

Abstract
Peptidergic neurones accumulate amines via an unusual uptake process, designated Transport-P. [3H]-prazosin binds to α1 adrenoceptors on these cells and is displaceable by unlabelled prazosin in concentrations up to 10−7 M. However, at greater concentrations of prazosin, there is a paradoxical accumulation of [3H]-prazosin which we have attributed to Transport-P. Uptake of prazosin via Transport-P is detectable at 10−10 M prazosin concentration, is linear up to 10−7 M and at greater concentrations becomes non-linear. In contrast, in noradrenergic neurones, noradrenaline uptake is linear and saturates above 10−7 M. In noradrenergic neurones and in non-neuronal cells, there is no uptake of prazosin in concentrations up to 10−6 M, suggesting that Transport-P is a specialised function of peptidergic neurones.
Using a mouse peptidergic (gonadotrophin-releasing hormone, GnRH) neuronal cell line which possesses Transport-P, we have studied the interaction of α1 adrenoceptors with Transport-P. Polymerase chain reactions and DNA sequencing of the products demonstrated that only the α1B sub-type of adrenoceptors is present in GnRH cells.
In COS cells transfected with α1b adrenoceptor cDNA and in DDT1 MF-2 cells which express native α1B adrenoceptors, [3H]-prazosin was displaced by unlabelled prazosin in a normal equilibrium process, with no prazosin paradox in concentrations up to 10−6 M. In DDT1 MF-2 cells, [3H]-prazosin was displaced likewise by a series of α1 adrenergic agonists, none of which increased the binding of [3H]-prazosin. Hence, the prazosin paradox is not due to some function of α1 adrenoceptors, such as internalization of ligand-receptor complexes.
In neurones which possess Transport-P, transfection with α1b adrenoceptor cDNA resulted in over-expression of α1B adrenoceptors, but the prazosin paradox was unaltered. Thus, α1 adrenoceptors and Transport-P mediate distinct functions in peptidergic neurones.
Keywords: Adrenergic receptors, alpha; biological transport; hypothalamus; prazosin
Introduction
Classically, a radiolabelled ligand binds to its receptor and can be displaced by added unlabelled ligand in a simple equilibrium process. When the α1 adrenergic ligand [3H]-prazosin binds to peptidergic neurones it is displaceable by unlabelled prazosin in concentrations up to 10−7 M. However, at greater concentrations of unlabelled prazosin, there is a paradoxical increase in the binding of [3H]-prazosin (Al-Damluji & Krsmanovic, 1992; Al-Damluji et al., 1993; Figure 1). The following hypothesis was formulated to explain these findings:
Figure 1.
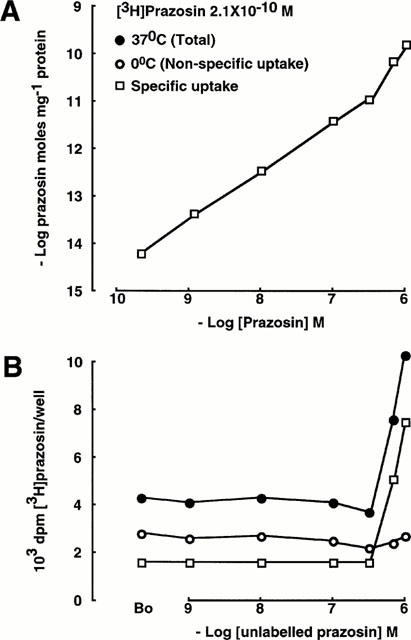
Uptake of prazosin in peptidergic neurones (GT1-1 GnRH cells). The lower panel demonstrates the uptake of radiolabelled ligand; the upper panel demonstrates uptake of total ligand (labelled and unlabelled), ie, it is corrected for the fall in specific activity of [3H]-prazosin due to isotope dilution. Non-specific uptake was defined as uptake at 0°C. In this and in subsequent Figures, standard error bars are not shown where they are smaller than the sizes of the symbols. ‘B0' is an abbreviation for binding of the radiolabelled ligand in the absence of unlabelled ligand.
(1) One component of the interaction with [3H]-prazosin is its normal equilibrium with α1 adrenoceptors on the peptidergic neurones. This equilibrium is the dominant process at lower concentrations of added unlabelled prazosin. Thus, the observed affinity at lower concentrations of prazosin for the binding sites is similar to its affinity for α1 adrenoceptors expressed in cultured cells transfected with α1 adrenoceptor cDNA (Al-Damluji & Kopin, 1996a).
(2) The paradoxical increase in [3H]-prazosin binding is due to an unusual uptake process which is activated by its ligand. This uptake process was designated Transport-P. A considerable amount of evidence supported this second part of the hypothesis. Thus: (a) the paradoxical increase in binding of [3H]-prazosin is inhibited competitively by antidepressants which are known to inhibit the pre-synaptic re-uptake of amines (Al-Damluji et al., 1993; Al-Damluji & Kopin, 1996b); (b) the paradoxical increase in accumulation of [3H]-prazosin was not seen in membrane preparations, indicating that it requires intact cells or storage organelles (Al-Damluji et al., 1993); (c) the prazosin paradox is an energy-dependent process, the source of energy being the electrochemical proton gradient which is generated by vacuolar-type ATPase (V-ATPase; Al-Damluji & Kopin, 1996a); (d) ligands for Transport-P must possess a specific chemical structure, consisting of a hydrophobic phenyl group, an alkyl side chain and a basic amine (Al-Damluji & Kopin, 1998); (e) a fluorescent analogue of prazosin accumulated in intracellular vesicles, providing visual evidence for uptake (Al-Damluji et al., 1997); (f) following accumulation by Transport-P, the amines can be released by an energy-dependent process (Shen & Al-Damluji, 1997).
We have now tested the first part of the hypothesis by examining gonadotrophin-releasing hormone (GnRH) peptidergic neurones, which express Transport-P, for the presence of RNA encoding α1 adrenoceptors. Three sub-types of α1 adrenoceptors have been defined in native tissues and by DNA cloning; the native receptors are now designated α1A, α1B and α1D and the corresponding cDNA clones are now designated α1a, α1b and α1d (Hieble et al., 1995). All three subtypes are present in the nervous system and at all three, prazosin is an antagonist with pA2 values of 9 – 10 (Lomasney et al., 1991). Hence, all three subtypes are candidates for an interaction with Transport-P.
We have tested the possibility that the prazosin paradox may be due to internalization of ligand-receptor complexes and we have examined the possible interaction of the relevant α1 adrenoceptor sub-type with Transport-P. Our findings indicate that the prazosin paradox is not a function of α1 adrenoceptors, and that Transport-P and α1 adrenoceptors mediate distinct functions in peptidergic neurones. Transport-P is likely to be encoded by an unusual transporter molecule which accumulates amines in peptidergic neurones.
