

Abstract
Long OT syndrome has many causes from both acquired and congenital disorders. For the congenital disorders, their presentation and disease course are not identical. We studied two pharmacological models of long QT syndrome (LQT) to identify differences in cellular electrophysiological properties that may account for this. LQT2 was simulated by suppression of the rapidly activating delayed rectifier potassium current (IKr) with the drug E-4031, and LQT3 was simulated by slowing of the sodium current (INa) decay with the toxin ATX II.
Single rabbit ventricular cell action potentials were studied using the amphotericin B perforated patch clamp technique. Action potential and early afterdepolarization (EAD) properties were rigorously defined by the frequency power spectra obtained with fast Fourier transforms.
The E-4031 (n=43 myocytes) and ATX II (n=50 myocytes) models produced different effects on action potential and EAD properties. The major differences are that ATX II, compared with E-4031, caused greater action potential prolongation, more positive plateau voltages, lower amplitude EADs with less negative take-off potentials, greater time to the EAD peak voltage, and longer duration EADs. Despite causing greater action potential prolongation, the incidence of EAD induction was much less with the ATX II model (28%) than with the E-4031 model (84%). Thus these two pharmacological models have strikingly different cellular electrophysiological properties.
Our findings provide cellular mechanisms that may account for some differences in the clinical presentation of LQT2 and LQT3.
Keywords: Long QT syndrome, early afterdepolarizations, E-4031, ATX II, fast Fourier transforms, rabbit ventricular myocytes
Introduction
In the congenital and acquired forms of the long QT syndrome (LQT) two ion channel targets for modification are the rapidly activating delayed rectifier K+ channel current (IKr) and the Na+ channel current (INa). Drug suppression of IKr, which has been used to model chromosome 7-linked congenital LQT (LQT2), is known to cause action potential prolongation and the induction of early afterdepolarizations (EADs) arising from plateau voltages, along with QT interval prolongation and the arrhythmia torsades de pointes (Roden, 1993; Colatsky & Argentieri, 1994; Zhou et al., 1995; Vorperian et al., 1996; January & Zhou, 1997; Shimizu & Antzelevitch, 2000). Similarly, modification of INa with sea anemone toxins such as ATX II, which has been used to model chromosome 3-linked congenital LQT (LQT3), slows the rate of channel inactivation to produce action potential prolongation and plateau EADs, QT interval prolongation and torsades de pointes (El-Sherif et al., 1988; Boutjdir & El-Sherif, 1991; Shimizu & Antzelevitch, 2000). In congenital LQT, mutations of these ion channels have been associated with phenotypically distinct electrocardiographic patterns (Moss et al., 1995) and with different clinical prognoses (Zareba et al., 1998). These findings suggest that the cellular electrophysiological properties associated with different ion channel gene defects or channel interventions may differ.
In the present work, we studied two pharmacological models of LQT in isolated rabbit ventricular myocytes to identify differences in underlying cellular electrophysiological properties. The isolated rabbit heart is an established model for studying mechanisms of torsades de pointes (D'Alonzo et al., 1997; Zabel et al., 1997; Eckhardt et al., 1999). In rabbit ventricular myocytes the delayed rectifier K+ channel current is thought to represent mostly IKr (Carmeliet, 1992; Clay et al., 1995; Zhou et al., 1995). We used the class III methanesulphonanilide antiarrhythmic drug E-4031 to block IKr and simulate LQT2, and the sea anemone toxin ATX II to delay Na+ channel inactivation and simulate LQT3. We studied action potential and EAD properties using the perforated patch clamp technique to avoid intracellular dialysis and ion channel current rundown that occurs with the conventional ruptured patch technique. We also employed fast Fourier transforms (FFTs) to rigorously define the EAD frequency power spectra and to separate EADs from irregular low amplitude membrane voltage oscillations found in the ATX II model. Our data show that these two LQT models produce strikingly different effects on action potential and EAD electrophysiological properties.
Preliminary reports of this work have appeared (Studenik et al., 1996; 1999).
Methods
Single rabbit ventricular cell isolation procedure
Rabbit ventricular cells were isolated by enzymatic dispersion (Verperian et al., 1996). Briefly, the rabbits were anaesthetized with Ketamine HCl (80 – 100 mg kg−1), Xylazine HCl (3 – 5 mg kg−1) and sodium pentobarbital (50 mg kg−1). Hearts were excised and perfused with normal Tyrode's solution for 4 min, nominally Ca2+-free Tyrode's solution for 5 min, and Tyrode's solution containing albumin (1 mg ml−1), protease (0.1 mg ml−1) and collagenase (0.6 mg ml−1) for 8 min. After enzyme perfusion the right and left ventricles were cut into small pieces and incubated in fresh enzyme solution containing collagenase (1 mg ml−1) and protease (0.1 mg ml−1) for 10 min at 37°C while being agitated in a shaking water bath. Regional differences in cellular electrophysiological properties were not studied. The isolated myocytes were stored in a solution containing (mM): K-glutamate 130, MgCl2 5.7, EGTA 0.1, and HEPES 10.
