

Abstract
Raised extracellular K+ relaxes some arteries, and has been proposed as Endothelium-Derived Hyperpolarizing Factor (EDHF). However, relaxation of rat small mesenteric arteries to K+ is highly variable. We have investigated the mechanism of K+-induced dilatation and relaxation of pressurized arteries and arteries mounted for measurement of isometric force.
Raising [K+]o from 5.88 – 10.58 mM did not dilate or relax pressurized or isometric arteries. Relaxation to raised [K+]o was revealed in the presence of 5-nitro-2-(3-phenylpropylamino) benzoic acid (NPPB); this effect of NPPB was concentration-dependent (IC50: 1.16 μM).
Relaxations to raised [K+]o in the presence of NPPB, were abolished by 30 μM Ba2+ or endothelial-denudation. Acetycholine (10 μM) relaxed endothelium-intact arteries in presence of raised [K+]o NPPB and Ba2+.
Relaxations to raised [K+]o were revealed in hyperosmotic superfusate (+60 mM sucrose). These relaxations were abolished by 30 μM Ba2+. In the presence of raised [K+]o, 60 mM sucrose and 30 μM Ba2+, 10 μM acetycholine still relaxed all arteries.
Fifty μM 18α-glycyrrhetinic acid (18α-GA), a gap junction inhibitor, depressed relaxations to both 10 μM acetylcholine and raised [K+]o, in the presence of 10 μM NPPB.
In summary, blockade of a volume-sensitive Cl− conductance in small rat mesenteric arteries, using NPPB or hyperosmotic superfusion, reveals a endothelium-dependent, Ba2+ sensitive dilatation or relaxation of rat mesenteric arteries to raised [K+]o. We conclude that inwardly rectifying potassium channels on the endothelium underlie relaxations to raised [K+]o in rat small mesenteric arteries.
Keywords: Potassium, EDHF, ouabain, barium, gap junction, 18α-glycyrrhetinic acid, mesenteric artery, rat
Introduction
Nitric oxide synthase- and prostacyclin-independent relaxation that is accompanied by an endothelium-dependent hyperpolarization of the vascular smooth muscle currently defines the involvement of an endothelial derived hyperpolarizing factor (EDHF) (Chen et al., 1988; Taylor & Weston, 1988; Feletou & Vanhoutte, 1988; Corriu et al., 1996; Ohlmann et al., 1997; Zygmunt et al., 1997). The identity of EDHF, and its mechanism of action remains elusive. It has been suggested that EDHF is K+, and that K+ efflux from the endothelium through apamin- and charybdotoxin (ChTx)-sensitive channels is sufficient to raise K+ in the myo-endothelial space (Edwards et al., 1998), leading to hyperpolarization and relaxation of the smooth muscle by stimulation of inward rectifier K+ channels (Edwards et al., 1988; Knot et al., 1996; McCarron & Halpern, 1990) and/or the electrogenic Na+/K+-ATPase on the smooth muscle (Prior et al., 1998). A test of this model is that raised extracellular potassium ([K+]o) should produce an endothelium-independent hyperpolarization and relaxation. Although hyperpolarization and relaxation to raised [K+]o has been reported in rat mesenteric and hepatic arteries (Edwards et al., 1998, 1999), several laboratories have demonstrated that raised [K+]o is a poor vasodilator in isolated rat mesenteric (Lacy et al., 2000; Doughty et al., 2000), porcine coronary (Quignard et al., 1999), and guinea-pig carotid arteries (Edwards et al., 1999), despite the presence of a robust, ACh-mediated, EDHF-type response.
We have tested the hypothesis that, in rat mesenteric arteries, Ba2+-sensitive relaxation to raised [K+]o is sensitive to the membrane potential of endothelial cells and is inhibited by blockade of gap junctions or removal of the endothelium. The ability of endothelial cells to sustain depolarized membrane potentials, coupled with the current-voltage relationship of strong inward rectifiers, may, in part, explain the inconsistency of raised [K]o as a vasodilator in isolated rat mesenteric arteries.
Methods
200 – 250 g male Wistar rats were killed by stunning and cervical dislocation. Third order superior mesenteric arteries were dissected in physiological saline solution (PSS) containing (mM): NaCl 119, KCl 4.7, NaHCO3 25, KH2PO4 1.18, CaCl2 1.8, MgSO4 1.2, glucose 11, EDTA 0.027, Nω-nitro-L-arginine methyl ester (L-NAME) 0.1, indomethacin 0.0028. The pH was 7.4 when gassed with 95% O2/5% CO2.
Pressure myography
Leak-free segments of artery, at least 1 mm in length, were mounted between two glass cannulae in an arteriograph (Living Systems Instrumentation, Burlington, VT, U.S.A.) at room temperature (18 – 21°C) and pressurized to 80 mmHg, under conditions of no lumenal flow. The mean relaxed arterial diameter at 80 mmHg was 283±6.9 μm (n=30±s.e.mean). The artery lumen was filled with the standard PSS. Constant pressure was maintained via a pressure servo control system (PS200, Living Systems Instrumentation). Pressure transducers at both ends of the artery allowed continual monitoring of intralumenal pressure. Arteries were viewed through a Nikon TMS inverted microscope and a measurement of the internal diameter was made from a video image using a video dimension analyser (V91, Living Systems Instrumentation). The arteriograph was continually superfused with the standard PSS at a rate of 25 ml min−1. The superfusing PSS was warmed to 37°C and no myogenic constriction of arteries was seen. Therefore, arteries were constricted with 0.3 – 1 μM phenylephrine (PE) applied in the superfusate. Pressure and diameter measurements were recorded to computer via a Digidata 1200B interface using Axoscope software version 8 (Axon Instruments, CA, U.S.A.). In experiments with endothelium-denuded arteries, removal of the endothelium was achieved by passing a bubble of air through the lumen of the artery, and was confirmed by loss of response to ACh.