Methods
Cell culture
Immortalized gonadotrophin-releasing hormone (GnRH) neurones (GT1-1 cells; Mellon et al., 1990), immortalized noradrenergic neurones (SK-N-SH neuroblastoma cells; Biedler et al., 1973), DDT1 MF-2 smooth muscle cells (Cornett & Norris, 1982), and COS-7 kidney cells (Gluzman, 1981) were cultured as previously described in detail (Al-Damluji et al., 1993). Briefly, the cells were grown in Corning 75 cm2 or 150 cm2 flasks in culture medium consisting of Dulbecco's modified Eagle's medium (DMEM) and Ham's F-12 (ratio 1 : 1) containing 10% fetal bovine serum (FBS) and sodium bicarbonate 3.7 g/l, in a humidified atmosphere containing 5% CO2 in air. Culture media were changed at 48 h intervals. When the cells reached confluence, they were dispersed in the presence of trypsin, deoxyribonuclease I (DNAseI) and ethylenediaminetetraacetic acid (EDTA) and incubated in Corning 12-well plates (2×106 cells/well) for uptake and binding studies.
Uptake and binding studies
Uptake and binding studies were performed on intact cells which were grown in 12-well plates coated with poly-D-lysine (2.5 μg cm2; Sigma P-6407; MW 70,000 – 150,000) and laminin (0.25 μg cm2; Sigma L-2020). Culture media were changed at 48 h intervals. Drugs were dissolved in buffer consisting of DMEM with 25 mM HEPES and 0.5 mM sodium ascorbate, pH 7.4. After 4 days in culture, the cells were washed twice with buffer at 25°C then incubated at 37°C or at 0°C for 60 min in the presence of [3H]-prazosin 2×10−10 M or [3H]-noradrenaline 2×10−10 M and unlabelled compounds in the indicated concentrations. At the end of the incubation period, the buffer was removed and the culture plates were placed on ice to inhibit the release of amines (Shen & Al-Damluji, 1997). The cells were then washed twice with buffer at 0°C. The buffer was then removed and the cells were solubilized with 2 ml of a warm solution of 0.1% sodium dodecyl sulphate and 0.1 M sodium hydroxide. Fifty microlitre aliquots were removed for protein assay and 10 ml of scintillation liquid was then added to the cell extract, mixed and radioactivity was measured in a scintillation spectrometer with an efficiency of 50%. Protein content was measured by the bicinchoninic acid modification of the biuret reaction (Smith et al., 1985) using albumin standards and reagents supplied by Pierce & Warriner (Chester, Cheshire, U.K.). The data are presented both as d.p.m. [3H]-prazosin or [3H]-noradrenaline, and as moles prazosin or noradrenaline (labelled and unlabelled), by accounting for the fall in specific activity of the radioligands consequent upon isotope dilution. Non-specific uptake was defined as the amount of cellular prazosin or noradrenaline accumulated at 0°C. Specific uptake was obtained by subtracting non-specific uptake from total uptake. Each experimental point was carried out in triplicate and each experiment was replicated at least once. The minimum number of estimations for each experimental point was therefore six. The data are expressed as the means±s.e.mean. Standard error bars are not shown where they are smaller than the sizes of the symbols. Half-maximal inhibitory concentrations (IC50) values were calculated from concentration-response curves. The affinity constant (KDpraz) of prazosin for α1 adrenoceptor binding sites was calculated from plots of bound vs bound/free ligand, in which the gradient=−1/KD (Scatchard, 1949). The affinities of other ligands were calculated using the equation:
where [Praz] is the concentration of [3H]-prazosin (Cheng & Prusoff, 1973).
Preparation of RNA and cDNA library from GnRH neurones
Preparation of total cellular RNA and poly(A)+ RNA was based on previously described procedures with minor modifications (Aviv & Leder, 1972; Glisin et al., 1974; Chirgwin et al., 1979; Okayama et al., 1987). Briefly, GT1-1 GnRH cells were grown in Falcon 150 mm dishes (3×107 cells/dish). After 10 days, the cells were washed with PBS then disrupted in ‘GTC solution' (5.5 M guanidinium thiocyanate, 25 mM sodium citrate and 0.5% lauryl sarcosine, pH 7.0) with 0.2 M 2-mercaptoethanol in DEPC-treated water. The viscous cell lysate was sheared with a Polytron then overlayed on a caesium TFA cushion (Pharmacia; density 1.51 g ml−1) and centrifuged at 25,000 r.p.m. for 26 h at 15°C. The pellet was redissolved in ‘GTC solution' and the RNA was precipitated with acetic acid and ethanol, then washed with 70% ethanol. Two more precipitation cycles with salt and ethanol were carried out to remove any remaining salts and the RNA was dissolved in TE buffer. RNA quality was tested by fractionation in a denaturing 1.2% agarose gel followed by visualization of ribosomal bands in u.v. light. Poly(A)+ RNA was isolated on an oligo(dT)-cellulose column (Collaborative Biomedical Products) followed by elution in a low salt buffer.
Using poly(A)+ RNA from the GnRH cells, a size-selected, directional cDNA library was constructed in the pSVSPORT1 vector, using the GIBCO BRL Superscript kit with modifications. Briefly, the first cDNA strand was synthesized using 5 μg poly(A)+ RNA, oligo(dT)-NotI primer-adapter (1 μg) and Superscript II reverse transcriptase (1000 units). The RNA was degraded and the second strand was synthesized by simultaneous addition of E. coli RNAse H, E. coli DNA polymerase and E. coli DNA ligase. A SalI/XhoI cohesive adapter was ligated to the 5′ end of the cDNA and the oligo(dT)-NotI primer-adapter at the 3′ end was digested with NotI to reveal the cohesive site. The cDNA was then fractionated in 0.8% agarose without ethidium bromide and DNA greater than 1000 bp was extracted from the gel using a Qiaex kit (Qiagen). The cDNA was then ligated to pSVSPORT1 which had been digested with NotI and SalI. The ligate was introduced into E. coli strain MC 1061 (Bio-Rad) by electroporation using a Bio-Rad GenePulser set at the following parameters: 2.5 kV; 25 uF; 200 ohms. The ratio of cDNA : plasmid in the ligation reaction was optimized to achieve the maximum number of transformants. Two million transformed bacteria were grown at 37°C in a shaking incubator for 1 h then transferred to a semi-solid medium (FMC Sea-Prep agarose 3 g in 1 litre of 2×LB medium with ampicillin 50 μg ml−1) and amplified by overnight growth in a static incubator. The semi-solid medium ensures representative amplification of the clones (Kriegler, 1991). The amplified bacteria were recovered from the semi-solid medium by centrifugation and divided into aliquots which were stored in liquid nitrogen. DNA was extracted from one of the aliquots using a Qiagen Maxiprep kit. Twenty colonies were picked at random and grown overnight in LB/ampicillin. Plasmid DNA was extracted, digested to separate the plasmid from the insert, then run in 0.8% agarose and cDNA insert sizes calculated. Analysis of 20 randomly selected clones indicated that average insert size was 1493±213 bp.