Perforated patch-clamp recording technique
Membrane voltage was recorded in whole cell current clamp configuration using the amphotericin B perforated patch method (Rae & Cooper, 1990; Zhou et al., 1995; Vorperian et al., 1996; Zhou & January, 1998). Amphotericin forms monovalent selective (cations>anions) channels in the cell membrane that provide electrical access to the cell interior. Amphotericin B (60 mg ml−1) was dissolved in DMSO and then added to internal pipette solution at final concentration of 240 μg ml−1. The internal pipette solution contained (in mM): K-glutamate 100, KCl 40, NaCl 0 or 5, EGTA 1, HEPES 10 (pH titrated to 7.2 with KOH). With the amphotericin B method, the access resistances usually were 4 – 10 MΩ. The external solution contained (in mM): NaCl 137, KCl 4.0, CaCl2 1.8, MgCl2 1.0, glucose 10, HEPES 10 (pH titrated to 7.4 with NaOH). A Dagan 3900 patch clamp amplifier was used to record action potentials which were elicited by applying 1 ms long depolarizing current pulses through the patch electrode at 0.5 Hz. All experiments were performed at 36±1°C.
Data analysis
Computer software (pCLAMP, version 6.02, Axon Instruments) was used to generate recording protocols. Data were digitized at 1 kHz and were stored for later analysis. Fast Fourier transforms (Bergland, 1969) were performed to analyse the repolarization phase of the action potential. The FFT method converts the time-domain into the frequency-domain and plots signal power (I2) versus frequency content (Hz).
Drugs and toxins
E-4031 was obtained from Eisai Ltd (Ibaraki, Japan) and was dissolved in distilled water to give a stock solution of 1 mM. ATX II was obtained from Sigma Chemicals (St. Louis, MO, U.S.A.) and was dissolved in distilled water to give a stock solution of 100 μM. In preliminary experiments, we identified the maximal drug and toxin concentrations that caused action potential prolongation but did not cause failure of the myocytes to repolarize to the resting potential. These drug concentrations (20 nM for E-4031 and 30 nM for ATX-II) were used in all subsequent experiments (n=43 myocytes for E-4031, n=50 myocytes for ATX II). Drug concentrations exceeding these (⩾30 nM E-4031 and 40 nM ATX II) frequently caused failure of rapid repolarization with the membrane voltage remaining at the plateau range. Ryanodine, thapsigargin and tetrodotoxin (TTX) were obtained from Sigma Chemicals (St. Louis, MO, U.S.A.).
Statistical methods
Where appropriate, data are given as the mean±standard deviation. Statistical significance was tested using a paired or Student's t-test.
Results
The effect of E-4031 on rabbit ventricular cell action potentials is shown in the Figure 1a. Initially it caused the control action potential (left trace) to prolong (middle trace) which was followed by the development of an EAD (right trace, arrow, take-off potential of −19 mV) that preceded repolarization. EADs in the E-4031 model were easily distinguished as secondary depolarizations at action potential plateau voltages. In this myocyte the control resting potential was −84 mV and action potential amplitude was 130 mV which did not change after drug exposure. The effect of ATX II on rabbit ventricular cell action potentials in a different myocyte is shown in Figure 1b. ATX II initially caused the control action potential (left trace) to markedly prolong with the simultaneous appearance of irregular low amplitude membrane voltage oscillations (middle trace, see also El-Sherif et al., 1990). ATX II consistently induced these irregular low amplitude membrane voltage oscillations during the plateau in all myocytes studied (n=50), whereas this type of activity was never observed in myocytes exposed to E-4031 (n=43). In some myocytes, ATX II exposure was followed by the development of a small amplitude EAD (right trace, arrow, take-off potential of −9 mV) which preceded repolarization. In this myocyte the control resting potential was −83 mV and action potential amplitude was 128 mV which did not change with toxin exposure.
Figure 1.
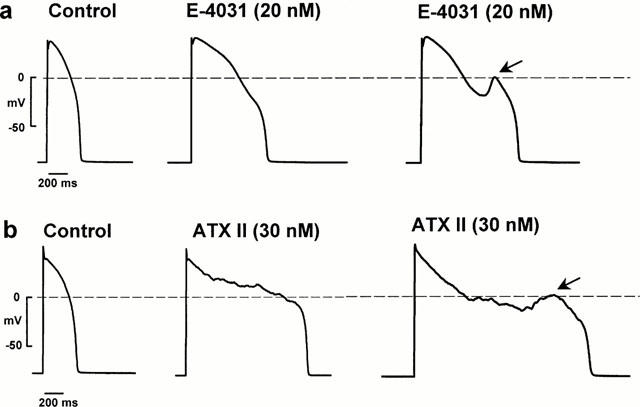
Effects of E-4031 and ATX II on rabbit ventricular action potentials recorded using the perforated patch method. (a) Shows the effect of E-4031 and (b) shows the effect of ATX II. Arrows indicate EADs.
