

Abstract
Exogenous ATP produces acute and localized pain in humans, and P2X receptor agonists elicit acute nociceptive behaviours in rodents following intradermal administration to the hindpaw. The predominant localization of P2X3 mRNA in sensory neurones has led to the hypothesis that activation of P2X3 and/or P2X2/3 receptors contributes to nociception.
The local administration of the P2X receptor agonist, BzATP (100 – 1000 nmol paw−1, s.c.) into the rat hindpaw produced an acute (<15 min) paw flinching response that was similar to that observed in the acute phase of the formalin (5%) test.
The co-administration of the potent P2X receptor antagonist, TNP-ATP (30 – 300 nmol paw−1), but not an inactive analogue, TNP-AMP, with BzATP into the rat hindpaw attenuated BzATP-induced nociception. Similarly, co-administration of TNP-ATP, but not TNP-AMP, with 5% formalin reduced both acute and persistent nociception in this test.
Co-administration of cibacron blue (30 and 100 nmol paw−1), a selective allosteric enhancer of P2X3 and P2X2/3 receptor activation, with BzATP (30 and 100 nmol paw−1) into the rat hindpaw produced significantly greater nociception as compared to the algogenic effects of BzATP alone. Intradermal co-administration of cibacron blue (30 and 100 nmol paw−1) with formalin (1 and 2.5%) into the rat hindpaw also produced significantly greater nociceptive behaviour as compared to formalin alone.
The ability of TNP-ATP and cibacron blue to respectively attenuate and enhance nociceptive responses elicited by exogenous BzATP and formalin provide further support for the hypothesis that activation of peripheral P2X3 containing channels contributes specifically to both acute and persistent nociception in the rat.
Keywords: BzATP, cibacron blue, formalin, pain, P2X3 receptor, TNP-ATP
Introduction
Extracellular ATP, acting at distinct families of ionotropic (P2X) and metabotropic (P2Y) receptors, is an important signalling molecule in excitatory neurotransmission, platelet aggregation, apoptosis, and cellular differentiation (Ralevic & Burnstock, 1998). The proposed role of ATP as a neurotransmitter (Burnstock, 1972) is supported by observations that ATP is released from sensory nerves (Houlton, 1959) and by subsequent demonstrations that ATP produces fast excitatory potentials in dorsal root ganglionic neurones (Jahr & Jessel, 1983). These data have led to the hypothesis that ATP, released from a variety of cell types and as a co-transmitter from sympathetic nerves, may be an important mediator of pain signalling (Burnstock, 1996; Burnstock and Wood, 1996). Consistent with this role, exogenous ATP elicits pain in a human blister base model (Bleehen & Keele, 1977) and produces pain following iontophoretic administration into the human forearm (Hamilton et al., 2000).
The nociceptive effects of exogenously administered ATP have also been demonstrated in laboratory animals where intradermal administration of P2X receptor agonists, including ATP and α,β-methyleneATP (α,β-meATP), into the rat hindpaw produce acute nociceptive paw flinching behaviour (Bland-Ward & Humphrey, 1997; Hamilton et al., 1999) and mechanical allodynia (Tsuda et al., 2000). A selective P2 receptor-mediated increase in ectopic neuronal excitability that is localised to damaged sensory afferents has also been recently reported in rats following chronic constriction nerve injury (Chen et al., 1999). Both the potency and effectiveness of locally administered P2X receptor agonists to elicit nociceptive responses are increased in situations of peripheral sensitization induced by intraplantar administration of chemical irritants like carrageenan, prostaglandin E2, or formalin (Sawynok & Reid, 1997; Hamilton et al., 1999). In addition to the peripheral nociceptive actions of P2X receptor activation, stimulation of spinal P2X receptors may also contribute to nociception as indicated by the ability of intrathecally (i.t.) administered P2 receptor agonists to increase sensitivity to acute and persistent noxious stimuli in rodents (Driessen et al., 1994; Tsuda et al., 1999a,1999b).
Nociception induced by exogenously administered P2X receptor agonists can be attenuated by systemically administered morphine (Bland-Ward & Humphrey, 1997), by the local co-administration of putative P2 receptor antagonists (Sawynok & Reid, 1997), by agonist-induced desensitization of P2X receptors (Bland-Ward and Humphrey, 1997), and by ablation of small diameter C-fibres (Hamilton et al. 1999). Interestingly, P2 receptor agonist-induced nociception can also be inhibited by i.t. administration of NMDA receptor antagonists (Tsuda et al., 1999a). A direct interaction between purinergic and excitatory amino acid systems in mediating nociceptive processing in the spinal cord is further supported by the demonstration that P2X receptor activation can stimulate glutamate release in spinal dorsal horn neurones (Gu & MacDermott, 1997).
The utility of available purinergic ligands to evaluate the role of individual P2 receptor subtypes in mammalian physiology has been complicated by the susceptibility of P2 receptor agonists to undergo enzymatic degradation, and by the lack of P2 receptor subtype-selective agonists and antagonists (King et al., 1998; Ralevic & Burnstock, 1998). However, the recent availability of recombinant mammalian P2 receptor subtypes has allowed for the systematic characterization of the pharmacology of specific P2 receptor subtypes (King et al., 1998; Bianchi et al., 1999) and led to further clarification of the pharmacological selectivity of ligands acting at P2X receptors. For example, 2′ and 3′-O-(4-benzoylbenzoyl)-ATP (BzATP), a putative P2X7 (P2Z) receptor (El-Moatassim & Dubyak, 1992) agonist (pEC50=5.33) has also been shown to activate potently recombinant human P2X1, P2X3 and P2X2/3 receptors (pEC50 values of 8.74, 7.10 and 6.10, respectively) (Bianchi et al., 1999). In addition, 2′,3′-O-(2,4,6-trinitrophenyl)-ATP (TNP-ATP), a fluorescent ATP analogue with antinociceptive actions following i.t. administration in mice (Tsuda et al., 1999a,1999b), has been found to be a potent nanomolar antagonist at the recombinant rat P2X1, P2X3, and P2X2/3 receptors (Virginio et al., 1998; Lewis et al., 1998; Thomas et al., 1998). The putative nonselective P2 receptor antagonist, cibacron blue (Reactive blue 2) (Ralevic & Burnstock, 1998) was also recently demonstrated to be a highly selective allosteric enhancer of P2X3 receptor activation at concentrations lower than are required for P2 receptor antagonism (Alexander et al., 1999).