Polymerase chain reactions, cloning and DNA sequence analysis
PCR primers were designed using the MacVector sequence analysis software. The following primer pairs were derived from regions which are homologous in the sequence of the cloned alpha-1 adrenergic receptors: A1BF2 upstream primer (ATCTTGGTCATCCTGTCGGTGG) and AIBB9 downstream primer (TGAAGAAGGGGAGCCAACATAAG); A1BF1 upstream primer (GAAATGTCCAACTCCAAGGAGCTGACCCTGAG) and AIBB1 downstream primer (CCAAGGTTTTGGCTGCTTTCTTTTCCCTGG); A1BF4 upstream primer (TCACCGAAGAACCCTTCTACGC) and A1BB4 downstream primer (TGTCATCCAGAGAGTCCTTCCG); A1BF5 upstream primer (AAATGAATCCCGATCTGGACACCG) and A1BB5 downstream primer (TCAATGGAGATGGCACATAGGCTC).
The PCR template was either GnRH cell total cellular RNA or the GnRH cell cDNA library. For RNA PCR, the reverse transcription reaction consisted of 250 ng RNA, A1BB9 downstream primer (0.2 μM), rTth DNA polymerase/reverse transcriptase (Perkin Elmer; 5 units), dNTPs (200 μM each), MnCl2 (1 mM) and reverse transcription buffer (1×) to a total volume of 20 μl. Reverse transcription was carried out in the thermal cycler at 60°C for 15 min. The reaction product was then amplified by adding to the reaction tube chelating buffer (Perkin Elmer) to 0.8×, MgCl2 (2.5 mM) and A1BF2 upstream primer (0.2 μM) to a total volume of 100 μl. Cycling parameters were as follows: 95°C for 1 min then 95°C for 10 s (denaturing) and 65°C for 15 s (annealing and extension); 35 cycles, followed by final extension at 65°C for 7 min. This first PCR was followed by a second amplification using the nested primer pair A1BF1/A1BB1 (0.2 μM each primer), 10 μl of the product of the first amplification as template, PCR buffer (Perkin Elmer; 1×; MgCl2 1.5 mM), dNTPs (200 μM each) and AmpliTaq Gold DNA polymerse (Perkin Elmer; 2.5 units) to a final volume of 75 μl. This second amplification utilized Ampliwax (Perkin Elmer) in a ‘hot start' technique, as recommended by the manufacturer. Cycling parameters were as follows: 95°C for 12 min to activate AmpliTaq Gold then 95°C for 30 s (denaturing), 65°C for 30 s (annealing) and 72°C for 1 min (extension); 35 cycles. Controls included reactions in which either a primer or the template were excluded. The reaction products were analysed in an ethidium bromide-stained agarose gel.
In the PCRs which used the cDNA library as template, the reactions which used the A1BF4/A1BB4 and A1BF5/A1BB5 primer pairs consisted of PCR buffer (Perkin Elmer; 1×; MgCl2 1.5 mM), dNTPs (200 μM each), primers (1 μM each), AmpliTaq Gold DNA polymerse (Perkin Elmer; 2.5 units) and template (0.5 μg) to a final volume of 75 μl. These reactions were carried out as ‘hot start' using Ampliwax. Cycling parameters were as follows: 95°C for 12 min then 95°C for 30 s (denaturing), 59°C for 30 s (annealing) and 72°C for 1 min (extension); 30 cycles. The reactions which used the A1BF2/A1BB9 primer pair were identical to the above except that the reaction buffer was PCR buffer II (Perkin Elmer; 1×) with MgCl2 2.5 mM and the annealing temperature was 65°C. Controls included reactions in which either a primer or the template were excluded. The reactions were analysed in agarose gels.
The reaction products were cloned using the TA technique. Briefly, PCR products were generated using the optimized conditions described above, except that the reactions included a final extension step of 72°C for 10 min to increase the likelihood of addition of 3′ adenosines by Taq polymerase. Immediately after the end of the reactions, 2 pmoles of reaction product was combined with 20 fmoles of the TA cloning vectors pCR2.1 or pCR3.1 (Invitrogen), T4 DNA ligase (4 Weiss units) and ligation buffer (Invitrogen; 1×) to a final volume of 10 μl and incubated overnight at 14°C. The ligation reaction was then used to transform competent E. coli strains INV-α F′ or TOP10F′ (Invitrogen) by heat shock according to the suppliers' protocol. The bacteria were grown overnight on LB agar plates with kanamycin 50 μg ml−1. Individual colonies were picked, amplified by overnight growth in liquid LB medium with kanamycin (50 μg ml−1) and the plasmid DNA was then extracted using Qiagen miniprep kits. An aliquot of each miniprep DNA was subjected to PCR using the primers and reaction conditions which were used to generate the product (see above) to confirm the correct identity of the inserts in the cloning vectors.
PCR products which had been correctly ligated to the cloning vectors were sequenced either using an automated ABI sequencer or manually. The latter employed the dsDNA Cycle Sequencing System (Life Technologies) in which a sequencing primer (1 pmole) was labelled with 33P using T4 polynucleotide kinase (1 unit) in a 5 μl reaction. The labelled primer was then used in a cycle sequencing reaction which employed 200 ng of template and 1.25 units of Taq polymerase. Cycling parameters were: 95°C for 30 s (denaturing), 55°C for 30 s (annealing) and 72°C for 1 min (extension); 30 cycles. The reaction products were then electrophoresed in a 6% polyacrylamide gel and autoradiographed using Kodak MR-2 film. Sequences were assembled and analysed using the AssemblyLign and MacVector sequence analysis software (Oxford Molecular Group). At least two clones were sequenced from each PCR product, in order to ensure accuracy of the data.
Expression of cDNA in mammalian cells
COS-7 cells or GnRH neurones were grown in Nunc 175 cm2 flasks (approximately 2×107 cells/flask) for 2 – 4 days and culture media were changed at 48 h intervals. The cells were then dispersed in the presence of trypsin, DNAseI and EDTA and washed twice in 10 ml culture medium. The cells were then concentrated to a density of 108 cells ml−1 of culture medium. An aliquot of the cell suspension (250 μl; 2.5×107 cells) was placed in Bio-Rad cuvettes (inter-electrode distance 0.4 cm) and 10 μg of plasmid DNA dissolved in TE buffer (1 μg μl−1) was added. In control experiments, TE buffer without DNA was added. The suspension was then mixed and electroporated at room temperature in a Bio-Rad Gene Pulser II device with a Capacitance Extender Plus and Pulse Controller Plus. Electroporation parameters were set as follows: GnRH neurones: 190 V, 1000 μF; COS-7 cells; 170 V, 950 μF. After 60 s, culture medium was added to the cuvette and the cell suspension was transferred to a total of 50 ml culture medium (0.5×106 electroporated cells ml−1). The cells were then incubated in 12-well culture plates (2 ml well−1) which had been coated with poly-D-lysine and laminin. The cells were grown at 37°C in the presence of 5% CO2 in air. Binding studies were carried out 2 days after electroporation using the methods described above.