Wire myography
Segments of mesenteric artery were mounted in PSS in a Mulvany-Halpern wire myograph for the recording of isometric tension. The mean relaxed arterial diameter at 100 mmHg (L100) was 180±14.7 μm (n=31±s.e.mean). Artery segments were sequentially stretched until the wall tension was equivalent to a transmural pressure of 100 mmHg (Mulvany & Halpern, 1977), the diameter was calculated and set to 90% of this value and the tissue allowed to equilibrate for 30 min. Arterial segments were first constricted with phenylephrine (10 μM) to give a maximal contraction. Subsequently PE was applied to give approximately 70% of maximal force (1 – 5 μM). Data are expressed as normalized force as percentage of the 70% maximal response (100%). In experiments with endothelium-denuded arteries, removal of the endothelium was achieved by abrasion using a hair and was confirmed by loss of response to ACh.
Drugs
All drugs were made up as stock solutions in milli-Q water, unless otherwise stated, diluted in the experimental solution and applied in the superfusate. NPPB and 18α-glycyrrhetinic acid were dissolved in dimethyl sulphoxide (DMSO), such that the concentration of DMSO did not exceed 0.1% in the final solution. Indomethacin was dissolved in 2% Na2CO3 or ethanol. All drugs were supplied by Sigma (Poole, Dorset, U.K.). All data are expressed as mean values±s.e.mean for ‘n' experiments. Statistical significance was tested using a Student's t-test on paired data, unless stated otherwise. P<0.05 was regarded as significant.
Results
Responses to acetylcholine and potassium
ACh (10 μM) dilated all pressurized arteries tested towards their passive diameter (n=30), whereas raising [K+]o in the superfusing PSS from 5.88 mM (normal) to 10.58 mM (high), by doubling the concentration of KCl in the standard PSS, dilated only nine out of 30 arteries towards their passive diameter. In arteries mounted for measurement of isometric force (wire myography) ACh (10 μM) relaxed all arteries tested, whereas raising [K+]o only relaxed two arteries (n=31).
Effects of chloride channel blockade on dilatation and relaxation
Where elevating [K+]o from 5.88 to 10.58 mM failed to dilate pressurized arteries, application of a low concentration of NPPB (20 μM) produced a partial reduction in tone. Subsequent elevation of [K+]o, from 5.88 to 10.58 mM, in the continued presence of NPPB, resulted in dilatation of all vessels tested. On washout of NPPB, raising [K+]o did not result in dilatation (n=4). The ability of NPPB to reveal dilatation to raised [K+]o was abolished on removal of the endothelium (n=3) (see Figure 1).
Figure 1.
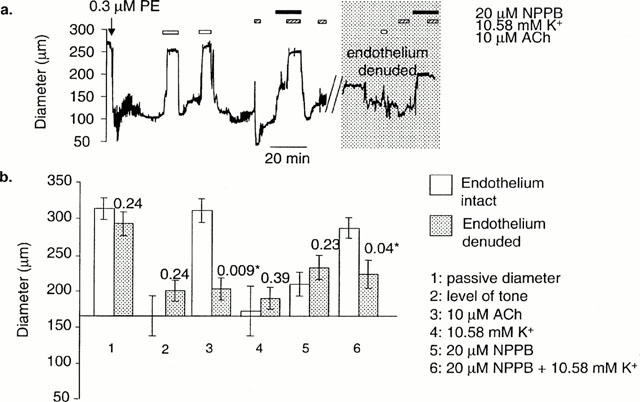
The effect of NPPB on dilatations to K+ in pressurized arteries. (a) In arteries where stepping [K+]o from 5.88 to 10.58 mM failed to produce dilatation, dilatation to K+ was induced in the presence of 20 μM NPPB. The effect of NPPB on the ability of K+ to dilate was fully reversible on washout. The gap in the data trace indicates a period of removal of the endothelium with an air bubble. In the absence of an endothelium, the effects of NPPB were abolished. (b) Meaned data (+E, n=4±s.e.mean; −E; n=3±s.e.mean). These data are not paired, and therefore P values are for an unpaired Student's t-test. * Shows significance (P<0.05).
If raising [K+]o from 5.88 to 10.58 mM did not relax isometric arteries, relaxation to raised [K+]o was revealed in the presence of NPPB (1 μM) (n=8) (Figure 2). The relaxation to raised [K+]o in the presence of NPPB was reversed by Ba2+ (30 μM) (Figure 2). In the continued presence of raised [K+]o, NPPB and Ba2+, subsequent addition of acetylcholine (10 μM) resulted in full relaxation (Figure 2). The ability of NPPB to reveal relaxations to raised [K+]o was lost after removal of the endothelium.
Figure 2.