Since subtype-selective ligands for the individual P2 receptors have yet to be identified, efforts to elucidate the specific P2X receptor subtypes involved in the transmission of nociceptive signals has been largely based on receptor localization and functional studies using immunohistochemical techniques. These studies have shown that both the homomeric P2X3 and heteromeric P2X2/3 receptor subtypes are selectively localized to the central and peripheral terminals of small diameter sensory neurones (Chen et al., 1995; Lewis et al., 1995; Vulchanova et al., 1997; 1998). The application of exogenous ATP directly to rat tooth pulp nociceptors elicits P2X3-like electrophysiological responses that are consistent with the immunohistochemical localization of P2X3 receptors on these neurones (Cook et al., 1997). Further, recent data has shown that P2X3 specific immunoreactivity is significantly increased in both the injured dorsal root ganglion and in the ipsilateral spinal dorsal horn following chronic constriction injury of the rat sciatic nerve (Novakovic et al., 1999).
Taken together, the functional and immunohistochemical localization of P2X3 containing receptors (P2X3 and/or P2X2/3) on sensory nerves indicates that these P2X3 containing channels may have a primary role in mediating the nociceptive effects of extracellular ATP. The present studies were conducted to evaluate the ability of peripherally administered TNP-ATP and cibacron blue to differentially modulate nociception in two animal pain models, P2X receptor agonist-induced acute nociception and a model of chemically-induced acute and persistent nociception, the formalin test (Tjosen et al., 1992; Abbott et al., 1995). Since BzATP has potent activity at P2X3 receptors in vitro (Bianchi et al., 1999), the nociceptive effects of intradermal BzATP were compared to those of other P2X receptor agonists and to formalin.
Methods
Subjects
Adult male Sprague-Dawley rats, 230 – 350 g, (Charles River, Wilmington, MA, U.S.A.) were housed in groups of five per cage and given free access to food and water. Animals were on a 12 h light-dark cycle of 0600 – 1800 h. Animals were only used once in each experiment. All experimental protocols and animal handling procedures were approved by an institutional animal care and use committee (IACUC).
Nociceptive testing
Nociceptive responses were assessed using procedures previously described for the formalin test of chemically-induced persistent pain (Abbott et al., 1995; Tjosen et al., 1992). Experimentally naive animals were placed in individual plexiglass cages and allowed 30 min to acclimatize to the testing environment. Following this period, animals received subcutaneous injections of either a formalin solution (1, 2.5, 5%), different doses of BzATP alone, or in combination with TNP-ATP or cibacron blue, into the dorsal surface of the right hind paw using an insulin gauge (29G1/2) needle. The volume of injection was 50 μl for all treatments. Saline (0.9%) was used as drug vehicle for all experiments. For co-administration studies, all test compounds were combined and administered in a single injection. To assess acute nociception, animals were observed immediately following drug injections and the number of flinch behaviours (paw withdrawals) was recorded over a 1-min period. Additional observations were conducted at sequential 5-min intervals during the first 15 – 20 min following drug injections (Phase I, acute phase of the formalin test). For some experiments, observations began 30 min after formalin injection and continued for 20 min thereafter (Phase II, persistent phase of the formalin test). For each individual experiment, six rats were used in separate experimental and control groups. Mean cumulative flinch responses were analysed by analysis of variance and post-hoc comparisons were conducted using Fisher's least significant difference test (GB-STAT, Dynamics Microsystems, Inc., Silver Spring, MD, U.S.A.). Statistical significance was determined at P<0.05.
Activation of rat P2X3 and P2X2/3 receptors in vitro
The recombinant rat P2X3 and rat P2X2/3 receptor cDNAs were identical to the previously published sequences used in the in vitro characterization of the pharmacology of the rat homomeric and heteromeric P2X3 receptors (Bianchi et al., 1999). 1321N1 human astrocytoma cells stably expressing rat P2X3, or rat P2X2/3 receptors were constructed using standard lipid-mediated transfection methods. All cell lines were maintained in D-MEM containing 10% FBS and antibiotics as follows: 300 μg ml−1 G418 for rat P2X3 containing cells; and 75 μg ml−1 hygromycin and 150 μg ml−1 G418 for rat P2X2/3 containing cells. Cells were grown at 37°C in a humidified atmosphere containing 5% CO2.
P2X receptor function was determined on the basis of agonist-mediated increases in cytosolic Ca2+ concentration as previously described (Bianchi et al., 1999). BzATP (10 μM) and α,β-meATP (10 μM) were used to activate rat P2X3 and P2X2/3 receptors, respectively. Briefly, a fluorescent Ca2+ chelating dye (Fluo-4) was used as an indicator of the relative levels of intracellular Ca2+ in a 96-well format using a Fluorescence Imaging Plate Reader (FLIPR, Molecular Devices, Sunnyvale, CA, U.S.A.). Cells were grown to confluence in 96-well black-walled tissue culture plates and loaded with the acetoxymethylester (AM) form of Fluo-4 (1 μM) in D-PBS for 1 – 2 h at 23°C. Fluorescence data was collected at 1 – 5 s intervals throughout each experimental run. Concentration response data were analysed using a four-parameter logistic Hill equation in GraphPad Prism (San Diego, CA, U.S.A.).
Drugs and reagents
Morphine sulphate was obtained from Mallinckrodt, Inc. (St. Louis, MC, U.S.A.) and was dissolved in a 0.9% saline solution. Adenosine 5′-triphosphate disodium (ATP), 2-methylthio-ATP tetrasodium (2-meSATP), and αβ-methylene ATP dilithium (αβ-meATP) were obtained from Research Biochemicals International (Natick, MA, U.S.A.). 2′ & 3′-O-(4-benzoylbenzoyl)-ATP tetraethylammonium salt (mixed isomers) (BzATP) and cibacron blue (Reactive blue-2) were obtained from Sigma Chemical Company (St. Louis, MO, U.S.A.). TNP-ATP and Fluo-4AM was purchased from Molecular Probes (Eugene, OR, U.S.A.). All compounds were freshly dissolved and diluted in 0.9% saline. G418 sulphate was obtained from Calbiochem-Novabiochem Corp. (La Jolla, CA, U.S.A.). Dulbecco's modified Eagle's medium (D-MEM) (with 4.5 mg ml−1 glucose and 4 mM L-glutamine) and foetal bovine serum (FBS) were obtained from Hyclone Laboratories, Inc. (Logan, UT, U.S.A). Dulbecco's phosphate-buffered saline (D-PBS) (with 1 mg ml−1 glucose and 3.6 mg l−1 Na pyruvate, without phenol red), hygromycin and Lipofectamine were obtained from Life Technologies (Grand Island, NY, U.S.A.).