Materials
Immortalized GT1-1 GnRH neuronal cells (Mellon et al., 1990) were generously provided by Dr R.I. Weiner. SK-N-SH cells, DDT1 MF-2 cells and COS-7 cells were from the American Type Culture Collection (Rockville, Maryland, U.S.A.; Catalogue numbers HTB-11, CRL-1701 and CRL-1651, respectively). Heat-inactivated FBS was from Life Technologies (Paisley, Scotland). [3H]-Prazosin was from Amersham (TRK.843; batch number 45; specific activity 78 Ci/mmole; 3H label in 7-methoxyl group). [3H]-Noradrenaline was from DuPont NEN (NET-678; lot number 3108213; specific activity 42 Ci/mmole; 3H label in ring positions 2, 5 and 6). Unlabelled compounds and culture media were from Sigma-Aldrich (Poole, Dorset, U.K.). Syrian hamster α1b adrenoceptor cDNA in the expression vector pBC was generously provided by Dr R.J. Lefkowitz (Cotecchia et al., 1988). Reagents for protein assay were from Pierce & Warriner (Chester, Cheshire, U.K.).
Results
Accumulation of prazosin in GnRH peptidergic neurones; comparison to the accumulation of noradrenaline in noradrenergic neurones
Figures 1 and 2 compare the accumulation of prazosin in GnRH neurones to the accumulation of noradrenaline in SK-N-SH noradrenergic neurones. The lower panels demonstrate the accumulation of radiolabelled ligand, and the upper panels demonstrate accumulation of total ligand (labelled and unlabelled) by correcting for the fall in specific activity consequent upon isotope dilution. At a concentration of 2.1×10−10 M, [3H]-prazosin associates with the peptidergic neurones and is displaced by unlabelled prazosin in concentrations up to 3.33×10−7 M (binding at 37°C: B0: 4285±50 d.p.m./well, unlabelled prazosin 3.33×10−7 M: 3791±38 d.p.m./well; Figure 1B). However, at greater concentrations of unlabelled prazosin, there was a paradoxical increase in the binding of [3H]-prazosin, as previously described (Al-Damluji et al., 1993). At 0°C, accumulation of prazosin is less than at 37°C but [3H]-prazosin still binds and is displaced by unlabelled prazosin (binding at 0°C: B0: 2885±88 d.p.m./well, unlabelled prazosin 3.33×10−7 M: 2185±47 d.p.m./well; Figure 1B). In addition, cooling the cells to inhibit transport processes, abolished the paradoxical increase in accumulation of [3H]-prazosin (Figure 1). Accumulation of prazosin in the peptidergic neurones at 0°C was linear with concentration up to at least 10−6 M, indicating that it represented non-specific accumulation. Hence, in these experiments, non-specific uptake was defined as uptake at 0°C. Specific uptake was obtained by subtracting non-specific uptake from total uptake. At concentrations of prazosin greater than 10−7 M, the specific accumulation of prazosin was non-linear (Figure 1).
Figure 2.
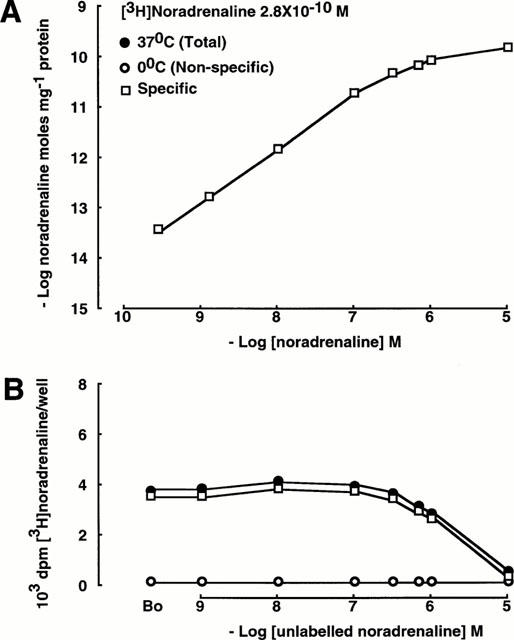
Uptake of noradrenaline in SK-N-SH noradrenergic neurones. As in Figure 1, the lower panel demonstrates uptake of the radiolabelled ligand and the upper panel demonstrates uptake of total ligand (labelled and unlabelled).
SK-N-SH noradrenergic neurones are known to possess the pre-synaptic re-uptake process for noradrenaline (Uptake1; Pacholczyk et al., 1991; Al-Damluji & Kopin, 1996a). At 37°C, [3H]-noradrenaline binds to the noradrenergic neurones and is displaced by unlabelled noradrenaline. There was no increase in the binding of [3H]-noradrenaline at concentrations of unlabelled noradrenaline greater than 10−7 M (Figure 2). At 0°C, accumulation of noradrenaline was less than at 37°C (B0 214±23 d.p.m./well vs 3832±73 d.p.m./well). Accumulation of noradrenaline in the noradrenergic neurones at 0°C was linear with concentration throughout, indicating that it represented non-specific accumulation. The concentration-dependance of specific accumulation of noradrenaline was hyperbolic and tended towards saturation at concentrations of noradrenaline greater than 10−7 M (Figure 2).
Molecular identification of α1B adrenoceptors in GnRH neurones
The PCR primers were derived from the regions of the coding sequence which were homologous between the three cloned subtypes of α1 adrenoceptors. Using GnRH neurone RNA as template, it was possible to detect a PCR product of the appropriate size in reactions which used the A1BF2/A1BB9 primer pair in a first PCR, followed by the A1BF1/A1BB1 nested primer pair in a second PCR (Figure 3). Further, using the GnRH neurone cDNA library as template, it was possible to detect PCR products of the appropriate sizes in reactions which used the A1BF2/A1BB9, A1BF4/A1BB4 and A1BF5/A1BB5 primer pairs (Figure 3). Analysis of the translated sequences of these three PCR products indicated that the sequences were 99.2% identical to the translated sequence of the mouse α1B adrenoceptor (GenBank accession number Y12738).
Figure 3.

The products (shown by arrows) of polymerase chain reactions, using as templates reverse-transcribed (RT) RNA from GnRH neurones or a cDNA library which we constructed from these GnRH neurones. The primers used are described in the text. In (A) (RT-RNA as template), the first PCR employed the A1BF2/A1BB9 primer pair and this was followed by a second PCR which employed the nested primer pair A1BF1/A1BB1. In (B – D) the template was the cDNA library. The primers were A1BF2/A1BB9 in lane B, A1BF4/A1BB4 in lane C and A1BF5/A1BB5 in lane D. Specific products of the predicted sizes were detected in all these reactions. Size markers are shown in parallel lanes in each panel.
GT1-1 is a mouse cell line. The mouse, rat and human α1B adrenergic receptor genes contain a large intron whose splice site is in the sixth transmembrane domain (Ramarao et al., 1992; Gao & Kunos, 1993; Zuscik et al., 1999). This splice site is flanked by the A1BF4/A1BB4 PCR primer pair. The sequence of the PCR product which was generated by this primer pair contained no intronic sequences, demonstrating that the reactions amplified mRNA rather than contaminating genomic DNA.
Binding of prazosin in COS-7 cells transfected with α1b adrenoceptor cDNA
In COS-7 cells which had been transfected by electroporation with α1b adrenoceptor cDNA, [3H]-prazosin bound to the cells and was displaced by unlabelled prazosin in the concentration range 10−10 to 10−7 M. The IC50 of prazosin for these binding sites was 2×10−9 M and the KD was 5×10−10 M. There was no increase in the binding of [3H]-prazosin at concentrations of unlabelled prazosin up to 10−6 M (Figure 4A). In these experiments, non-specific binding was defined as the amount of prazosin accumulated in control cells which had been electroporated in the absence of DNA. Specific binding of prazosin to COS-7 cells which had been transfected with α1b adrenoceptor cDNA reached saturation at approximately 10−8 M.