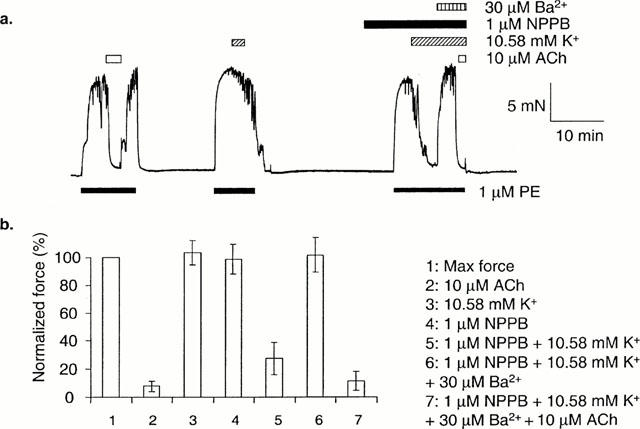
The effect of NPPB on dilatations to K+ in arteries mounted for isometric measurement of force. (a) In arteries where stepping [K+]o from 5.88 to 10.58 mM failed to produce relaxation, relaxation to K+ was induced in the presence of 1 μM NPPB. This effect was reversed by 30 μM Ba2+. Ten μM ACh was still able to relax fully in the presence of K+, NPPB and Ba2+. (b) Mean data (n=8±s.e.mean).
Concentration-dependence of NPPB-induced relaxations to raised [K+]o
NPPB concentration-dependently relaxed arteries mounted for measurement of isometric force, both in the presence (+E) and the absence of (−E) endothelium (+E: IC50=7.20 μM, n=6; −E: IC50=7.43 μM, n=4). In the presence of raised [K+]o the NPPB concentration-effect curve was shifted leftwards, but only if the endothelium was intact (+E: IC50=1.16 μM, n=10; −E: 8.22 μM, n=5). This leftward shift was inhibited in the presence of 30 μM Ba2+ (IC50=4.69 μM, n=6). These data are summarized in Figure 3.
Figure 3.
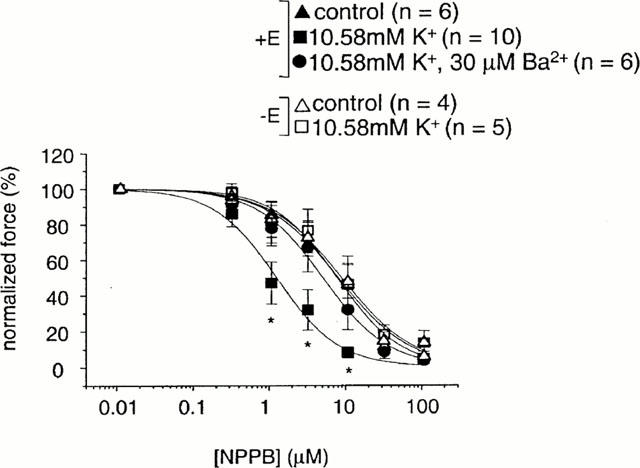
The concentration-effect curve for NPPB in arteries mounted for isometric measurement of force. In endothelium-intact arteries, NPPB relaxed with an IC50 of 7.20 μM (n=6±s.e.mean). In the presence of 10.58 mM K+, the concentration-effect curve for relaxation of arteries by NPPB was shifted left (IC50: 1.16 μM) (n=10±s.e.mean). This leftward shift was inhibited in the presence of 30 μM Ba2+ (IC50: 4.69 μM) (n=6±s.e.mean). P values are for an unpaired Student's t-test. * Show a significant difference from the control curve (P<0.05). In endothelium-denuded arteries, NPPB relaxed with an IC50 of 7.43 μM (n=4±s.e.mean). In the presence of 10.58 mM K+, the concentration-effect curve for relaxation of arteries by NPPB was not significantly affected (IC50: 8.22 μM) (n=5±s.e.mean).
Effect of anisosmotic solutions on responses to raised [K+]o
In arteries mounted for measurement of isometric force, where stepping [K+]o from 5.88 to 10.58 mM failed to produce relaxation, relaxation to K+ was revealed during superfusion of a hyper-osmotic PSS solution (60 mM sucrose added). This effect was reversed by 30 μM Ba2+. Ten μM ACh was still able to relax fully in the presence of 30 μM Ba2+ and 60 mM sucrose (n=4) (see Figure 4).
Figure 4.
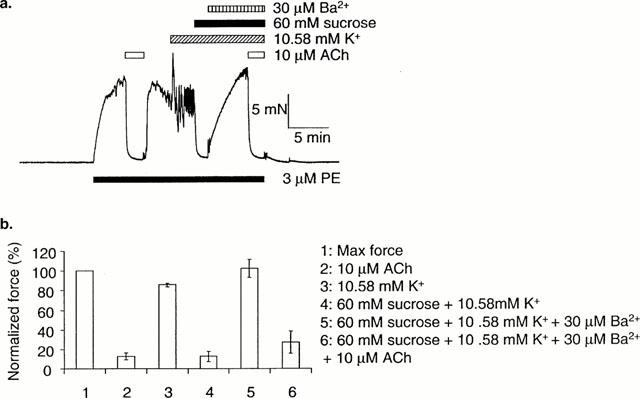
Hyper-osmotic stress mimics the effects of NPPB to reveal relaxation of isometric arteries to K+. (a) In arteries where raising [K+]o from 5.88 to 10.58 mM did not produce relaxation, relaxation to K+ was induced in the presence of a hyper-osmotic solution (60 mM sucrose added). This effect was reversed by 30 μM Ba2+. (b) Mean data (n=6±s.e.mean). 10 μM ACh was still able to relax fully in the presence of Ba2+.