Results
Nociceptive effects of P2X receptor agonists
The intradermal administration of BzATP (100 – 1000 nmol paw−1) into the dorsal surface of the rat hindpaw produced a dose-dependent paw flinching response (Figure 1a). The magnitude of nociceptive paw flinching following 1000 nmol paw−1 BzATP was equivalent to that observed following the acute intradermal administration of 5% formalin (Phase I of the formalin test). The duration of this effect was short-lasting with the majority of paw flinch responses occurring in the first 5-min interval following drug injection. BzATP did not produce a second phase of prolonged nocifensive paw flinching behaviour (data not shown) like that which is characteristically observed following intradermal formalin administration (Phase II of the formalin test) (Tjosen et al., 1992; Sawynok & Reid, 1997). The ability of intradermal BzATP to produce nocifensive behaviour in the rat was supported by the ability of systemically administered morphine to reduce dose-dependently (ED50=4 mg Kg−1, s.c.) BzATP (1000 nmol paw−1) induced hindpaw flinching (data not shown). Other nucleotide agonists including ATP, α,β-meATP, and 2meSATP also produced acute nociceptive paw flinching (Figure 1b), but the maximal responses at the doses tested were significantly less than that observed for BzATP. ADP has been previously shown not to activate P2X3 receptors (Bianchi et al., 1999) and intradermal administration of ADP did not produce any nociceptive responding (P>0.05).
Figure 1.

(a) Time course for the acute nociceptive effects of intradermal BzATP in the rat. BzATP or 5% formalin (50 μl) was injected into the dorsal surface for the rat hindpaw at the doses indicated (n=6 per group) and nociceptive responses were recorded in 5-min bins for 20 min post-injection. BzATP dose-dependently increased the number of paw flinch responses (F (4,25)=25.9, P<0.05). Values represent mean±s.e.mean, *P<0.05 as compared to vehicle (saline, 50 μl)-treated rats. (b) Dose-response determinations for the acute nociceptive effects of P2X receptor agonists following intradermal administration into the rat hindpaw (n=6 per dose group). Values represent mean±s.e.mean for the cumulative nociceptive paw flinching responses occurring 15 min post-injection. *P<0.05 as compared to vehicle-treated rats.
Antinociceptive effects of TNP-ATP
The novel P2X receptor antagonist, TNP-ATP inhibited potently agonist-stimulated calcium flux in 1321N1 cells expressing either the rat P2X3 or P2X2/3 receptors (Table 1). As has been shown for the human P2X3 receptor (Virginio et al., 1998), sequential removal of the terminal phosphate groups significantly reduces antagonist potency at both P2X3 and P2X2/3 receptors. In particular, TNP-AMP shows little inhibitory activity at rat P2X3 receptors at concentrations up to 30 μM.
Table 1.
Antagonism of rat P2X3 and P2X2/3 receptor activation by TNP nucleotides

Co-administration of intradermal TNP-ATP (30 – 300 nmol paw−1) with BzATP (1000 nmol paw−1) into the dorsal surface of the rat hind paw, produced a significant (P<0.05) and dose-dependent reduction in nociceptive paw flinching behaviour (Figure 2a). The antinociceptive effects of TNP-ATP appear to be pharmacologically specific since co-administration of TNP-AMP with BzATP did not reduce BzATP-induced paw flinching behaviour. TNP-ATP also was equally effective to attenuate the acute nociceptive effects of α,β-meATP (Figure 2b). These effects were mediated at a local peripheral site of action since intradermal administration of TNP-ATP into the contralateral paw was ineffective in reducing nociception (Figure 2b).
Figure 2.
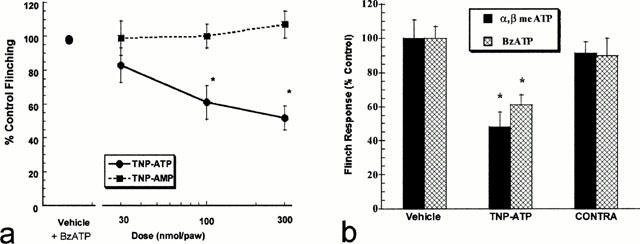
(a) Intradermal co-administration of TNP-ATP (F(3,20)=8.20, P<0.05), but not TNP-AMP (F(3,20)=0.30, P>0.05), dose-dependently attenuates BzATP (1000 nmol paw−1) induced acute nociceptive paw flinching in the rat (n=6 per dose group). Values represent mean±s.e.mean for the cumulative nociceptive paw flinching responses occurring 15 min post-injection. *P<0.05 as compared to vehicle-treated rats. (b) TNP-ATP (300 nmol paw−1) significantly (P=0.05) attenuates the nociceptive effects of intradermal α,β meATP and BzATP (1000 nmol paw−1) in the rat. Administration of TNP-ATP into the paw contralateral (CONTRA) to P2X agonist injection was ineffective. Values represent mean±s.e.mean, *P<0.05 as compared to vehicle-treated rats.
Co-administration of TNP-ATP with 5% formalin into the dorsal surface of the rat hindpaw also dose-dependently reduced nocifensive behaviours in the acute (Phase I) portion of the formalin test (Figure 3). In addition, the antinociceptive effects of TNP-ATP were also evident in the persistent phase (Phase II) of the formalin test where a significant 30% reduction in formalin-induced paw flinching was observed at both doses (30 and 100 nmol paw−1) of TNP-ATP. In agreement with its antinociceptive activity against BzATP, TNP-ATP, but not TNP-AMP, attenuated nociceptive responses in both the acute (Phase I) and persistent (Phase II) components of the formalin test.
Figure 3.
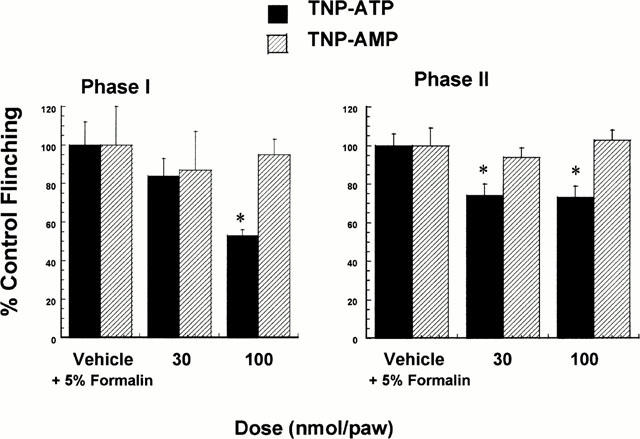
Intradermal co-administration of TNP-ATP, but not TNP-AMP, with 5% formalin attenuated acute nociceptive paw flinching in the rat (n=6 per dose group). Phase I represents cumulative acute nociceptive responses occurring 15 min immediately following intradermal administration (F(2,27)=5.15, P<0.05). Phase II represents cumulative nociceptive responses recorded for a 20-min period beginning 30 min post formalin injection (F(2,27)=6.97, P<0.05). Values represent mean±s.e.mean, *P<0.05 as compared to vehicle-treated rats.