Figure 4.
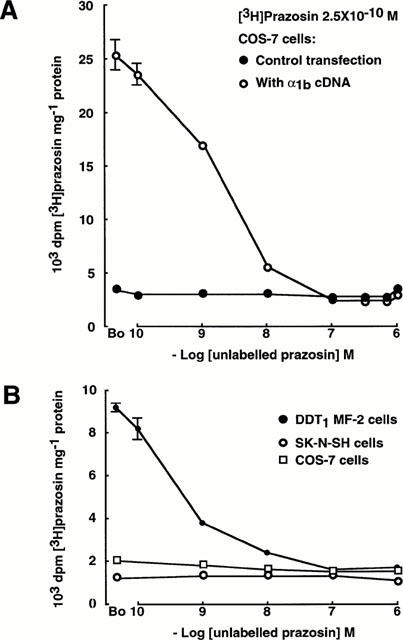
(A) Binding of prazosin in COS-7 cells transfected with α1b adrenoceptor cDNA: [3H]-prazosin is displaced by unlabelled prazosin (IC50 2×10−9 M, KD 5×10−10 M) and there is no increase in the binding of [3H]-prazosin at concentrations of unlabelled prazosin up to 10−6 M. In the control transfection, COS cells were electroporated in the absence of DNA. (B) Binding of prazosin in DDT1 MF-2 cells which express native α1B adrenoceptors, in SK-N-SH noradrenergic neurones and in COS-7 kidney cells. The concentration dependence of the binding of [3H]-prazosin and its displacement by unlabelled prazosin (KD 4.8×10−10 M) is seen in the DDT1 MF-2 cells. Binding in the other two cell lines is at very low levels and is unaffected by desipramine (not shown). There is no increase in the binding of [3H]-prazosin at concentrations of unlabelled prazosin up to 10−6 M in any of the cell lines.
Binding of prazosin in DDT1 MF-2 cells, SK-N-SH cells and COS-7 cells
In DDT1 MF-2 smooth muscle cells which are known to express native α1B adrenoceptors, [3H]-prazosin bound to the cells and was displaced by unlabelled prazosin in the concentration range 10−10 to 10−7 M (KD 4.8×10−10 M). There was no increase in the binding of [3H]-prazosin at concentrations of unlabelled prazosin up to 10−6 M (Figure 4B).
SK-N-SH neurones and COS-7 kidney cells took up only small amounts of prazosin, and this was unaffected by 10−5 M desipramine. Further, there was no increase in the uptake of [3H]-prazosin at concentrations of unlabelled prazosin up to 10−6 M in either type of cell (Figure 4B).
Effects of adrenergic agonists on [3H]-prazosin binding in DDT1 MF-2 cells
In DDT1 MF-2 cells, bound [3H]-prazosin was displaced by the selective α1 adrenergic agonists methoxamine and phenylephrine (KD 2×10−4 M and 1.9×10−5 M, respectively) and by the non-selective endogenous agonists adrenaline and noradrenaline (KD 6.2×10−6 M and 10−5 M). None of these agonists caused an increase in the binding of [3H]-prazosin (not shown).
Over-expression of α1B adrenoceptors in peptidergic neurones which possess Transport-P
In GT1-1 GnRH cells which had been transfected by electroporation with α1b adrenoceptor cDNA, [3H]-prazosin (2.5×10−10 M) binding was greater than in control (mock-transfected) cells (B0: control transfection 4262±107 d.p.m./mg protein; with α1b transfection 7416±69 d.p.m./mg protein; Figure 5A). In both groups of cells, [3H]-prazosin was displaced by unlabelled prazosin in concentrations up to 3.33×10−7 M (control 3080±64 d.p.m./mg protein; with α1b transfection 3120±104 d.p.m./mg protein; Figure 5A). At greater concentrations of unlabelled prazosin, the binding of [3H]-prazosin increased in both the control cells and in the cells which had been transfected with α1b adrenoceptor cDNA. There was no difference in the increase between the two groups of cells (Figure 5A).
Figure 5.
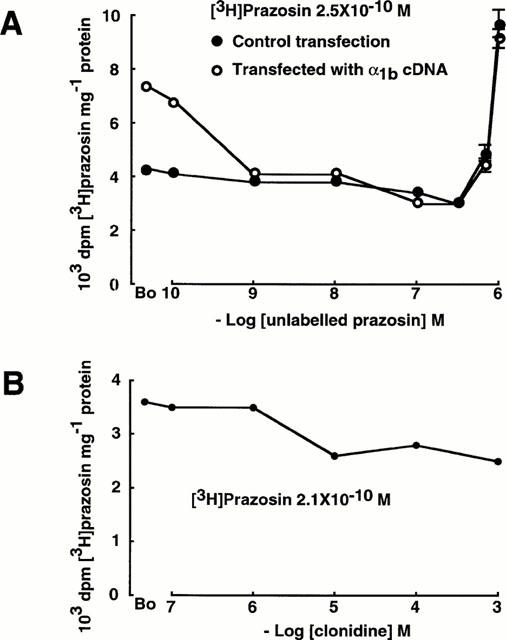
(A) Over-expression of α1B adrenoceptors in peptidergic neurones which possess Transport-P: GT1-1 GnRH cells were transfected with α1b adrenoceptor cDNA. In the control transfection, GT1-1 cells were electroporated in the absence of DNA. There was no difference in the prazosin paradox between control and transfected cells. (B) Effect of the α2 adrenergic agonist clonidine on the binding of [3H]-prazosin in GT1-1 GnRH cells.
The α2 adrenergic agonist clonidine displaced [3H]-prazosin (2.1×10−10 M) from GT1-1 cells (B0 3610±94 d.p.m./mg protein; clonidine 10−3 M 2487±50 d.p.m./mg protein; Figure 5B). There was no increase in the binding of [3H]-prazosin at concentrations of clonidine up to 10−3 M (Figure 5B).