Only two out of 31 isometrically mounted arteries relaxed to both 10 μM ACh and raised [K+]o under control conditions. In these arteries, reduction of the osmolarity of the superfusate by approximately 80 mosM (omission of 40 mM NaCl) reversed the relaxation to raised [K+]o. Subsequent restoration of normal osmolarity resulted in a complete and sustained relaxation in [K+]o, which was reversed again by a return to hypo-osmotic PSS (see Figure 5).
Figure 5.
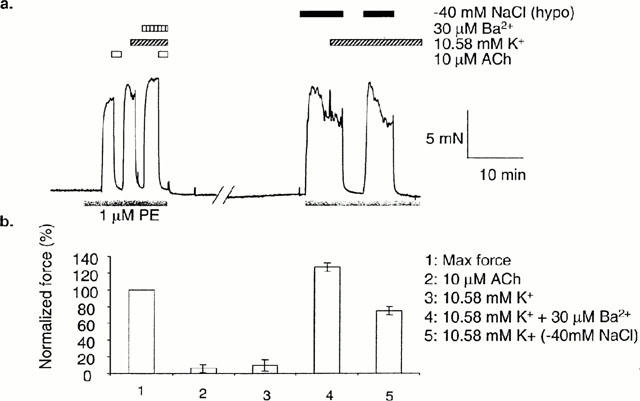
Hypo-osmotic stress prevents relaxation of isometric arteries to K+. In arteries where raising [K+]o from 5.88 to 10.58 mM produced relaxation, relaxation to K+ was abolished in the presence of 30 μM Ba2+ or a hypo-osmotic solution (40 mM NaCl omitted). (b) Mean data (n=2±s.e.mean).
Effect of gap junction inhibitors on responses to raised [K+]o
Relaxation to raised [K+]o, when revealed by NPPB (10 μM), was depressed in the presence of the gap junction inhibitor, 18α-GA (50 μM). In some arteries, a transient relaxation to raised [K+]o was observed before addition of NPPB. This transient relaxation was also depressed on application 18α-GA, and an example is shown in Figure 6a. Relaxation to ACh (10 μM) was also depressed by 18α-GA. Arteries dilated to the potassium channel opener, levcromakalim (2 μM) in the presence of 18α-GA. Mean data is summarized in Figure 6b.
Figure 6.
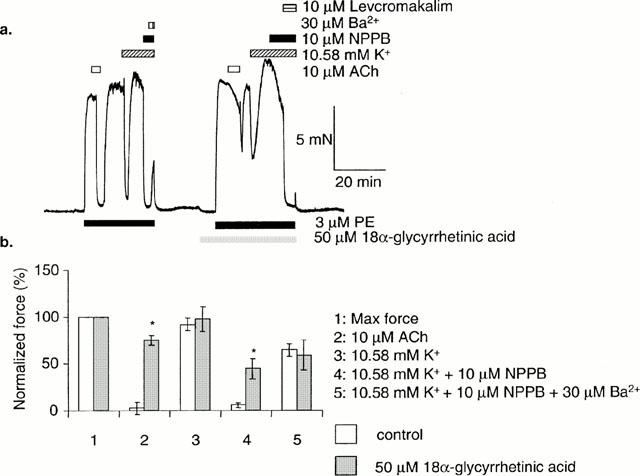
After a transient relaxation to K+, sustained relaxation to K+ was induced in the presence of 10 μM NPPB. This effect was reversed by 30 μM Ba2+. Relaxation to 10 μM ACh, and sustained relaxation to K+ was reduced in the presence of 50 μM 18α-glycyrrhetinic acid (b) Mean data (n=8±s.e.mean). P values are for a paired Student's t-test. * Show a significant difference between the control data, and in the presence of 18α-glycyrrhetinic acid (P<0.05).
Discussion
Inwardly rectifying potassium channels: endothelium
We have shown previously, in pressurized arteries, that dilatation to raised [K+]o only occurs in a small proportion of arteries, and this can be blocked by low concentrations of Ba2+ (Doughty et al., 2000), suggesting that hyperpolarization by inwardly rectifiying potassium channels is sufficient to account for K+-induced dilatation (Nelson & Quayle, 1995). However, in endothelium-denuded arteries, Ba2+-sensitivity was absent, suggesting that the inwardly rectifying potassium channels are present on the endothelial cells and not the smooth muscle. Endothelial inwardly rectifying potassium channels have been suggested to underlie the relaxation of mesenteric arteries in response to hypercapnia (Okazaki et al., 1998).
Our data suggest that the major source of Ba2+-sensitive hyperpolarizing current/factor is the endothelium, and this is transmitted to smooth muscle through myo-endothelial gap junctions. Accumulation of [K+]o in the myo-endothelial space, generated by K+ efflux through endothelial ChTx- and apamin-sensitive K+ channels (Doughty et al., 1999), will tend to hyperpolarize the endothelium by increasing outward current through the inward rectifier. Hyperpolarization of smooth muscle would also occur via Na+/K+-ATPase activity (Lacy et al., 2000; Doughty et al., 2000). Inwardly rectifying potassium channels have not been measured in mesenteric endothelial cells to our knowledge, but inwardly rectifying potassium channels have been shown to be a major determinant of endothelial membrane potential in cultured bovine pulmonary endothelial cells (Voets et al., 1996; Nilius et al., 1997).