With the ATX II model, the irregular low amplitude membrane voltage oscillations could be difficult to distinguish from EADs. To develop quantitative criteria for the presence of EADs, we used FFTs to analyse the frequency power spectra of all myocytes exposed to E-4031 and ATX II. For calculation of the FFT, each recorded action potential data set was truncated from the peak voltage of the action potential normally to −30 mV of repolarization. This region contains the action potential plateau and EADs, and in the ATX II model it also contained the irregular low amplitude membrane voltage oscillations. The resting potential, action potential upstroke and rapid repolarization phase negative to −30 mV were excluded from analysis since the EADs we studied do not arise during these portions of the action potential, and contamination of FFTs by the frequency content of these signals was avoided. To perform the FFT, each data set was fit with a nonlinear regression line. These were subtracted to give a difference signal to minimize frequency interference, and the frequency content was analysed. Figure 2 shows an individual FFT power spectrum record obtained following ATX II exposure in a myocyte, with the truncated action potential record and nonlinear regression fit to it shown in the inset. The FFT contained a discrete power peak at around 5 Hz.
Figure 2.
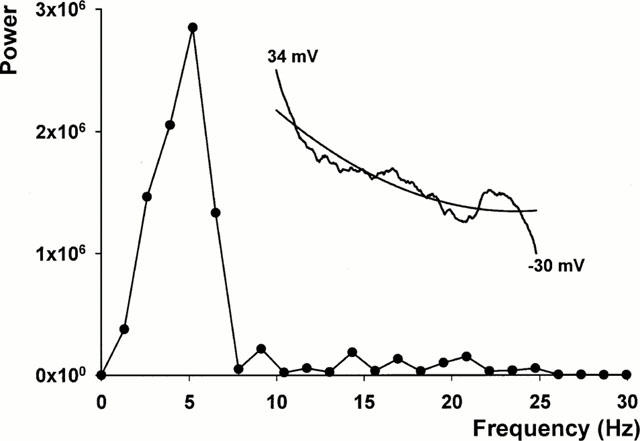
Fast Fourier transform analysis of the frequency power spectrum of an action potential plateau in an ATX II treated myocyte. The FFT shows a discrete power peak at about 5 Hz along with low amplitude signals at higher frequencies. The inset shows the original truncated action potential recording from its peak voltage of 34 to −30 mV and the nonlinear regression fit. The truncated action potential record contains irregular low amplitude membrane voltage oscillations and an EAD.
Figure 3 shows averaged FFTs for the E-4031 and ATX II treated myocytes. Of 43 myocytes exposed to E-4031, 36 (84%) developed EADs (see example truncated action potential record in the inset in Figure 3a). The averaged FFT for these myocytes shows a discrete peak in the frequency power spectrum at about 5 Hz. In every E-4031 treated myocyte that developed EADs, the power spectrum peak amplitude exceeded 2.2×106 units. In the myocytes (n=7) not generating EADs, there was no discrete peak in the FFT and the power spectrum maximum amplitude was less than 0.5×106 units (data not shown). For ATX II treated myocytes, EAD activity (see truncated action potential record in the inset in Figure 3b) was observed in 14 of 50 myocytes (28%). The individual FFTs for each myocyte with EADs showed a discrete peak in the frequency power spectrum between 4 – 5 Hz and the peak power amplitude exceeded 2.8×106 units. The averaged FFT for these myocytes is shown in Figure 3b and it contains a discrete peak in the frequency power spectrum at about 5 Hz, similar to that found in myocytes with E-4031 induced EADs. The data from these 14 myocytes were then re-analysed after truncating the repolarization record of each action potential to before the onset of the EAD (see truncated action potential record in the inset in Figure 3c, same record as used in inset in Figure 3b), thus eliminating the EAD from the reanalysis. The resulting averaged FFT is plotted in Figure 3c, and shows a frequency power spectrum with no discrete power peak and a low power amplitude. Finally, we analysed the remaining 36 myocytes treated with ATX II that developed irregular low amplitude membrane voltage oscillations but no EAD activity. The averaged FFT is plotted in Figure 3d with the inset showing an example truncated action potential record. The FFT shows a low amplitude frequency power spectrum and it lacked a discrete peak at about 5 Hz, similar to that shown in Figure 3c. We conclude from these data that the presence of a discrete peak in the FFT power spectrum between 4 – 5 Hz with a peak amplitude exceeding 2.2×106 units represents power added from EADs occurring during the terminal portion of the action potential plateau. Particularly for the ATX II model, it provides a quantitative approach for identifying and separating EADs from ATX II induced irregular low amplitude membrane voltage oscillations which lack these properties.
Figure 3.