Discussion
Possible relationship of the prazosin paradox to α1 adrenoceptors
In previous work, we had demonstrated that immortalized GnRH neurones possess α1 adrenoceptors, as indicated by binding of prazosin and its displacement by unlabelled prazosin (Al-Damluji et al., 1993). Those binding studies were carried out in the presence of the antidepressant desipramine, in order to inhibit the accumulation of prazosin via Transport-P. In the present work, we confirm that these neurones express an α1 adrenoceptor gene and we identify its molecular sub-type as being α1B. The presence of these receptors in GnRH neurones is sufficient to account for the displacement of [3H]-prazosin by unlabelled prazosin in the concentration range 10−9 to 10−7 M. However, a simple binding process at these receptors would obviously not explain the paradoxical increase above that concentration range. We therefore investigated the possibility that this prazosin paradox in peptidergic neurones may be due to some post-binding event at α1 adrenoceptors, such as internalization of ligand-receptor complexes. COS-7 cells transfected with α1b adrenoceptor cDNA expressed abundant α1B adrenoceptors, as indicated by the great increase in [3H]-prazosin binding. However, there was no prazosin paradox in these transfected cells; the accumulation of prazosin was in accord with simple equilibrium and reached saturation at approximately 10−8 M (Figure 4A). It therefore seemed unlikely that the prazosin paradox is due to some function of α1 adrenoceptors. Nevertheless, the α1B adrenoceptors in these COS-7 cells were not in a native environment; we therefore studied DDT1 MF-2 cells which express native α1B adrenoceptors and which are known to internalize these receptors upon exposure to the agonists adrenaline, noradrenaline and phenylephrine (Cornett & Norris, 1982; Fratelli & DeBlasi, 1987; Cotecchia et al., 1988). As in the transfected COS-7 cells, there was no prazosin paradox in DDT1 MF-2 cells (Figure 4B). In any case, receptor internalization seemed an unlikely explanation since prazosin is an α1 adrenoceptor antagonist but internalization of ligand-receptor complexes usually follows binding of an agonist to its receptor. We therefore examined the effects of a series of adrenergic agonists which included both the endogenous non-selective neurotransmitters adrenaline and noradrenaline, and synthetic selective agonists. All these agonists displaced [3H]-prazosin from α1B adrenoceptors in DDT1 MF-2 cells, but the prazosin paradox was not seen in any of these experiments. We conclude that receptor internalization is not a factor in our experiments.
The prazosin paradox is unaffected by an increase in α1 adrenoceptors
The prazosin paradox was observed in peptidergic neurones but DDT1 MF-2 are smooth muscle cells which may not possess the specialized neuronal components which are required for function of neuronal receptors. We therefore over-expressed α1B adrenoceptors in the GnRH peptidergic neurones, to see if this would enhance the prazosin paradox in these neurones. However, the prazosin paradox was unaffected by over-expression of α1B adrenoceptors, indicating that it is mediated by a different component of the GnRH neurones (Figure 5A). From all of the foregoing, we conclude that the prazosin paradox is unlikely to be due to some function of α1 adrenoceptors. The paradoxical increase in binding of [3H]-prazosin in the presence of greater concentrations of unlabelled prazosin is likely to be due, instead, to an unusual uptake process whose functional properties are distinct from those of α1 adrenoceptors. Further, the structural properties of ligands for Transport-P are different from those of ligands for α1 adrenoceptors (Al-Damluji & Kopin, 1998).
Possible role of α2 adrenoceptors
In our previous work on GT1-1 cells, we were unable to detect the presence of α2 adrenoceptor binding sites using the ligand [3H]-idazoxan (RX781094; Al-Damluji et al., 1993). However, Lee et al. (1995) subsequently used another ligand, RX821002, and demonstrated the presence of α2A binding sites in a related cell line, GT1-7 cells. They corroborated the finding by demonstrating RNA and immunoreactivity which is appropriate for α2A adrenergic receptors in the GT1-7 cells. At this stage, it is unclear whether these apparent differences are due to technical factors or to some genuine difference between the GT1-1 and the GT1-7 cells; in the mouse and rat brain, many cells which are immunoreactive to GnRH do not possess α2A adrenoceptors (Lee et al., 1995). It is therefore possible that the GT1-1 and the GT1-7 cell lines may have been derived from subsets of GnRH neurones which differ in the expression of α2 adrenoceptors. In any case, presence of α2 adrenoceptors does not account for the functional properties of Transport-P for the following reasons: (a) The structural properties of ligands for Transport-P are different from the properties of ligands for α2 adrenoceptors (Al-Damluji & Kopin, 1998); (b) The α2 adrenergic agonist clonidine does not cause a paradoxical increase in accumulation of [3H]-prazosin in the GnRH neurones (Figure 5B). The displacement of [3H]-prazosin by clonidine is attributable to the known agonist action of clonidine at α1 adrenoceptors (Nichols & Ruffolo, 1991).
Functional properties of Transport-P
The present work includes a new analysis of the uptake of prazosin in GnRH neurones (Figure 1). Transport-P accumulates prazosin when this ligand is present in concentrations of 10−10 M, as evidenced by blockade of the accumulation of prazosin by lowering the temperature (Figure 1) and by desipramine (Al-Damluji et al., 1993). Accumulation of prazosin in peptidergic neurones is linear up to 10−7 M but at greater concentrations of prazosin, uptake increases in a non-linear manner, i.e., out of proportion to the increase in the extracellular concentration of prazosin (Figure 1). This indicates that the uptake process is being activated. This property of Transport-P has not been described for any other membrane transport system. The activation of uptake may be due to positive co-operativity of prazosin molecules which may bind to more than one site in the transporter molecule. The activation has characteristics of an energy-dependent process; the paradoxical increase is abolished at 0°C, at which temperature the accumulation of prazosin is linear with concentration up to 10−6 M, indicating that it is non-specific. A feasible source of energy for the activation is the electrochemical proton gradient which is generated by V-ATPase; accumulation of prazosin at 10−6 M is inhibited by the organic base chloroquine, by the carboxylic ionophore monensin, by the V-ATPase inhibitor bafilomycinA1 and by increasing extracellular pH (Al-Damluji & Kopin, 1996a). As these pharmacological manoeuvres increase intracellular pH by different mechanisms, their inhibitory effect on the accumulation of prazosin in peptidergic neurones indicates that Transport-P is dependent on intracellular acidity which is generated by V-ATPase. Further, following accumulation by Transport-P, retention of the amines in peptidergic neurones requires maintenance of intracellular acidity (Shen & Al-Damluji, 1997). A microscopic method for detection of Transport-P revealed that a fluorescent analogue of prazosin accumulated in peptidergic neurones in a granular distribution and that this process could be inhibited by chloroquine (Al-Damluji et al., 1997). This indicated that these amines accumulate in acidified intracellular vesicles.
In the present study, cooling the cells to 0°C was used as an experimental control, in order to inhibit uptake without affecting binding to the α1 adrenoceptors (Figure 1B). The same experimental control was used to study the uptake of noradrenaline in noradrenergic neurones (Figure 2), in order to enable comparison of Transport-P in peptidergic neurones to Uptake1 in noradrenergic neurones. The results distinguish Transport-P from Uptake1; in noradrenergic neurones, there was no increase in the accumulation of [3H]-noradrenaline in the presence of concentrations of unlabelled noradrenaline greater than 10−7 M. Uptake of noradrenaline is linear up to 10−7 M and begins to saturate at greater concentrations of the amine (Figure 2). Further, we found no evidence for specific accumulation of prazosin in the noradrenergic neurones (Figure 4B), which further distinguishes Transport-P from Uptake1. Inability to accumulate prazosin in SK-N-SH noradrenergic neurones is explicable by the presence in prazosin of phenolic methoxyl groups which inhibit Uptake1 (Iversen, 1967). In contrast, Transport-P is unaffected by phenolic methoxyl groups (Al-Damluji & Kopin, 1998). Non-neuronal cells such as muscle and glia possess an uptake process for amines (Uptake2) which is insensitive to antidepressants and reserpine but is blocked by steroid hormones (Iversen, 1965; Russ et al., 1996). Our studies so far suggest that Transport-P does not exist in non-neuronal cells. Thus, there was no antidepressant-blockable prazosin paradox in COS-7 kidney cells or in DDT1 MF-2 smooth muscle cells (Figure 4B). Other features which distinguish Transport-P from Uptake1, Uptake2 and from vesicular uptake have been discussed (Al-Damluji & Kopin, 1998).