Inwardly rectifying potassium channels: smooth muscle
There is a paucity of evidence for inwardly rectifying potassium channels in mesenteric artery myocytes, despite several attempts to demonstrate their presence (Nelson & Quayle, 1995). The presence of messenger RNA encoding an inwardly rectifying K+ channel family (Kir 2.1) in rat mesenteric cells has been demonstrated (Bradley et al., 1999). However, no recordings of the native conductance in isolated mesenteric smooth muscle cells are shown or described. Indeed, inwardly rectifying potassium channels have only been identified in one voltage-clamp study of intact mesenteric arterioles of the guinea-pig (Edwards & Hirst, 1988), and it is unclear from this study if the recorded currents were generated by the endothelium as well as the smooth muscle. Evidence for inwardly rectifying potassium channels is also limited in other arterial smooth muscle where EDHF is present (Quignard et al., 1999; Quayle et al., 1997).
Membrane potential and inwardly rectifying potassium channel current
Inwardly rectifying potassium channels hyperpolarize cells by passing outward current in the voltage range just positive to Ek (Hille & Schwarz, 1978). This behaviour can be attributed to the mechanism of block by intracellular polyamines that is thought to underlie strong inward rectification of Kir channels (Nichols & Lopatin, 1997; Lopatin et al., 1995). Thus, the membrane potential of the endothelium will be critically important for a mechanism of hyperpolarization involving this channel.
The apparent absence of relaxation to raised [K+]o or Ba2+-sensitivity in some arteries can be explained by Voets et al. (1996), who demonstrated that the majority of bovine cultured pulmonary endothelial cells had a relatively positive membrane potential (mean −26 mV with range −80 to +5 mV) that was unaffected by external Ba2+. A small proportion of cells, however, had a more negative membrane potential that could be depolarized by Ba2+. Moreover, in cells that had relatively positive membrane potentials, inhibition of a volume-regulated Cl− conductance using hyperosmotic superfusion, NPPB or quinine, hyperpolarized towards Ek. These observations suggest that membrane potential will determine the influence that inward rectifier potassium channels can have on membrane potential.
Membrane potential and blockade of chloride channels: endothelium
In our experiments, raising [K+]o will change the reversal potential for potassium and so shift the current-voltage relationship of Kir current rightwards along the voltage axis. Only endothelial cells with a membrane potential just positive to the new EK will hyperpolarize. At more positive potentials polyamine block would prevent outward current through the inwardly rectifying potassium channel. ECl is approximately 40 mV positive to EK in these experiments, so if endothelial cell membrane potential is dominated by a chloride conductance, there will be little or no outward current through inward rectifier potassium channels, and arteries will not constrict to Ba2+ or dilate to raised [K+]o. Our data, using the chloride channel blocker, NPPB, supports this hypothesis. Arteries that failed to dilate to raised [K+]o under control conditions, consistently dilated to raised [K+]o in the presence of a low concentration of NPPB. This effect of NPPB was dependent on an intact endothelium, concentration-dependent and reversible on washout.
Membrane potential and blockade of chloride channels: smooth muscle
NPPB can have non-specific effects in resistance arteries (Kato et al., 1999), and may cause relaxation by blocking L-type calcium channels on smooth muscle (Doughty et al., 1998). However, at the concentration used in this study, it is unlikely that these non-chloride channel effects of NPPB can account for the data. However, there are reports of NPPB-sensitive, volume-regulated Cl− channels in both vascular endothelium and smooth muscle and so it is possible that blockade of Cl− channels located on smooth muscle could potentiate the ability of the endothelium to hyperpolarize smooth muscle.
Separating the effects of NPPB on endothelium and smooth muscle
NPPB can cause relaxation of myogenic tone by inhibition of voltage-gated calcium channel current in myocytes isolated from rat cerebral arteries (Doughty et al., 1998). To enable identification of a Ba2+-sensitive component of relaxation to-raised potassium, independently of a non-specific, concentration-dependent relaxation to NPPB, concentration-effect curves for relaxation of PE-induced contraction by NPPB were compared. For all experimental paradigms the concentration-effect curve could be described by a Hill equation with slopes that were close to 1. The control experiment, using an artery with intact endothelium, was well fitted by a Hill equation with an IC50 of 7.2 μM. Repeating the experiment after removing the endothelium was without effect (IC50=7.43 μM). This suggests that the effects of NPPB alone on force are independent of the endothelium, and are consistent with a site of action on smooth muscle. This lack of endothelium-dependence contrasts sharply with the effect of raised potassium: the concentration-effect curve to NPPB was left-shifted to raised potassium for endothelium intact arteries (IC50=1.16 μM) but not endothelium denuded arteries (IC50=8.22 μM). Finally, the left-shift induced by raised potassium in endothelium intact arteries could be inhibited by Ba2+ (30 μM; IC50=4.69 μM). Taken together these data are consistent with a model that places a Ba2+-sensitive mechanism of relaxation in the endothelium. This mechanism appears to operate only when potassium is raised above control levels.
Does the level of tone influence the ability of raised [K+]o to relax?
In some experiments NPPB was used at moderate concentrations (10 – 20 μM) which caused a partial relaxation of tone. It is generally true that it is easier to relax smaller contractions compared with larger contractions. Thus, the partial relaxation by NPPB may predispose the arteries to relax to raised potassium. It is unlikely that partial relaxation to NPPB in itself explains the subsequent relaxation to raised potassium. Low concentrations of NPPB (1 μM) were without effect on PE-induced force were sufficient to reveal relaxations to potassium in some arteries. Thus partial relaxation to NPPB is not required in order to observe relaxation to raised potassium.