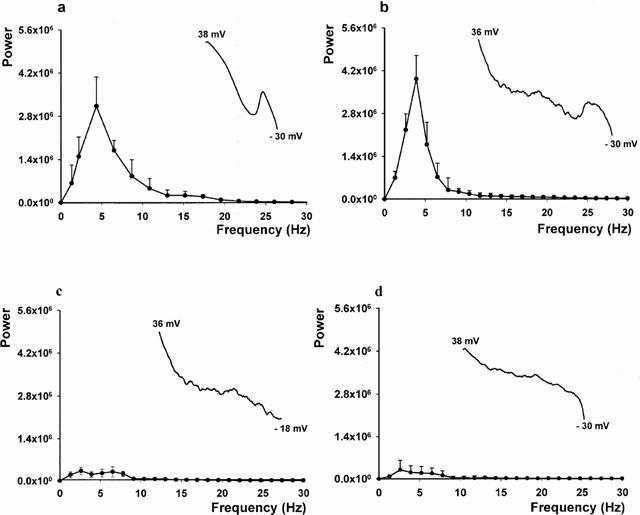
Averaged frequency power spectra of myocytes exposed to E-4031 and ATX II. (a) Shows the FFTs of 36 myocytes exposed to E-4031 that developed EADs. (b) Shows the FFTs from 14 myocytes exposed to ATX II that developed EADs. (c) Shows the FFTs of the same 14 myocytes after truncating the repolarization record of each action potential before the onset of EADs. (d) Shows the FFTs of 36 myocytes treated with ATX II that developed irregular low amplitude membrane voltage oscillations but without EAD activity. The inset in each panel shows a representative action potential record truncated from the peak action potential voltage to −30 mV (a,b,d) or to the EAD take-off potential (c).
Using the FFT approach to confirm the presence of EADs, we then analysed properties of the initial action potential that contained an EAD following exposure to E-4031 or ATX II. These data are summarized in Table 1. The data show that the resting potential and action potential amplitudes were similar for control conditions and after the development of EADs in both the E-4031 (n=36 myocytes) and ATX II (n=14 myocytes) groups. However, for E-4031 treated cells compared to ATX II treated cells, the EAD take-off potential was more negative, EAD amplitude was larger, EAD peak voltage was reached earlier, and the EAD duration was less. The action potential plateau voltages also differed between the two models as previously shown in Figure 1. Figure 4a shows averaged data for control action potentials and the last action potential before the development of an EAD in the same groups of myocytes (E-4031, n=36, and ATX II, n=14). Action potential plateau voltages were measured at 100 ms increments following the action potential upstroke. For the two groups of myocytes, the control action potential voltages at 100, 200 and 300 ms following the upstroke were similar (P>0.05 at each time point), and action potentials had repolarized by 400 ms. Following exposure to E-4031 or ATX II, the action potentials prolonged prior to developing EADs, however, the plateau voltages and durations preceding the onset of repolarization were different as shown in Figure 4b. For an example, following E-4031 exposure the plateau voltage was −18.9±4.1 mV at an action potential duration of 600 ms (n=36 myocytes). Following this, the action potentials began to rapidly repolarize. For ATX II the action potential plateau voltage was more positive and the action potential durations were longer. For example, the plateau voltage at an action potential duration of 600 ms (n=14 myocytes) was 7.8±3.2 mV, and it reached −10.1±3.7 mV at an action potential duration of 1200 ms (n=14 myocytes). Following this, the action potentials began to rapidly repolarize.
Table 1.
Resting and action potential amplitudes and EAD parameters in control and after addition of E-4031 and ATX II

Figure 4.
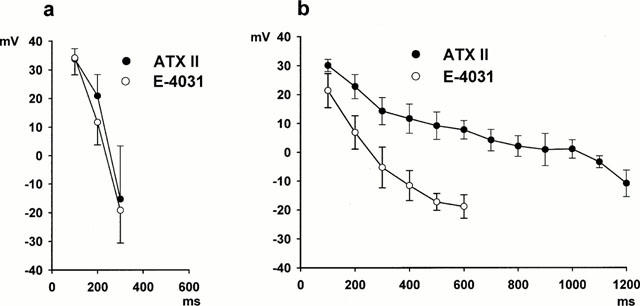
Averaged action potential plateau voltages measured at 100 ms increments following the action potential upstroke. (a) Shows data for the control action potentials recorded before exposure to E-4031 (n=36 myocytes) or ATX II (n=14 myocytes). For control conditions repolarization was complete by 400 ms. (b) Shows data for the same myocytes for action potentials prolonged by exposure to E-4031 (n=36 myocytes) and ATX II (n=14 myocytes) just prior to the appearance of EADs.
A possibility is that the marked action potential prolongation at more positive voltages found with ATX II could cause an increase in intracellular Na+ and Ca2+. In turn, this could lead to activation of Ca2+ -dependent currents which might cause the irregular low amplitude membrane voltage oscillations and EADs. To test for this, the sarcoplasmic reticulum modulator ryanodine (5 μM) and the sarcoplasmic reticulum Ca2+ release inhibitor thapsigargin (1 μM) were added to the bath of five cells with ATX II-induced EADs. Their addition rapidly abolished cell shortening, but did not antagonize the ATX II effects on the action potentials as shown in Figure 5. Similar findings were obtained in the four additional cells studied. In contrast, the Na+ channel blocker TTX (0.5 μM) shortened action potential duration and suppressed the irregular low amplitude membrane voltage oscillations (Figure 6) with this finding confirmed in four cells.
Figure 5.
