI] [PubMed] [Google Scholar]
- VIGNE P., PACAUD P., URBACH V., FEOLDE E., BREITTMAYER J.P., FRELIN C. The effect of PPADS as an antagonist of inositol (1,4,5)trisphosphate induced intracellular calcium mobilization. Br. J. Pharmacol. 1996;119:360–364. doi: 10.1111/j.1476-5381.1996.tb15994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VIRGINIO C., ROBERTSON G., SURPRENANT A., NORTH R.A. Trinitrophenyl-substituted nucleotides are potent antagonists selective for P2X1, P2X3, and heteromeric P2X2/3 receptors. Mol. Pharmacol. 1998;53:969–973. [PubMed] [Google Scholar]
- VIZI E.S. Role of high-affinity receptors and membrane transporters in non-synaptic communication and drug action in the central nervous system. Pharmacol. Rev. 2000;52:63–90. [PubMed] [Google Scholar]
- VIZI E.S., SPERLAGH B. Receptor- and carrier-mediated release of ATP of postsynaptic origin: cascade transmission. Prog. Brain. Res. 1999;120:159–169. doi: 10.1016/s0079-6123(08)63553-0. [DOI] [PubMed] [Google Scholar]
- VIZI E.S., SPERLAGH B., BARANYI M. Evidence that ATP released from the postsynaptic site by noradrenaline, is involved in mechanical responses of guinea-pig vas deferens: cascade transmission. Neurosci. 1992;50:455–465. doi: 10.1016/0306-4522(92)90437-7. [DOI] [PubMed] [Google Scholar]
- VON KUGELGEN I., KURZ K., STARKE K. Axon terminal P2-purinoceptors in feedback control of sympathetic transmitter release. Neurosci. 1993;56:263–267. doi: 10.1016/0306-4522(93)90330-i. [DOI] [PubMed] [Google Scholar]
- VON KUGELGEN I., NÖRENBERG W., KOCH A., MEYER A., ILLES P., STARKE K. P2 receptors controlling neurotransmitter release from postganglionic sympathetic neurones. Prog. Brain. Res. 1999;120:173–183. doi: 10.1016/s0079-6123(08)63554-2. [DOI] [PubMed] [Google Scholar]
- VON KUGELGEN I., STOFFEL D., STARKE K. P2-purinoceptor-mediated inhibition of noradrenaline release in rat atria. Br. J. Pharmacol. 1995;115:247–254. doi: 10.1111/j.1476-5381.1995.tb15870.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XIANG Z., BO X., BURNSTOCK G. Localization of ATP-gated P2X receptor immunoreactivity in rat sensory and sympathetic ganglia. Neurosci. Lett. 1998;256:105–108. doi: 10.1016/s0304-3940(98)00774-5. [DOI] [PubMed] [Google Scholar]
- YANG S., CHEEK D.J., WESTFALL D.P., BUXTON I.L. Purinergic axis in cardiac blood vessels. Agonist-mediated release of ATP from cardiac endothelial cells. Circ. Res. 1994;74:401–407. doi: 10.1161/01.res.74.3.401. [DOI] [PubMed] [Google Scholar]
- ZHANG Y.X., YAMASHITA H., OHSHITA T., SAWAMOTO M., NAKAMURA S. ATP induces release of newly synthesized dopamine in the rat striatum. Neurochem. Int. 1996;28:395–400. doi: 10.1016/0197-0186(95)00105-0. [DOI] [PubMed] [Google Scholar]
- ZHONG Y., DUNN P.M., BURNSTOCK G. Guinea-pig sympathetic neurons express varying proportions of two distinct P2X receptors. J. Physiol. (Lond.) 2000a;523:391–402. doi: 10.1111/j.1469-7793.2000.t01-1-00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHONG Y., DUNN P.M., BURNSTOCK G. Pharmacological comparison of P2X receptors on rats coeliac, mouse coeliac and mouse pelvic ganglion neurons. Neuropharmacol. 2000b;39:172–180. doi: 10.1016/s0028-3908(99)00145-8. [DOI] [PubMed] [Google Scholar]