Pronociceptive effects of cibacron blue
Consistent with its ability to selectively enhance human P2X3 receptor activation (Alexander et al., 1999), cibacron blue produced a concentration-dependent increase in both BzATP (1 μM) and α,β-meATP (10 μM) stimulated calcium flux (EC50 values=580 and 720 nM, respectively) in 1321N1 cells expressing the recombinant rat P2X3 receptor (Figure 4a).
Figure 4.
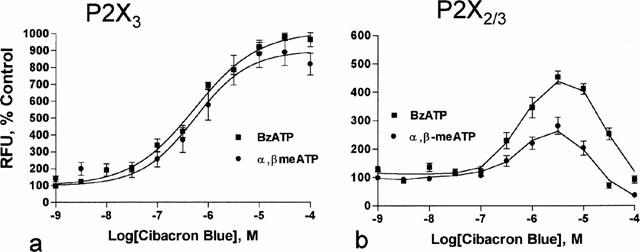
Concentration-effect determinations for the effects of cibacron blue on agonist activation of rat P2X3 and P2X2/3 receptors. (a) Representative concentration-effect curves for cibacron blue (EC50=0.7 μM) to enhance BzATP (1 μM) and α,β-meATP (10 μM) activation of rat P2X3 receptors. (b) Representative concentration-effect curves for cibacron blue to enhance BzATP (1 μM) and α,β-meATP (10 μM) activation of rat P2X2/3 receptors. RFU=relative fluorescence units.
Cibacron blue over the concentration range of 0.3 – 10 μM also enhanced the activation of the rat P2X2/3 receptor by BzATP (1 μM) and α,β-meATP (10 μM) (Figure 4b). However, these effects were biphasic with concentrations of cibacron blue greater than 10 μM producing less enhancement of agonist-mediated activation of the P2X2/3 receptor. Additionally, concentrations of cibacron blue greater than 30 μM antagonized the activation of the rat P2X2/3 receptor by α,β-meATP. Consistent with these observations, cibacron blue was found to inhibit only ATP-induced activation of the rat P2X2 receptor (IC50=8 μM) (data not shown).
Since specific concentrations of cibacron blue were shown to enhance the activation of the rat P2X3 and P2X2/3 receptors in vitro, additional studies were conducted to investigate the potential of cibacron blue to facilitate nociception in the rat. Consistent with the data shown in Figure 1b, the intradermal administration of BzATP alone (10 – 300 nmol paw−1) into the rat hindpaw produced a dose-dependent increase in nociceptive behaviour (Figure 5a – d). The intradermal administration of cibacron blue alone (10 – 300 nmol paw−1) produced only a mild, but statistically significant acute nociceptive response at a dose of 100 nmol paw−1 (Figure 5a – d).
Figure 5.
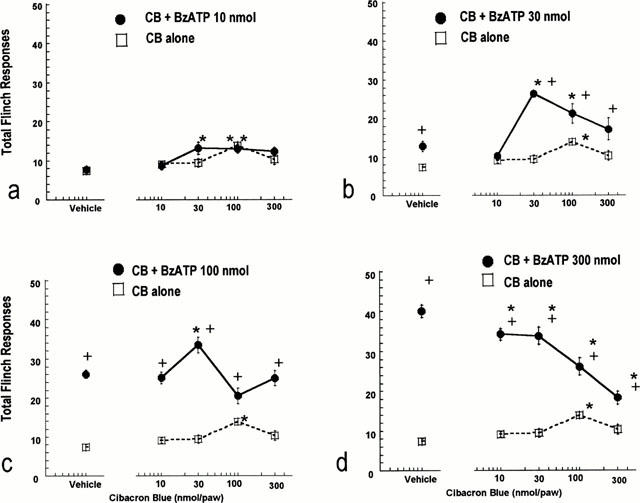
Nociceptive effects of intradermal co-administration of BzATP and cibacron blue into the hindpaw of the rat (F(16,352)=7.30, P<0.05). Vehicle responses indicate the effects of saline (open square) or co-administration of saline and BzATP (filled circles) on acute (cumulative responses for the first 15 min post-injection) paw flinching behaviour. The nociceptive effects of cibacron blue alone are indicated by the open squares and dotted lines. The effects of co-administration of cibacron blue and BzATP are indicated by the filled circles and solid lines. Values represent mean±s.e.mean from three separate experiments (n=6 per dose group), *P<0.05 as compared to the nociceptive effects of BzATP alone,+P<0.05 as compared to the nociceptive effects of cibacron blue alone.
The intradermal co-administration of cibacron blue with BzATP into the rat hindpaw produced significant and biphasic effects in nociceptive paw flinching behaviour relative to the nociceptive effects of BzATP alone (Figure 5a – d). At a low dose of BzATP (10 nmol paw−1), cibacron blue produced a small, but statistically significant (P<0.05) enhancement in nociceptive responding as compared to the effects of BzATP alone (Figure 5a). The pronociceptive effects of cibacron blue were significantly greater when combined with a minimally nociceptive dose of BzATP (30 nmol paw−1) (Figure 5b). At a higher dose of BzATP (100 nmol paw−1), the ability of cibacron blue to enhance nociception was observed only at the dose of 30 nmol paw−1 (Figure 5c). In contrast, the intradermal co-administration of cibacron blue with a high dose of BzATP (300 nmol paw−1) produced a dose-dependent inhibition of paw flinching responses as compared to the nociceptive effects of BzATP alone (Figure 5d). As indicated in Figure 6, the ability of cibacron blue to enhance the nociceptive effects of both BzATP and α,β-meATP was mediated at the local site of administration since cibacron blue administered into the contralateral paw was ineffective.
Figure 6.