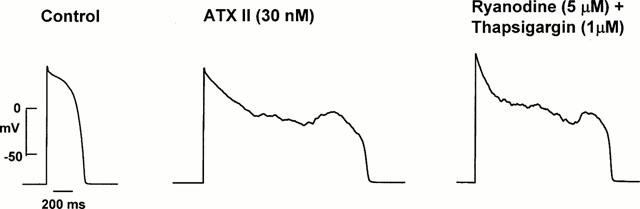
Effect of ryanodine (5 μM) and thapsigargin (1 μM) on the action potential and an EAD. The left panel shows the control action potential. The middle panel shows action potential prolongation along with irregular low amplitude membrane voltage oscillations and an EAD after exposure to ATX II (30 nM). The right panel shows that ryanodine and thapsigargin did not suppress the ATX II-induced activity. Data from the same cell.
Figure 6.
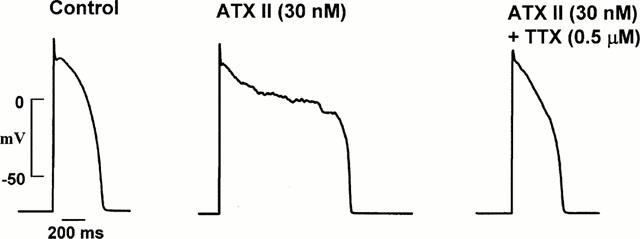
Effect of TTX (0.5 μM) on the action potential. The left panel shows the control action potential. The middle panel shows ATX II-induced action potential prolongation accompanied by irregular low amplitude membrane voltage oscillations and an EAD. The addition of TTX shortened action potential duration and suppressed the irregular low amplitude membrane voltage oscillations as shown in the right panel. Data from the same cell.
Discussion
This is the first report comparing the E-4031 and ATX II LQT models studied in single myocytes. The most significant finding of this work is that the E-4031 and ATX II models produced strikingly different effects on action potential and EAD properties. The major differences are that ATX II, compared with E-4031, caused: (1) greater action potential prolongation; (2) more positive plateau voltages; (3) lower amplitude EADs with less negative take-off potentials; (4) greater time to the EAD peak voltage; and (5) longer duration EADs. Despite causing greater action potential prolongation, the frequency of EAD induction was much less with the ATX II model (28%) than with the E-4031 model (84%).
These experiments used a perforated patch clamp technique which is likely to be important in our findings. This patch clamp method avoids intracellular dialysis with pipette contents (such as Ca2+ buffers), and the diffusion of molecules and soluble proteins out of cells. In turn, this minimizes the rundown of several ion channel currents, drug sensitivity is increased, and changes in cell Ca2+ and cell contraction are maintained (see Zhou & January, 1998 for discussion). Since L-type Ca2+ channels are postulated to serve as the charge carrier for plateau EADs (January & Riddle, 1989; Zeng & Rudy, 1995), its preservation particularly important when studying EAD models. This may help to explain the infrequent reports of EAD properties studied using the conventional ruptured patch method.
E-4031 is a methanesulphonanilide antiarrhythmic drug that selectively blocks IKr and the human-ether-a-go-go-related gene (HERG) channel that encodes the pore-forming subunit (Sanguinetti & Jurkiewicz, 1990; Sanguinetti et al., 1995; Trudeau et al., 1995). The drug concentration used in these experiments reduced IKr tail current amplitude in rabbit ventricular cells by about 50% (Vorperian et al., 1996). These channels generate their maximal repolarizing current at voltages near the action potential plateau (Zhou et al., 1998), and their suppression with drugs such as E-4031 is a common mechanism for prolonging action potential duration and for initiating plateau EADs (Zhou et al., 1994). In contrast, ATX II binds to the Na+ channel to slow inactivation from the open state resulting in prolonged channel bursting. This generates a maintained or slowly decaying inward Na+ current present over a broad range of voltages (Lazdunski et al., 1980; Lawrence & Catterall, 1981; Warashina & Fujita, 1983; Isenberg & Ravens, 1984), which may account for ATX II induced action potential prolongation at more positive plateau voltages. The action potential prolongation by ATX II was uniformly accompanied by irregular low amplitude membrane voltage oscillations. This activity was previously reported to represent EADs (El-Sherif et al., 1990; Boutjdir et al., 1994). Our findings using FFTs show that this activity lacked the frequency power spectrum pattern typical of plateau EADs. This activity also occurred at voltages positive to the recovery range of L-type Ca2+ window current (above 0 mV) needed for EAD depolarization (January & Riddle, 1989; Hirano et al., 1992; Zeng & Rudy, 1995). The irregular low amplitude membrane voltage oscillations were not suppressed by ryanodine and thapsigargin, suggesting that Ca2+ overload and activation of Ca2+ -dependent currents was not essential. It was suppressed, however, by TTX which is consistent with a central role for Na+ channels. We conclude that the irregular low amplitude membrane voltage oscillations do not represent plateau EADs, rather they are likely to arise from current flowing with ATX II induced Na+ channel bursting during the action potential plateau. Our findings also provide evidence that the voltage of the action potential plateau, and not just its duration, is important in regulating EAD generation, and suggests that the QT interval on an ECG may not be the ideal measurement to define risks for arrhythmia provocation in LQT models.