Modulating chloride channels with osmolarity
The effect of anisosmotic solutions was investigated as an alternative to NPPB block. Voets et al. (1996) demonstrated that the endothelial chloride conductance was volume-sensitive, being activated by hyposmotic and depressed by hyperosmotic superfusion. Similar volume-sensitive anion-selective conductances have been reported in a variety of endothelial and smooth muscle preparations (Nilius et al., 1999; Clapham, 1998; Yamazaki et al., 1998; Nelson et al., 1997). In recordings of isometric force, superfusion with PSS made hyperosmotic by the addition of 60 mM sucrose had little effect on constrictions to PE, but revealed relaxations to raised [K+]o in arteries that had previously not responded to [K+]o. This relaxation to raised [K+]o in the presence of hyperosmotic solutions was reversed by 30 μM Ba2+ suggesting that relaxation was dependent upon inwardly rectifying potassium channel mediated hyperpolarization. Figure 5 shows data from one of a small number of arteries that exhibited relaxations to raised [K+]o under control conditions. Under control conditions both ACh and raised [K+]o resulted in relaxation. Reduction of osmolarity (by approximately 80 mosM) did not prevent constriction to PE, but abolished the relaxation to raised [K+]o. Subsequent restoration of normal osmolarity resulted in a sustained relaxation to raised [K+]o that was reversed by a return to hyposmotic PSS. Together, these data suggest strongly that Ba2+-sensitive relaxations to raised [K+]o reflect hyperpolarization mediated by an inwardly rectifying potassium current. Hyperpolarization by an inwardly rectifying potassium current depends upon the magnitude of a volume- and NPPB-sensitive chloride current.
Other models of EDHF
EDHF-mediated relaxation, stimulated by ACh, is dependent on hyperpolarization of the endothelium, mainly by toxin-sensitive K+ conductances, but an inwardly rectifying K+ conductance may also be important. Endothelial hyperpolarization will lead to hyperpolarization of smooth muscle if functional myoendothelial gap junctions exists (Sandow & Hill, 2000). Gap junction inhibitors have been demonstrated to be very effective inhibitors of EDHF in rabbit conduit, rabbit iliac, guinea-pig carotid, rat hepatic and rat mesenteric arteries (Chaytor et al., 1998; Dora et al., 1999; Taylor et al., 1998; Edwards et al., 1999; Doughty et al., 2000). This is confirmed in these experiments using 18α-glycyrrhetinic acid, and is consistent with the hypothesis that endothelial hyperpolarization is transmitted to smooth muscle via myo-endothelial gap junctions. The gap junction inhibitor, 18α-glycyrrhetinic acid, is also an effective inhibitor of relaxations to raised [K+], in the presence of NPPB. This strengthens the evidence that K+ hyperpolarizes the endothelium by increasing the outward current passed through inwardly rectifying potassium, and this is coupled to smooth muscle hyperpolarization and relaxation through myo-endothelial gap junctions.
Conclusions
Endothelium-dependent, Ba2+-sensitive dilatations (and relaxations) to raised [K+]o could be measured from rat mesenteric artery only following reduction in chloride channel activity using NPPB or hyperosmotic PSS. We conclude that inwardly rectifying potassium channels, expressed on the endothelium, underlie the relaxation to raised [K+]o in this preparation, and that the membrane potential of endothelium is critical in this response.
Figure 7.
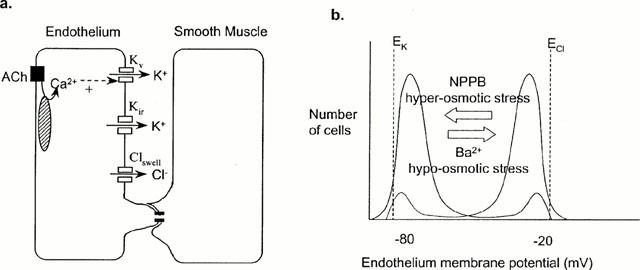
Model. (a) Inwardly rectifying potassium channels (Kir) and volume-sensitive chloride channels (Clswell) are the major determinants of the endothelial cell membrane potential. Potassium efflux through calcium-activated potassium channels (Kv) increases outward current through inwardly rectifying potassium channels (Kir) if membrane potential is close to EK, thus hyperpolarizing the endothelium. Hyperpolarization will be transmitted to smooth muscle through myoendothelial gap junctions. (b) The membrane potential of most endothelial cells is depolarized towards ECl, and these arteries do not relax/hyperpolarize to K+. NPPB or hyperosmotic stress can be used to block chloride channels, shifting the membrane potential towards EK, such that arteries relax/hyperpolarize to K+. If membrane potential close to EK, such that K+ relaxes/hyperpolarizes the artery, hypo-osmotic stress, which activates chloride channels, or Ba2+, which blocks Kir, will depolarize the endothelial membrane potential towards ECl and block relaxation.
Acknowledgments
This work was supported by British Heart Foundation grant number PG/97182 (J.M. Doughty and P.D. Langton) and Medical Research Council grant number G9609076 (J.P. Boyle). We thank Ms Lisa Grant for assistance with some of these experiments, and Dr Jane M. Hinton for comments on the manuscript.