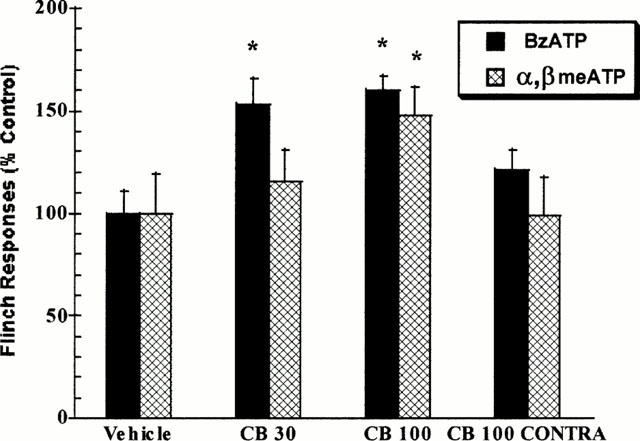
Intradermal cibacron blue (CB, 30 and 100 nmol paw−1) significantly (P<0.05) enhances the nociceptive effects of minimally effective doses (30 nmol paw−1) of intradermal BzATP and α,β meATP in the rat. Administration of cibacron blue (CB 100 CONTRA) into the paw contralateral to P2X agonist injection was ineffective. Values represent mean±s.e.mean, *P<0.05 as compared to vehicle-treated rats.
Since intermediate doses of cibacron blue (30 and 100 nmol paw−1) were found to be most effective in enhancing the nociceptive effects of intradermal BzATP, these doses of cibacron blue were also examined for their ability to enhance nociception in the formalin test. In the acute phase (Phase I) of the formalin test, intradermal cibacron blue alone produced a significant and dose-dependent nociceptive response (Figure 7). Co-administration of intradermal cibacron blue (30 and 100 nmol paw−1) with various concentrations of formalin (1, 2.5 and 5%) also produced greater nociception in the acute phase (Phase I) of the formalin test as compared to the effects of formalin alone (Figure 7). However, these effects appeared to be additive with formalin since a significant interaction between the nociceptive effects of formalin and cibacron blue was not observed (P>0.05) (Figure 7).
Figure 7.
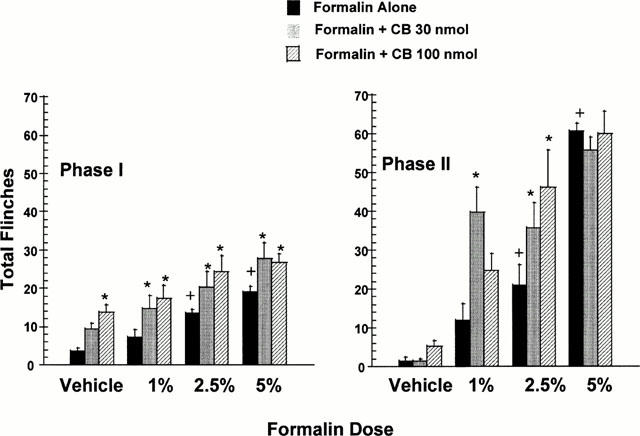
Cibacron blue increases nociceptive paw flinching in both Phase I and Phase II of the rat formalin test. Cibacron blue (CB, 30 and 100 nmol paw−1) was co-administered with intradermal formalin (1 – 5%) into the rat hind paw. Phase I nociceptive responses were recorded for the first 15 min following administration. Phase II nociceptive responses were recorded for a 20-min period beginning 30 min after administration. Intradermal administration of increasing doses of formalin alone produced significant increases in nociceptive responses (+P<0.05) as compared to vehicle injections. Values represent mean±s.e.mean (n=6 per dose group), *P<0.05 as compared to formalin alone.
During the persistent nociceptive component (Phase II) of the formalin test, intradermal cibacron blue alone did not produce a significant (P>0.05) nociceptive response (Figure 7). However, co-administration of cibacron blue (30 and 100 nmol paw−1) with formalin (1 and 2.5%) produced significantly greater paw flinching behaviour relative to the nociceptive effects of either formalin or cibacron blue administered alone (Figure 7). In this persistent portion of the formalin test, cibacron blue significantly potentiated the nociceptive effects of 1% and 2.5% formalin as indicated by a significantly (P<0.05) greater than additive interaction between the nociceptive effects of formalin and cibacron blue. At a minimally nociceptive dose of formalin (1%), the pronociceptive effects of cibacron blue were biphasic with 30 nmol paw−1 of cibacron blue producing a significantly larger enhancement of persistent nociception as compared to the higher dose of cibacron blue (100 nmol paw−1) (Figure 7). The intradermal administration of 5% formalin produced significantly greater nociception relative to lower doses of formalin, however, co-administration of cibacron blue with this dose of formalin did not produce a further enhancement of paw flinching behaviour.
The pro-nociceptive enhancing effects of cibacron blue appear to be pharmacologically specific since a structurally similar cibacron blue analogue reactive orange which does not allosterically modulate P2X3 receptor activation (Alexander et al., 1999), did not produce nociception alone following intradermal administration (30 and 100 nmol paw−1) and had no effect on acute or persistent nociceptive paw flinching when co-administered with formalin (Figure 8).
Figure 8.

Reactive orange does not increase nociceptive paw flinching in either Phase I and Phase II of the rat formalin test. Reactive Orange (RO, 30 and 100 nmol paw−1) was co-administered with intradermal formalin (2.5%) into the rat hind paw. Phase I nociceptive responses were recorded for the first 15 min following administration. Phase II nociceptive responses were recorded for a 20-min period begining 30 min after administration. Intradermal administration of increasing doses of formalin alone produced significant increases in nociceptive responses. Values represent mean±s.e.mean (n=6 per dose group), *P<0.05 as compared to vehicle alone.
Discussion
The present data demonstrating that the local administration of prototypic P2X receptor agonists produce acute paw flinching behaviour is consistent with and extends other recent findings that implicate a role for P2X3 containing channels in nociception (Bland-Ward & Humphrey, 1997; Dowd et al., 1998; Hamilton et al., 1999). In particular, the present studies show that a putative P2X7 receptor agonist, BzATP (El-Moatassim & Dubyak, 1992), which is in fact a more potent agonist for P2X3, and P2X2/3 receptors (Bianchi et al., 1999), can produce nociceptive responses in the rat that are similar in magnitude to that produced by the acute intradermal administration of 5% formalin. The pattern of in vivo activity for P2X receptor agonists to induce nociception is consistent with previous pharmacological evaluations of prototypic nucleotide agonists to activate recombinant homomeric P2X3 and heteromeric P2X2/3 receptors in vitro (Bianchi et al., 1999).
Similar to other reports (Bland-Ward & Humphrey, 1997; Hamilton et al., 1999), relatively high intradermal doses of the P2 receptor agonists were required to elicit acute nociceptive responses in naive rats. This situation may reflect the susceptibility of these agonists to be rapidly degraded in vivo and/or penetrate to sites of action (Bland-Ward & Humphrey, 1997; Hamilton et al., 1999). As the present data show, however, intradermal BzATP was a significantly more efficacious nocifensive stimulus as compared to other P2X receptor agonists like α,βmeATP and ATP such that a high dose of BzATP (1000 nmol paw−1) produced a similar degree of acute nociceptive behaviour as compared to 5% formalin.