Clinical implications and limitations
Extrapolation of our cellular electrophysiological data to the clinical syndrome is limited by incomplete understanding of LQT, in part due to the relatively small number of patients reported (particularly for LQT3), and because the phenotype of specific mutations within a gene defect may not be identical. None-the-less, our results may provide insight into previous clinical and experimental observations. In congenital LQT, the longest QT intervals usually are found in patients with LQT3 (Zareba et al., 1998; Shimizu & Antzelevitch, 1999). These patients, however, have a lower frequency and cumulative probability of cardiac events when compared to patients with K+ channel mutations. Our findings provide a potential mechanism in that EADs occurred less frequently in the ATX II model, presumably because of the more positive plateau voltages that preceded repolarization. EADs are thought to be a critical cellular trigger for the initiation of torsades de pointes with the arrhythmia maintained by a re-entrant mechanism that depends on cellular and transmural differences in repolarization properties (El-Sherif et al., 1996; 1997; Antzelevitch et al., 1996). Although the cumulative probability of a cardiac event is lower in LQT3 patients, it also has been suggested that the risk of death during a cardiac event is higher in these patients (Zareba et al., 1998). Our data show that EADs in the ATX II model occurred later in the action potential than those found with the E-4031 model. Because gradients of dispersion of repolarization are increased with increasing QT interval (Shimizu & Antzelevitch, 1999), the initiation of an EAD at a longer QT interval in LQT3 potentially could produce a more favourable substrate for a sustained arrhythmia and sudden death. Thus, differences in the cellular electrophysiological properties may account for differences in action potential prolongation and the induction of EADs, which may contribute to the clinical presentation of LQT.
The use of pharmacological models as surrogates for congenital models of LQT has been widely used (Shimizu & Antzelevitch, 1999; Shimizu et al., 1999; Burashnikov & Antzelevitch, 2000) but must be done with caution. Pharmacological models of LQT are likely to differ from congenital LQT in several ways: (1) most drugs bind and unbind to ion channels with voltage- and frequency-dependent properties; (2) kinetic differences and voltage shifts found with some LQT mutant channels are not mimicked by drugs; and (3) drugs do not mimic intracellular protein processing abnormalities found with some LQT mutant channels. Thus multiple phenotypes are likely to exist that can not be mimicked by a single drug. Pharmacological models, however, provide qualitative similarities to the respective clinical syndromes and remain essential tools in enhancing knowledge of the cellular electrophysiological properties.
Acknowledgments
We gratefully acknowledge and thank the late Stephen H. Nellis, Ph.D. for expert assistance with the FFT analysis. We also thank Timothy J. Kamp, M.D., Ph.D. for reviewing the manuscript. This work was supported in part by HL60723 and the Oscar Rennebohm Foundation. Dr Zhou is a recipient of an American Heart Association Scientist Development Grant.
Abbreviations
- EAD
early afterdepolarization
- FFT
fast Fourier transform
- HERG
human-ether-a-go-go-related gene
- IKr
rapidly activating delayed rectifier potassium current
- INa
sodium current
- LQT
long QT syndrome
References
- ANTZELEVITCH C., SUN Z.Q., ZHANG Z.Q., YAN G.X. Cellular and ionic mechanisms underlying erythromycin-induced long QT intervals and torsade de pointes. J. Am. Coll. Cardiol. 1996;28:1836–1848. doi: 10.1016/S0735-1097(96)00377-4. [DOI] [PubMed] [Google Scholar]
- BERGLAND G.D. A guided tour of the fast Fourier transform. IEEE Spectrum. 1969;2:41–52. [Google Scholar]
- BOUTJDIR M., EL-SHERIF N. Pharmacological evaluation of early afterdepolarizations induced by sea anemone toxin (ATX II) in dog heart. Cardiovasc. Res. 1991;25:815–819. doi: 10.1093/cvr/25.10.815. [DOI] [PubMed] [Google Scholar]
- BOUTJDIR M., RESTIVO M., WEI Y., STERGIOPOULOS K., EL-SHERIF N. Early afterdepolarization formation in cardiac myocytes: analysis of phase plane patterns, action potential, and membrane currents. J. Cardiovasc. Electrophysiol. 1994;5:609–620. doi: 10.1111/j.1540-8167.1994.tb01302.x. [DOI] [PubMed] [Google Scholar]