The absence of a desipramine-sensitive prazosin paradox in noradrenergic neurones, in kidney cells and in smooth muscle cells suggests that Transport-P is a specialized function of peptidergic neurones. The acidified vesicles which accumulate amines in peptidergic neurones are therefore unlikely to be some acidified compartment which exists in all mammalian cells (such as mitochondria or lysosomes), but is more likely a specialized anatomical feature of peptidergic neurones.
Significance for GnRH neurones
Under the present culture conditions, the GT1-1 GnRH cells express the α1 adrenergic receptors at relatively low levels, possibly due to the absence of physiological factors which are known to induce the expression of α1 adrenergic receptors, such as glucocorticoids and gonadal steroids (Sakaue & Hoffman, 1991; Petitti et al., 1992; Karkanias et al., 1996). In vivo, GnRH neurones are densely innervated by noradrenergic nerve terminals which modulate reproductive function via their action on α1 adrenoceptors in the hypothalamus (for review see Al-Damluji, 1993; Rosie et al., 1994; Le et al., 1997). Previously, we demonstrated the existence of α1 adrenoceptor binding sites in GnRH neurones (Al-Damluji & Krsmanovic, 1992; Al-Damluji et al., 1993) and we now identify these receptors as being of the α1B sub-type. In the rat, GnRH neurones are located in the pre-optic area of the hypothalamus, and this part of the hypothalamus has been shown to contain α1B adrenoceptors (Petitti et al., 1992; Karkanias et al., 1996). The density of these α1B adrenoceptors in the pre-optic area is modulated by gonadal steroids, suggesting that at least some of the α1B adrenoceptors in the pre-optic area are located on GnRH neurones (Petitti et al., 1992; Karkanias et al., 1996). Thus, the finding in the present study that immortalized GnRH neurones possess α1B adrenoceptors is consistent with pharmacological evidence obtained in the native state in rat brain slices. Identification of the molecular subtype of α1 adrenoceptors in GnRH neurones should help to further future work on the molecular mechanisms of the adrenergic control of puberty and reproduction.
In conclusion, the GnRH peptidergic neurones of the hypothalamus possess α1B adrenoceptors and an unusual uptake process for amines, designated Transport-P. Prazosin interacts with both of these neuronal components: at low concentrations, prazosin binds to the receptors and is also accumulated by Transport-P; at greater concentrations it activates Transport-P, resulting in accumulation of the amine in acidified neuronal vesicles. The functional properties of the accumulation of prazosin in peptidergic neurones suggest that it is mediated by an unusual transporter molecule in these neurones.
Acknowledgments
We thank Professor Anthony Winder. Dr Michael Thomas and the Department of Clinical Biochemistry for supporting Miss Susan White as a Trainee Clinical Biochemist. We also thank Mr Christopher Odell at the Ludwig Institute, University College London for automated DNA sequence analysis.
Abbreviations
- B0
Binding of the radiolabelled ligand in the absence of unlabelled ligand
- DMEM
Dulbecco's modified Eagle's medium
- DNAseI
deoxyribonuclease I
- EDTA
ethylenediaminetetraacetic acid
- FBS
foetal bovine serum
- GnRH
gonadotrophin-releasing hormone
- HEPES
N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulphonic acid) IC50, half-maximal inhibitory concentration
- KD
affinity constant for binding
- TE buffer
10 mM Tris.HCl, 1 mM EDTA
- V-ATPase
vacuolar-type ATPase
References
- AL-DAMLUJI S.Adrenergic control of the secretion of anterior pituitary hormones Bailliere's Clinical Endocrinology and Metabolism 7 1993355–392.(part 2) [DOI] [PubMed]
- AL-DAMLUJI S., KOPIN I.J. Functional properties of the uptake of amines in immortalised peptidergic neurones (transport-P) Br. J. Pharmacol. 1996a;117:111–118. doi: 10.1111/j.1476-5381.1996.tb15162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AL-DAMLUJI S., KOPIN I.J. Binding and competitive inhibition of amine uptake at post-synaptic neurones (transport-P) by tricyclic antidepressants. Br. J. Pharmacol. 1996b;117:811–816. doi: 10.1111/j.1476-5381.1996.tb15265.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AL-DAMLUJI S., KOPIN I.J. Structural properties of phenylethylamine derivatives which inhibit transport-P in peptidergic neurones. Br. J. Pharmacol. 1998;124:693–702. doi: 10.1038/sj.bjp.0701894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AL-DAMLUJI S., KRSMANOVIC L.High-affinity uptake of noradrenaline by GnRH cells Abstr. Endo. Soc. 199274197(abstract) [Google Scholar]
- AL-DAMLUJI S., KRSMANOVIC L., CATT K.J. High affinity uptake of noradrenaline in post-synaptic neurones. Br. J. Pharmacol. 1993;109:299–307. doi: 10.1111/j.1476-5381.1993.tb13570.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AL-DAMLUJI S., PORTER D., KRSMANOVIC L., KNUTSON J.R., KOPIN I.J. Visual detection of transport-P in peptidergic neurones. Br. J. Pharmacol. 1997;120:876–882. doi: 10.1038/sj.bjp.0700970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AVIV H., LEDER P. Purification of biologically active globin messenger RNA by chromatography on oligothymidylic acid-cellulose. Proc. Natl. Acad. Sci. U.S.A. 1972;69:1408–1412. doi: 10.1073/pnas.69.6.1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BIEDLER J.L., HELSON L., SPENGLER B.A. Morphology and growth, tumorigenicity, and cytogenetics of human neuroblastoma cells in continuous culture. Cancer Res. 1973;33:2643–2652. [PubMed] [Google Scholar]
- CHENG Y.C., PRUSOFF W.H. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- CHIRGWIN J.M., PRZYBYLA A.E., MACDONALD R.J., RUTTER W.J. Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry. 1979;18:5294–5299. doi: 10.1021/bi00591a005. [DOI] [PubMed] [Google Scholar]
- CORNETT L.E., NORRIS J.S. Characterization of the alpha-1-adrenergic receptor subtype in a smooth muscle cell line. J. Biol. Chem. 1982;257:694–697. [PubMed] [Google Scholar]