Since the potent P2X receptor antagonist, TNP-ATP may also be rapidly degraded in vivo (Lewis et al., 1997), the potential antinociceptive effects of TNP-ATP were assessed following peripheral intradermal administration into the rat hindpaw in an attempt to maximize the local concentration of TNP-ATP at a relevant site of action. The present data demonstrate that TNP-ATP, administered locally, can effectively attenuate nociception produced by either exogenously administered P2X receptor agonist (BzATP and α,βmeATP) or formalin. The antinociceptive effects of TNP-ATP appear to be pharmacologically specific since the less active analogue, TNP-AMP, was ineffective in reducing the nociceptive effects of either BzATP (1000 nmol paw−1) or 5% formalin. These antinociceptive effects of intradermal TNP-ATP extend other recent data, demonstrating that the i.t. administration of TNP-ATP blocks acute thermal hyperalgesia produced by i.t. administration of P2X receptor agonists (Tsuda et al., 1999b) and reduces acute nociception produced by intradermally administered formalin and capsaicin (Tsuda et al., 1999b).
The ability of TNP-ATP to attenuate dose-dependently acute nociception produced by either BzATP or formalin was similar. Additionally, the antinociceptive effects of peripherally administered TNP-ATP were also evident during the persistent portion (Phase II) of the formalin test. This latter result contrasts with a recent report indicating that i.t. administration of TNP-ATP attenuates Phase I, but not Phase II, nociception in a murine formalin test regardless of whether TNP-ATP administration occurred before or after formalin administration (Tsuda et al., 1999b). However, another more stable P2 receptor antagonist, PPADS, was equally effective in reducing nociception in both phases of the formalin test following i.t. administration (Tsuda et al., 1999b), further supporting the idea that TNP-ATP may have a short half-life in vivo. The ability of peripherally administered TNP-ATP to attenuate formalin-induced persistent nociception in the present study may also have resulted from the use of significantly higher doses as compared to the doses used for i.t. administration (Tsuda et al., 1999b). Taken together, these results suggest that activation of both peripheral and spinal P2X receptors may contribute to the expression of nociception following tissue injury. Further, the ability of intradermal TNP-ATP to attenuate paw flinching in the formalin model indicates that activation of peripheral P2X receptors contributes to nociception in both the acute afferent barrage and persistent central sensitization phases of this model (Coderre & Melzack, 1992).
Since TNP-ATP is a potent antagonist that is selective for the P2X1, P2X3 and P2X2/3 receptors as compared to other P2X receptors (Lewis et al., 1998), the analgesic actions of TNP-ATP against both BzATP and formalin induced nociception support a role for the activation of these P2X receptor subunits in nociceptive signalling. There is evidence that P2X3 and P2X2/3 receptors are localized to peripheral and central processes of sensory afferent nerves (Vulchanova et al., 1997) and P2X3 and P2X2/3 responses are present on dorsal root ganglion neurones (Burgard et al., 1999). However, significant involvement of P2X1 receptors in nociceptive signalling does not appear likely since P2X1 mRNA is not highly expressed in dorsal root ganglion neurones (Lewis et al., 1995; Kidd et al., 1995; Collo et al., 1996), P2X1-mediated electrophysiological responses are not detected in rat dorsal root ganglion neurones (Rae et al., 1998), and i.t. administration of a putative P2X1 selective agonist, β,γ-(l)-meATP does not produce thermal hyperalgesia in rats (Tsuda et al., 1999a).
A specific contribution of P2X3 containing channels to the nociceptive effects of exogenously administered P2X receptor agonists and formalin is indicated further by the present demonstration that cibacron blue can selectively enhance the nociceptive effects of these agents. Previous work has shown that cibacron blue selectively enhances human P2X3 receptor activation by increasing agonist potency and efficacy, as well as facilitating receptor resensitization following acute agonist-induced desensitization (Alexander et al., 1999). In the present studies, the ability of cibacron blue to enhance agonist activation of the rat P2X3 receptor in vitro was essentially identical to its allosteric effects on the human P2X3 receptor (Alexander et al., 1999). Additionally, the present data show that cibacron blue also enhances agonist activation of the rat heteromeric P2X2/3 receptor in vitro. However, the allosteric effects of cibacron blue on rat P2X2/3 receptors are complex in that biphasic enhancement of receptor activation was observed as the concentration of cibacron blue was increased. A contribution of homomeric P2X2 receptors to these biphasic effects appears unlikely since cibacron blue produced similar effects on both BzATP- and α,β-meATP-mediated activation of rat P2X2/3 receptors and α,β-meATP does not activate P2X2 receptors (Lewis et al., 1995; Bianchi et al., 1999). Additionally, cibacron blue has been previously shown not to enhance agonist activation of the human P2X1, P2X2, and P2X7 receptors (Alexander et al., 1999). Thus, cibacron blue, at low micromolar concentrations, is highly selective for enhancing agonist activation of homomeric P2X3 and heteromeric P2X2/3 receptors as compared to its nonselective antagonist activity at other P2X receptor subtypes at higher concentrations (Ralevic & Burnstock, 1998; Alexander et al., 1999).
While the exact reasons for these apparent differences in the effects of cibacron blue on homomeric P2X3 and heteromeric P2X2/3 receptors in vitro remain unclear, the intrinsic antagonist properties of cibacron blue (Ralevic & Burnstock, 1998) may be more pronounced for the P2X2/3 receptor as compared to the P2X3 receptor. In this regard, the allosteric effects of cibacron blue are analogous to pharmacological actions of other allosteric modulators. For example, the selective allosteric enhancer of adenosine A1 receptors, PD 81,723 also shows intrinsic antagonist properties at high concentrations (Bruns & Fergus, 1990; Linden, 1997; Jarvis et al., 1999). Regardless, both the in vitro and in vivo data are consistent in that the allosteric enhancement effects of cibacron blue on P2X3 and P2X2/3 receptors are manifested at specific concentrations (and doses). At higher concentrations (and doses), the allosteric effects of cibacron blue are minimised and/or receptor antagonism may predominate due to its intrinsic and nonselective P2 receptor antagonist properties (Ralevic & Burnstock, 1998). The pronociceptive effects of cibacron blue in vivo appear to be pharmacologically specific since another anthraquinone sulphonic acid derivative, reactive orange, which does not alter P2X3 receptor function (Alexander et al., 1999), did not enhance the nociceptive effects of intradermal formalin.