- BURASHNIKOV A., ANTZELEVITCH C. Block of I(Ks) does not induce early afterdepolarization activity but promotes beta-adrenergic agonist-induced delayed afterdepolarization activity. J. Cardiovasc. Electrophysiol. 2000;11:458–465. doi: 10.1111/j.1540-8167.2000.tb00342.x. [DOI] [PubMed] [Google Scholar]
- CARMELIET E. Voltage- and time-dependent block of the delayed K+ current in cardiac myocytes by dofetilide. J. Pharmacol. Exp. Ther. 1992;262:809–817. [PubMed] [Google Scholar]
- CLAY J.R., OGBAGHEBRIEL A., PASQUETTA T., SASYNIUK B.I., SHRIER A. A quantitative description of the E-4031-sensitive repolarization current in rabbit ventricular myocytes. Biophys. J. 1995;69:1830–1837. doi: 10.1016/S0006-3495(95)80053-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COLATSKY T.J., ARGENTIERI T.M. Potassium channel blockers as antiarrhythmic drugs. Drug Dev. Res. 1994;33:235–249. [Google Scholar]
- D'ALONZO A.J., ZHU J.L., DARBRENZIO R.B. Effects of class III antiarrhythmic agents in an in vitro rabbit model of spontaneous torsades de pointes. Eur. J. Pharmacol. 1997;369:57–64. doi: 10.1016/s0014-2999(99)00057-6. [DOI] [PubMed] [Google Scholar]
- ECKHARDT L., HAVERKAMP W., BORGGREFE M., BREITHARDT G. Experimental models of torsades de pointes. Cardiovasc. Res. 1999;39:178–193. doi: 10.1016/s0008-6363(98)00043-1. [DOI] [PubMed] [Google Scholar]
- EL-SHERIF N., CAREF E.B., YIN H., RESTIVO M. The electrophysiological mechanism of ventricular arrhythmias in the long QT syndrome. Tridimensional mapping of activation and recovery patterns. Circ Res. 1996;79:474–492. doi: 10.1161/01.res.79.3.474. [DOI] [PubMed] [Google Scholar]
- EL-SHERIF N., CHINUSHI M., CAREF E.B., RESTIVO M. Electrophysiological mechanism of the characteristic electrocardiographic morphology of torsade de pointes tachyarrhythmias in the long-QT syndrome: detailed analysis of ventricular tridimensional activation patterns. Circulation. 1997;96:4392–4399. doi: 10.1161/01.cir.96.12.4392. [DOI] [PubMed] [Google Scholar]
- EL-SHERIF N., CRAELIUS W., BOUTJDIR M., GOUGH B. Early afterdepolarizations and arrhythmogensis. J. Cardiovasc. Electrophysiol. 1990;2:145–158. [Google Scholar]
- EL-SHERIF N., ZEILER R.H., CRAELIUS W., GOUGH W.B., HENKIN R. QTU prolongation and polymorphic ventricular tachyarrhythmias due to bradycardia-dependent early afterdepolarizations. Afterdepolarizations and ventricular arrhythmias. Circ. Res. 1988;63:286–305. doi: 10.1161/01.res.63.2.286. [DOI] [PubMed] [Google Scholar]
- HIRANO Y., MOSCUCCI A., JANUARY C.T. Direct measurement of L-type Ca2+ window current in heart cells. Circ. Res. 1992;70:445–455. doi: 10.1161/01.res.70.3.445. [DOI] [PubMed] [Google Scholar]
- ISENBERG G., RAVENS U. The effects of the Anemone sulcata toxin (ATX II) on membrane currents of isolated mammalian myocytes. J. Physiol. 1984;357:127–149. doi: 10.1113/jphysiol.1984.sp015493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JANUARY C.T., RIDDLE J.M. Early afterdepolarizations: Mechanism of induction and block. A role for L-type Ca2+ current. Circ. Res. 1989;64:977–990. doi: 10.1161/01.res.64.5.977. [DOI] [PubMed] [Google Scholar]
- JANUARY C.T., ZHOU Z.Early afterdepolarizations: Mechanisms and possible role in arrhythmias Electrocardiology ‘96: From the Cell to the Body Surface 1997Singapore: World Scientific Inc; 231–240.ed. Liebman, J. pp [Google Scholar]
- LAWRENCE J.C., CATTERALL W.A. Tetrodotoxin-insensitive sodium channels. Binding of polypeptide neurotoxins in primary cultures of rat muscle cells. Biol. Chem. J. 1981;256:6223–6229. [PubMed] [Google Scholar]
- LAZDUNSKI M., BALERNA M., BARHANIN J., CHICHEPORTICHE R., FOSSET M., FRELIN C., JACQUES Y., LOMBET A., POUYSSEGUR J., RENAUD J.F., ROMEY G., SCHWEITZ H., VINCENT J.P. Molecular aspects of the structure and mechanism of the voltage-dependent sodium channel. Ann. NY Acad. Sci. 1980;358:169–182. doi: 10.1111/j.1749-6632.1980.tb15395.x. [DOI] [PubMed] [Google Scholar]
- MOSS A.J., ZAREBA W., BENHORIN J., LOCATI E.H., HALL W.J., ROBINSON R.J., SCHWARTZ P.J., TOWBIN J.A., VINCENT G.M., LEHMANN M.H. ECG T-wave patterns in genetically distinct forms of the hereditary long QT syndrome. Circulation. 1995;92:2929–2934. doi: 10.1161/01.cir.92.10.2929. [DOI] [PubMed] [Google Scholar]