- COTECCHIA S., SCHWINN D.A., RANDALL R.R., LEFKOWITZ R.J., CARON M.G., KOBILKA B.L. Molecular cloning and expression of the cDNA for the hamster alpha-1-adrenergic receptor. Proc. Natl. Acad. Sci. U.S.A. 1988;85:7159–7163. doi: 10.1073/pnas.85.19.7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FRATELLI M., DEBLASI A. Agonist-induced α1-adrenergic receptor changes. Evidence for receptor sequestration. FEBS Lett. 1987;212:149–153. doi: 10.1016/0014-5793(87)81575-2. [DOI] [PubMed] [Google Scholar]
- GAO B., KUNOS G. Isolation and characterization of the gene encoding the rat alpha-1B adrenergic receptor. Gene. 1993;131:243–247. doi: 10.1016/0378-1119(93)90300-r. [DOI] [PubMed] [Google Scholar]
- GLISIN V., CRKVENJAKOV R., BYUS C. Ribonucleic acid isolated by cesium chloride centrifugation. Biochemistry. 1974;13:2633–2637. doi: 10.1021/bi00709a025. [DOI] [PubMed] [Google Scholar]
- GLUZMAN Y. SV40-transformed simian cells support the replication of early SV40 mutants. Cell. 1981;23:175–182. doi: 10.1016/0092-8674(81)90282-8. [DOI] [PubMed] [Google Scholar]
- HIEBLE J.P., BYLUND D.B., CLARKE D.E., EIKENBURG D.C., LANGER S.Z., LEFKOWITZ R.J., MINNEMAN K.P., RUFFOLO R.R. International Union of Pharmacology X. Recommendation for nomenclature of alpha-1-adrenoceptors: consensus update. Pharmacol. Rev. 1995;47:267–270. [PubMed] [Google Scholar]
- IVERSEN L.L. The uptake of catechol amines at high perfusion concentrations in the rat isolated heart: a novel catechol amine uptake process. Br. J. Pharmacol. 1965;25:18–33. doi: 10.1111/j.1476-5381.1965.tb01753.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IVERSEN L.L. The Uptake and Storage of Noradrenaline in Sympathetic Nerves. London: Cambridge University Press; 1967. [Google Scholar]
- KARKANIAS G.B., ANSONOFF M.A., ETGEN A.M. Estradiol regulation of alpha-1b adrenoceptor mRNA in female rat hypothalamus-preoptic area. J. Neuroendocrinol. 1996;8:449–455. doi: 10.1046/j.1365-2826.1996.04716.x. [DOI] [PubMed] [Google Scholar]
- KRIEGLER M. Gene Transfer and Expression: a Laboratory Manual. New York: W.H. Freeman & Co; 1991. [Google Scholar]
- LE W.W., BERGHORN A., SMITH M.S., HOFFMAN G.E. Alpha-1-adrenergic receptor blockade blocks LH secretion but not LHRH cFos activation. Brain Res. 1997;747:236–245. doi: 10.1016/s0006-8993(96)01269-3. [DOI] [PubMed] [Google Scholar]
- LEE A., TALLEY E., ROSIN D.L., LYNCH K.R. Characterization of α2A-adrenergic receptors in GT1 neurosecretory cells. Neuroendocrinology. 1995;62:215–225. doi: 10.1159/000127007. [DOI] [PubMed] [Google Scholar]
- LOMASNEY J.W., COTECCHIA S., LORENZ W., LEUNG W.Y., SCHWINN D.A., YANG-FENG T.L., BROWNSTEIN M., LEFKOWITZ R.J., CARON M.G. Molecular cloning and expression of the cDNA for the alpha-1A-adrenergic receptor. J. Biol. Chem. 1991;266:6365–6369. [PubMed] [Google Scholar]
- MELLON P.L., WINDLE J.L., GOLDSMITH P.C., PADULA C.A., ROBERTS J.L., WEINER R.I. Immortalization of hypothalamic GnRH neurons by genetically targeted tumorigenesis. Neuron. 1990;5:1–10. doi: 10.1016/0896-6273(90)90028-e. [DOI] [PubMed] [Google Scholar]
- NICHOLS A.J., RUFFOLO R.R.Structure-activity relationships for α-adrenoceptor agonists and antagonists α-Adrenoceptors: Molecular Biology, Biochemistry and Pharmacology 1991Progress in Basic and Clinical Pharmacology. Basel: Karger; Ruffolo, R.R. (ed) [Google Scholar]
- OKAYAMA H., KAWAICHI M., BROWNSTEIN M., LEE F., YOKOTA T., ARAI K. High-efficiency cloning of full-length cDNA: construction and screening of cDNA expression libraries for mammalian cells. Meth. Enzym. 1987;154:3–28. doi: 10.1016/0076-6879(87)54067-8. [DOI] [PubMed] [Google Scholar]
- PACHOLCZYK T., BLAKELY R.D., AMARA S.G. Expression cloning of a cocaine- and antidepressant-sensitive human noradrenaline transporter. Nature. 1991;350:350–354. doi: 10.1038/350350a0. [DOI] [PubMed] [Google Scholar]
- PETITTI N., KARKANIAS G.B., ETGEN A.M. Estradiol selectively regulates alpha-1b noradrenergic receptors in the hypothalamus and preoptic area. J. Neuroscience. 1992;12:3869–3876. doi: 10.1523/JNEUROSCI.12-10-03869.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAMARAO C.S., KINCADE-DENKER J.M., PEREZ D.M., GAIVIN R.J., RIEK R.P., GRAHAM R.M. Genomic organization and expression of the human α 1B-adrenergic receptor. J. Biol. Chem. 1992;267:21936–21945. [PubMed] [Google Scholar]
- ROSIE R., SUMNER E.H., FINK G. An α-1 adrenergic mechanism mediates estradiol stimulation of LHRH mRNA synthesis and estradiol inhibition of POMC mRNA synthesis in the hypothalamus of the prepubertal female rat. J. Steroid Biochem. Mole. Biol. 1994;49:399–406. doi: 10.1016/0960-0760(94)90286-0. [DOI] [PubMed] [Google Scholar]
- RUSS H., STAUDT K., MARTEL F., GLIESE M., SCHOMIG E. The extraneuronal transporter for monoamine transmitters exists in cells derived from human central nervous system glia. Eur. J. Neurosci. 1996;8:1256–1264. doi: 10.1111/j.1460-9568.1996.tb01294.x. [DOI] [PubMed] [Google Scholar]
- SAKAUE M., HOFFMAN B.B. Glucocorticoids induce transcription and expression of the α1B adrenergic receptor gene in DDT1 MF-2 smooth muscle cells. J. Clin. Invest. 1991;88:385–389. doi: 10.1172/JCI115315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCATCHARD G. The attractions of proteins for small molecules and ions. Ann. N.Y. Acad. Sci. 1949;51:660–672. [Google Scholar]
- SHEN W.B., AL-DAMLUJI S.Release of amines from acidified stores in peptidergic neurones J. Endocrinol. 1997152suppl.P151abstract [Google Scholar]
- SMITH P.K., KROHN R.I., HERMANSON G.T., MALLIA A.K., GARTNER F.H., PROVENZANO M.D., FUJIMOTO E.K., GOEKE N.M., OLSON B.J., KLENK D.C. Measurement of protein using bicinchoninic acid. Analyt. Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- ZUSCIK M.J., PIASCIK M.T., PEREZ D.M. Cloning, cell-type specificity, and regulatory function of the mouse α1B-adrenergic receptor promoter. Mol. Pharmacol. 1999;56:1288–1297. doi: 10.1124/mol.56.6.1288. [DOI] [PubMed] [Google Scholar]