The pronociceptive effects of cibacron blue may also depend on the nociceptive state of the animal. Specifically, the ability of cibacron blue to enhance BzATP-induced nociception was optimal at an intermediate dose of 30 nmol paw−1. This dose of cibacron blue was also found to potentiate significantly the persistent nociceptive effects of intradermal 1% and 2.5% formalin. Higher doses of cibacron blue produced less enhancement and/or antagonism of nociception induced by high doses of BzATP. Cibacron blue was also ineffective in enhancing the persistent portion (Phase II) of the standard (5%) formalin test. These data suggest that under situations where the intradermal nociceptive stimulus engenders a high rate of nocifensive paw flinching behaviour, the ability of cibacron blue to provide a further increase in the nocifensive behaviour may be reduced.
The ability of cibacron blue to enhance the nociceptive responses produced by the local administration of an exogenous P2X receptor agonist, or formalin, coupled with the ability of TNP-ATP to attenuate effectively the nociceptive effects of these noxious stimuli, provides further pharmacological evidence that activation of both the homomeric P2X3 and heteromeric P2X2/3 receptors contribute specifically to pain signalling. The present data, however, do not directly address the relative contributions of each of these P2X3-containing channels to the initiation and maintenance of nociception. Since both receptors share a highly similar pharmacological profile (Bianchi et al., 1999), delineation of specific nociceptive contributions by each receptor subtype has been based on functional responses. Demonstrations that P2X receptor-mediated nociceptive responses can undergo agonist-induced desensitization may indicate that such responses are mediated by the fast desensitizing P2X receptor subtypes, such as the P2X3 receptor (Bland-Ward & Humphrey, 1997; Tsuda et al., 1999Tsuda et al., 1999). A recent report has demonstrated that nociceptive paw flinching induced by P2X receptor agonists and formalin are mediated by capsaicin-sensitive C-fibre neurones that predominately express fast-desensitizing P2X3-like currents in response to ATP (Tsuda et al., 2000). Interestingly, this study also showed that local injection of P2X receptor agonists into the rat paw produced tactile allodynia that was mediated by capsaicin-insensitive medium-sized neurones that express predominately slow-desensitizing P2X2/3-like currents (Tsuda et al., 2000). These findings suggest that the pharmacological actions of TNP-ATP and cibacron blue in modulating acute nociceptive paw flinching behaviour may be mediated predominately by specific actions at homomeric P2X3 receptors localized to small diameter nociceptors. However, both P2X3 and P2X2/3 receptor activation may mediate longer-lasting nociceptive responses, as well as nociception involving mechanical hyperalgesia and allodynia (Tsuda et al., 2000). Further clarification of the relative roles of P2X3 and P2X2/3 receptors in these nociceptive responses will be dependent on the development of ligands that show pharmacological selectivity between the homomeric and heteromeric P2X3 containing channels.
Acknowledgments
The authors woud like to thank Dr Prisca Honore for comments on an earlier version of this manuscript.
Abbreviations
- ATP
adenosine-5′-triphosphase
- BzATP
2′ and 3′-O-(4-benzoylbenzoyl)-ATP
- TNP-ATP
2′,3′-O-(2,4,6-trinitrophenyl)-ATP
- α,β-meATP
α,β-methyleneATP
- 2meSATP
2-methylthioATP
- ADP
adenosine-5′-diphosphate
References
- ABBOTT F.V., FRANKLIN K.B.J., WESTBROOK R.F. The formalin test: scoring properties of the first and second phases of the pain response in rats. Pain. 1995;60:91–102. doi: 10.1016/0304-3959(94)00095-V. [DOI] [PubMed] [Google Scholar]
- ALEXANDER K., NIFORATOS W., BIANCHI B., BURGARD E.C., LYNCH K.J., KOWALUK E.A., JARVIS M.F., VAN BIESEN T. Allosteric modulation and accelerated resensitization of human P2X3 receptors by cibacron blue. J. Pharmacol. Exp. Ther. 1999;291:1135–1142. [PubMed] [Google Scholar]
- BIANCHI B.R., LYNCH K.J., TOUMA E., NIFORATOS W., BURGARD E.C., ALEXANDER K.M., PARK H.S., YU H., METZGER R., KOWALUK E.A., JARVIS M.F., VAN BIESEN T. Eur. J. Pharmacol. 1999. pp. 127–138. [DOI] [PubMed]
- BLAND-WARD P.A., HUMPHREY P.P.A. Acute nociception mediated by hindpaw P2X receptor activation in the rat. Br. J. Pharmacol. 1997;122:366–371. doi: 10.1038/sj.bjp.0701371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BLEEHEN T., KEELE C.A. Observation on the algogenic actions of adenosine compounds on the human blister base preparation. Pain. 1977;3:367–377. doi: 10.1016/0304-3959(77)90066-5. [DOI] [PubMed] [Google Scholar]
- BRUNS R.F., FERGUS J.H. Allosteric enhancement of adenosine A1 receptor binding and function by 2-amino-3-benzoylthiophenes. Mol. Pharmacol. 1990;38:939–949. [PubMed] [Google Scholar]
- BURGARD E.C., NIFORATOS W., VAN BIESEN T., LYNCH K.J., TOUMA E., METZGER R.E., KOWALUK E.A., JARVIS M.F. P2X receptor-mediated ionic currents in dorsal root ganglion neurons. J. Neurophysiol. 1999;82:1590–1598. doi: 10.1152/jn.1999.82.3.1590. [DOI] [PubMed] [Google Scholar]
- BURNSTOCK G. Purinergic nerves. Pharmacol. Rev. 1972;24:509–581. [PubMed] [Google Scholar]
- BURNSTOCK G. A unifying purinergic hypothesis for the initiation of pain. Lancet. 1996;347:1604–1605. doi: 10.1016/s0140-6736(96)91082-x. [DOI] [PubMed] [Google Scholar]
- BURNSTOCK G., WOOD J.N. Purinergic receptors: their role in nociception and primary afferent neurotransmission. Curr. Opinion Neurobiol. 1996;6:526–532. doi: 10.1016/s0959-4388(96)80060-2. [DOI] [PubMed] [Google Scholar]