Abbreviations
- ACh
acetylcholine
- ChTx
charybdotoxin
- DMSO
dimethyl sulphoxide
- +E
endothelium intact
- −E
endothelium denuded
- EDHF
Endothelium-Derived Hyperpolarizing Factor
- L-NAME
Nω-nitro-L-arginine methyl ester
- NPPB
5-nitro-2-(3-phenylpropylamino) benzoic acid
- PE
phenylephrine
- PSS
physiological saline solution
References
- BRADLEY K.K., JAGGAR J.H., BONEV A.D., HEPPNER T.J., FLYNN E.R.M., NELSON M.T., HOROWITZ B. K(ir)2.1. encodes the inward rectifier potassium channel in rat arterial smooth muscle cells. J. Physiol. 1999;515:639–651. doi: 10.1111/j.1469-7793.1999.639ab.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAYTOR A.T., EVANS W.H., GRIFFITH T.M. Central role of heterocellular gap junctional communication in endothelium-dependent relaxations of rabbit arteries. J. Physiol. 1998;508:561–573. doi: 10.1111/j.1469-7793.1998.561bq.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN G., SUZUKI H., WESTON A.H. Acetylcholine releases endothelium-derived hyperpolarizing factor and EDRF from rat blood vessels. Br. J. Pharmacol. 1988;95:1165–1174. doi: 10.1111/j.1476-5381.1988.tb11752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLAPHAM D.E. The list of potential volume-sensitive chloride currents continues to swell (and shrink) J. Gen. Physiol. 1998;111:623–624. doi: 10.1085/jgp.111.5.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CORRIU C., FELETOU M., CANET E., VANHOUTTE P.M. Endothelium-derived factors and hyperpolarization of the carotid artery of the guinea-pig. Br. J. Pharmacol. 1996;119:959–964. doi: 10.1111/j.1476-5381.1996.tb15765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DORA K.A., MARTIN P.E.M., CHAYTOR A.T., EVANS W.H., GARLAND C.J., GRIFFITH T.M. Role of heterocellular gap junctional communication in endothelial-dependent smooth muscle hyperpolarization: inhibition by a connexinmimetic peptide. Biochem. Biophys. Res. Com. 1999;254:27–31. doi: 10.1006/bbrc.1998.9877. [DOI] [PubMed] [Google Scholar]
- DOUGHTY J.M., BOYLE J.P., LANGTON P.D. Potassium does not mimic EDHF in rat mesenteric arteries. Br. J. Pharmacol. 2000;130:1174–1182. doi: 10.1038/sj.bjp.0703412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOUGHTY J.M., MILLER A.L., LANGTON P.D. Non-specificity of chloride channel blockers in rat cerebral arteries: block of the L-type calcium channel. J. Physiol. 1998;507:433–439. doi: 10.1111/j.1469-7793.1998.433bt.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOUGHTY J.M., PLANE F., LANGTON P.D. Charybdotoxin and apamin block EDHF in rat mesenteric artery if selectively applied to the endothelium. Am. J. Physiol. 1999;276:H1107–H1112. doi: 10.1152/ajpheart.1999.276.3.H1107. [DOI] [PubMed] [Google Scholar]
- EDWARDS F.R., HIRST G.D.S. Inward rectification in submucosal arterioles of guinea-pig ileum. J. Physiol. 1988;404:437–454. doi: 10.1113/jphysiol.1988.sp017298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDWARDS F.R., HIRST G.D.S., SILVERBERG G.D. Inward rectification in rat cerebral arterioles; involvement of potassium ions in autoregulation. J. Physiol. 1988;404:455–466. doi: 10.1113/jphysiol.1988.sp017299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDWARDS G., DORA K.A., GARDENER M.J., GARLAND C.J., WESTON A.H. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- EDWARDS G., FELETOU M., GARDENER M.J., THOLLON C., VANHOUTTE P.M., WESTON A.H. Role of gap junctions in the responses to EDHF in rat and guinea-pig small arteries. Br. J. Pharmacol. 1999;128:1788–1794. doi: 10.1038/sj.bjp.0703009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDWARDS G., GARDENER M.J., FELETOU M., BRADY G., VANHOUTTE P.M., WESTON A.H. Further investigations of endothelium-derived hyperpolarizing factor (EDHF) in rat hepatic artery: studies using 1-EBIO and ouabain. Br. J. Pharmacol. 1999;128:1064–1070. doi: 10.1038/sj.bjp.0702916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FELETOU M., VANHOUTTE P.M. Endothelium-dependent hyperpolarization of canine coronary smooth muscle. Br. J. Pharmacol. 1988;93:515–524. doi: 10.1111/j.1476-5381.1988.tb10306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HILLE B., SCHWARZ G. Potassium channels as multi-ion single-file pores. J. Gen. Physiol. 1978;72:409–442. doi: 10.1085/jgp.72.4.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KATO K., EVAN A.M., KOZLOWSKI R.Z. Relaxation of Endothelin-1-induced pulmonary arterial constriction by niflumic acid and NPPB: mechanism(s) independent of chloride channel block. J. Pharmacol. Exp. Therapeut. 1999;288:1242–1250. [PubMed] [Google Scholar]