- RAE J.L., COOPER K. New techniques for the study of lens electrophysiology. Exp. Eye Res. 1990;50:603–614. doi: 10.1016/0014-4835(90)90101-y. [DOI] [PubMed] [Google Scholar]
- RODEN D.M. Current status of class III antiarrhythmic drug therapy. Am. J. Cardiol. 1993;72:44B–49B. doi: 10.1016/0002-9149(93)90040-j. [DOI] [PubMed] [Google Scholar]
- SANGUINETTI M.C., JIANG C., CURRAN M.E., KEATING M.T. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- SANGUINETTI M.C., JURKIEWICZ N.K. Two components of cardiac delayed rectifier K+ current. Differential sensitivity to block by class III antiarrhythmic agents. J. Gen. Physiol. 1990;96:195–215. doi: 10.1085/jgp.96.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIMIZU W., ANTZELEVITCH C. Differential effects of β-adrenergic agonists and antagonists in LQT1, LQT2 and LQT3 models of the long QT Syndrome. J. Am. Coll. Cardiol. 2000;35:778–786. doi: 10.1016/s0735-1097(99)00582-3. [DOI] [PubMed] [Google Scholar]
- SHIMIZU W., MCMAHON B., ANTZELEVITCH C. Sodium pentobarbital reduces transmural dispersion of repolarization and prevents torsades de pointes in models of acquired and congenital long QT syndrome. J. Cardiovasc. Electrophysiol. 1999;10:154–164. doi: 10.1111/j.1540-8167.1999.tb00656.x. [DOI] [PubMed] [Google Scholar]
- STUDENIK C., ZHOU Z., JANUARY C.T. Pharmacological models of long QT syndromes: Implications for drug therapy and ion channel defects (abstract) Circulation. 1996;94:I–642. [Google Scholar]
- STUDENIK C., ZHOU Z., JANUARY C.T. Differences in cellular electrophysiological and early afterdepolarization properties in two pharmacological models of LQTS (abstract) Biophys. J. 1999;76:A366. [Google Scholar]
- TRUDEAU M.C., WARMKE J.W., GANETZKY B., ROBERTSON G.A. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science. 1995;269:92–95. doi: 10.1126/science.7604285. [DOI] [PubMed] [Google Scholar]
- VORPERIAN V.R., ZHOU Z., MOHAMMAD S., HOON T.J., STUDENIK C., JANUARY C.T. Torsades de pointes with an antihistamine metabolite: Potassium channel block with desmethylastemizole. J. Am. Coll. Cardiol. 1996;28:1556–1561. doi: 10.1016/s0735-1097(96)00352-x. [DOI] [PubMed] [Google Scholar]
- WARASHINA A., FUJITA S. Effect of sea anemone toxins on the sodium inactivation process in crayfish axons. J. Gen. Physiol. 1983;81:305–323. doi: 10.1085/jgp.81.3.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZABEL M., HOHNLOSER S.H., BEHRENS S., LI Y.G., WOOSLEY R.L., FRANZ M.R. Electrophysiologic features of torsades de pointes: Insights from a new isolated rabbit heart model. J. Cardiovasc. Electrophysiol. 1997;8:1148–1158. doi: 10.1111/j.1540-8167.1997.tb01001.x. [DOI] [PubMed] [Google Scholar]
- ZAREBA W., MOSS A.J., SCHWARTZ P.J., VINCENT G.M., ROBINSON J.L., PRIORI S.G., BENHORIN J., LOCATI E.H., TOWBIN J.A., KEATING M.T., LEHMANN M.H., HALL W.J. Influence of genotype on the clinical course of the long-QT syndrome. International Long-QT Syndrome Registry Research Group. New Engl. J. Med. 1998;339:960–965. doi: 10.1056/NEJM199810013391404. [DOI] [PubMed] [Google Scholar]
- ZENG J., RUDY Y. Early afterdepolarizations in cardiac myocytes: mechanism and rate dependence. Biophys. J. 1995;68:949–964. doi: 10.1016/S0006-3495(95)80271-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHOU Z., GONG Q., YE B., FAN Z., MAKIELSKI J.C., ROBERTSON G.A., JANUARY C.T. Properties of HERG channels stably expressed in HEK 293 cells studied at physiological temperature. Biophys. J. 1998;74:230–241. doi: 10.1016/S0006-3495(98)77782-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHOU Z., JANUARY C.T. Both T- and L-type Ca2+ channels can contribute to excitation-contraction coupling in cardiac Purkinje cells. Biophys. J. 1998;74:1830–1839. doi: 10.1016/S0006-3495(98)77893-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHOU Z., STUDENIK C., JANUARY C.T. Mechanisms of early afterdepolarizations induced by block of IKr (abstract) Circulation. 1994;92:I–435. [Google Scholar]
- ZHOU Z., STUDENIK C., JANUARY C.T.Properties of E-4031-induced early afterdepolarizations in rabbit ventricular myocytes: Studies using a perforated patch method Potassium Channels in Normal and Pathological Conditions 1995Leuven: Leuven University Press; 375–379.eds. Vereecke, J., van Bogaert, P.P. & Verdonck, F.P. pp [Google Scholar]