- KNOT H.J., ZIMMERMANN P.A., NELSON M.T. Extracellular K+-induced hyperpolarizations and dilatations of rat coronary and cerebral arteries involve inward rectifier K+ channels. J. Physiol. 1996;492:419–430. doi: 10.1113/jphysiol.1996.sp021318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LACY P.S., PILKINGTON G., HANVESAKUL R., FISH H.J., BOYLE J.P., THURSTON H. Evidence against potassium as an endothelium-derived hyperpolarizing factor in rat mesenteric small arteries. Br. J. Pharmacol. 2000;129:605–611. doi: 10.1038/sj.bjp.0703076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOPATIN A.N., MAKHINA E.N., NICHOLS C.G. The mechanism of inward rectification of potassium channels: long- pore plugging by cytoplasmic polyamines. J. Gen. Physiol. 1995;106:923–955. doi: 10.1085/jgp.106.5.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCCARRON J.G., HALPERN W. Potassium dilates rat cerebral arteries by two independent mechanisms. Am. J. Physiol.–Heart Circ. Physiol. 1990;259:H902–H908. doi: 10.1152/ajpheart.1990.259.3.H902. [DOI] [PubMed] [Google Scholar]
- MULVANY M.J., HALPERN W. Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Cir. Res. 1977;41:19–26. doi: 10.1161/01.res.41.1.19. [DOI] [PubMed] [Google Scholar]
- NELSON M.T., CONWAY M.A., KNOT H.J., BRAYDEN J.E. Chloride channel blockers inhibit myogenic tone in rat cerebral arteries. J. Physiol. 1997;502:259–264. doi: 10.1111/j.1469-7793.1997.259bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NELSON M.T., QUAYLE J.M. Physiological roles and properties of potassium channels in arterial smooth muscle. Am. J. Physiol.–Cell Physiol. 1995;268:C799–C822. doi: 10.1152/ajpcell.1995.268.4.C799. [DOI] [PubMed] [Google Scholar]
- NICHOLS C.G., LOPATIN A.N. Inward rectifier potassium channels. Ann. Rev. Physiol. 1997;59:171–191. doi: 10.1146/annurev.physiol.59.1.171. [DOI] [PubMed] [Google Scholar]
- NILIUS B., VIANA F., DROOGMANS G. Ion channels in vascular endothelium. Ann. Rev. Physiol. 1997;59:145–170. doi: 10.1146/annurev.physiol.59.1.145. [DOI] [PubMed] [Google Scholar]
- NILIUS B., VOETS T., PRENEN J., BARTH H., AKTORIES K., KAIBUCHI K., DROOGMANS G., EGGERMONT J. Role of Rho and Rho kinase in the activation of volume-regulated anion channels in bovine endothelial cells. J. Physiol. 1999;516:67–74. doi: 10.1111/j.1469-7793.1999.067aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OHLMANN P., MARTINEZ M.C., SCHNEIDER F., STOCLET J.C., ANDRIANTSITOHAINA R. Characterization of endothelium-derived relaxing factors released by bradykinin in human resistance arteries. Br. J. Pharmacol. 1997;121:657–664. doi: 10.1038/sj.bjp.0701169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OKAZAKI K., ENDOU M., OKUMURA F. Involvement of barium-sensitive K+-channels in endothelium-dependent vasodilation produced by hypercapnia in rat mesenteric vascular beds. Br. J. Pharmacol. 1998;125:168–174. doi: 10.1038/sj.bjp.0702048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRIOR H.M., WEBSTER N., QUINN K., BEECH D.J., YATES M.S. K+ induced dilation of a small renal artery: no role for inward rectifier K+ channels. Cardiovasc. Res. 1998;37:780–790. doi: 10.1016/s0008-6363(97)00237-x. [DOI] [PubMed] [Google Scholar]
- QUAYLE J.M., NELSON M.T., STANDEN N.B. ATP-sensitive and inwardly rectifying potassium channels in smooth muscle. Physiol. Rev. 1997;77:1165–1232. doi: 10.1152/physrev.1997.77.4.1165. [DOI] [PubMed] [Google Scholar]
- QUIGNARD J.-F., FELETOU M., THALLON C., VILAINE J.-P., DUHAULT J., VANHOUTTE P.M. Potassium ions and endothelium-derived hyperpolarizing factor in guinea-pig carotid and porcine coronary arteries. Br. J. Pharmacol. 1999;127:27–34. doi: 10.1038/sj.bjp.0702493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SANDOW S.L., HILL C.E. Incidence of myoendothelial gap junctions in the proximal and distal mesenteric arteries of the rat is suggestive of a role in endothelium-derived hyperpolarizing factor-mediated responses. Circ. Res. 2000;86:341–346. doi: 10.1161/01.res.86.3.341. [DOI] [PubMed] [Google Scholar]
- TAYLOR H.J., CHAYTOR A.T., EVANS W.H., GRIFFITH T.M. Inhibition of the gap junctional component of endothelium-dependent relaxations in rabbit iliac artery by 18-alpha glycyrrhetinic acid. Br. J. Pharmacol. 1998;125:1–3. doi: 10.1038/sj.bjp.0702078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAYLOR S.G., WESTON A.H. Endothelium-derived hyperpolarizing factor: A new endogenous inhibitor from the vascular endothelium. Trends Pharmacol. Sci. 1988;9:272–274. doi: 10.1016/0165-6147(88)90003-x. [DOI] [PubMed] [Google Scholar]
- VOETS T., DROOGMANS G., NILIUS B. Membrane currents and the resting membrane potential in cultured bovine pulmonary artery endothelial cells. J. Physiol. 1996;497:95–107. doi: 10.1113/jphysiol.1996.sp021752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAMAZAKI J., DUAN D., JANIAK R., KUENZLI K., HOROWITZ B., HUME J.R. Functional and molecular expression of volume-regulated chloride channels in canine vascular smooth muscle cells. J. Physiol. 1998;507:729–736. doi: 10.1111/j.1469-7793.1998.729bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZYGMUNT P.M., EDWARDS G., WESTON A.H., LARSSON B., HOGESTATT E.D. Involvement of voltage-dependent potassium channels in the EDHF-mediated relaxation of rat hepatic artery. Br. J. Pharmacol. 1997;121:141–149. doi: 10.1038/sj.bjp.0701108. [DOI] [PMC free article] [PubMed] [Google Scholar]