- CHEN C.-C., AKOPIAN A.N., SIVILOTTI L., COLQUHOUN D., BURNSTOCK G., WOOD J.N. A P2X purinoceptor expressed by a subset of sensory neurons. Nature. 1995;377:428–431. doi: 10.1038/377428a0. [DOI] [PubMed] [Google Scholar]
- CHEN Y., SHU Y., ZHAO Z. Ectopic purinergic sensitivity develops at sites of chronic nerve constriction injury in rat. NeuroReport. 1999;10:2779–2782. doi: 10.1097/00001756-199909090-00015. [DOI] [PubMed] [Google Scholar]
- CODERRE T.J., MELZACK R. The contribution of excitatory amino acids to central sensitization and persistent nociception after formalin-induced tissue injury. J. Neurosci. 1992;12:3665–3670. doi: 10.1523/JNEUROSCI.12-09-03665.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COLLO G., NORTH R.A., KAWASHIMA R., MERLO-PICH E., NEIDHART S., SURPRENANT A. Cloning of P2X5 and P2X6 receptors and the distribution and properties of an extended family of ATP-gated ion channels. J. Neurosci. 1996;16:2495–2507. doi: 10.1523/JNEUROSCI.16-08-02495.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COOK S.P., VULCHANOVA L., HARGREAVES K.M., ELDE R., MCCLESKEY E.W. Distinct ATP receptors on pain-sensing and stretch-sensing neurons. Nature. 1997;387:505–508. doi: 10.1038/387505a0. [DOI] [PubMed] [Google Scholar]
- DOWD E., MCQUEEN D.S., CHESSELL I.P., HUMPHREY P.P.A. P2X receptor-mediated excitation of nociceptive afferents in the normal and arthritic rat knee joint. Br. J. Pharmacol. 1998;125:341–346. doi: 10.1038/sj.bjp.0702080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DRIESSEN B., REIMANN W., SELVE N., FRIDERICHS E., BULTMANN R. Antinociceptive effect of intrathecally administered P2-purinoceptor antagonists in rats. Brain Res. 1994;666:182–188. doi: 10.1016/0006-8993(94)90770-6. [DOI] [PubMed] [Google Scholar]
- EL-MOATASSIM C., DUBYAK G.R. A novel pathway for the activation of phospholipase D by P2Z purinergic receptors in BAC1.2F5 macrophages. J. Biol. Chem. 1992;267:23664–23673. [PubMed] [Google Scholar]
- GU J.G., MACDERMOTT A.B. Activation of ATP P2X receptors elicits glutamate release from sensory neuron synapses. Nature. 1997;389:749–753. doi: 10.1038/39639. [DOI] [PubMed] [Google Scholar]
- HAMILTON S.G., WADE A., MCMAHON S.B. The effects of inflammation and inflammatory mediators on nociceptive behavior induced by ATP analogues in the rat. Br. J. Pharmacol. 1999;126:326–332. doi: 10.1038/sj.bjp.0702258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAMILTON S.G., WARBURTON J., BHATTACHARJEE A., WARD J., MCMAHON S.B. ATP in human skin elicits a dose-related pain response under conditions of hyperalgesia. Brain. 2000;123:1238–1246. doi: 10.1093/brain/123.6.1238. [DOI] [PubMed] [Google Scholar]
- HOULTON P. The liberation of adenosine triphosphate on antidromic stimulation of sensory nerves. J. Physiol. 1959;145:494–504. doi: 10.1113/jphysiol.1959.sp006157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JAHR C.E., JESSELL T.M. ATP excites a subpopulation of rat dorsal horn neurones. Nature. 1983;304:730–733. doi: 10.1038/304730a0. [DOI] [PubMed] [Google Scholar]
- JARVIS M.F., GESSNER G., COX B.F., SHAPIRO G., MERKEL L., MYERS M., MARTIN G.E. Differential effects of the adenosine A1 receptor allosteric enhancer PD 81, 723 on agonist binding to brain and adipocyte membranes. Brain Res. 1999;840:75–83. doi: 10.1016/s0006-8993(99)01747-3. [DOI] [PubMed] [Google Scholar]
- KIDD E.J., GRAHAEMES C.B.A., SIMON J., MICHEL A.D., BARNARD E.A., HUMPHREY P.P.A. Localization of P2x purinoceptor transcripts in the rat nervous system. Mol. Pharmacol. 1995;48:569–573. [PubMed] [Google Scholar]
- KING B.F., TOWNSEND-NICHOLSON A., BURNSTOCK G. Metabotropic receptors for ATP and UTP: exploring the correspondence between native and recombinant nucleotide receptors. Trends Pharmacol. Sci. 1998;19:506–514. doi: 10.1016/s0165-6147(98)01271-1. [DOI] [PubMed] [Google Scholar]
- LEWIS C., NEIDHART S., HOLY C., NORTH R.A., BUELL G., SURPRENANT A. Coexpression of P2X2 and P2X3 receptor subunits can account for ATP-gated currents in sensory neurones. Nature. 1995;377:432–435. doi: 10.1038/377432a0. [DOI] [PubMed] [Google Scholar]
- LEWIS C.J., SURPRENANT A., EVANS R.J. 2′,3′-O-(2,4,6-trinitrophenyl) adenosine 5′-triphosphate (TNP-ATP)-a nanomolar affinity antagonist at rat mesenteric artery P2X receptor ion channels. Br. J. Pharmacol. 1998;124:1463–1466. doi: 10.1038/sj.bjp.0702001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LINDEN J.Allosteric enhancement of adenosine receptors Purinergic Approaches in Experimental Therapeutics 1997Wiley-Liss, Inc.: New York; 85–97.eds. Jacobson, K.A., Jarvis, M.F. [Google Scholar]
- NOVAKOVIC S.D., KASSOTAKIS L.C., OLGESBY I.B., SMITH J.A.M., EGLEN R.M., FORD A.P.D.W., HUNTER J.C. Immunocytochemical localization of P2X3 purinoceptors in sensory neurons in naïve rats and following neuropathic injury. Pain. 1999;80:273–282. doi: 10.1016/s0304-3959(98)00225-5. [DOI] [PubMed] [Google Scholar]
- RAE M.G., ROWAN E.G., KENNEDY C. Pharmacological properties of P2X3-receptors present in neurons of the rat dorsal root ganglia. Br. J. Pharmacol. 1998;124:176–180. doi: 10.1038/sj.bjp.0701803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RALEVIC V., BURNSTOCK G. Receptors for purines and pyrimidines. Pharmacol. Rev. 1998;50:413–492. [PubMed] [Google Scholar]
- SAWYNOK J., REID A. Peripheral adenosine 5′-triphosphate enhances nociception in the formalin test via activation of a purinergic P2X receptor. Eur. J. Pharmacol. 1997;330:115–121. doi: 10.1016/s0014-2999(97)01001-7. [DOI] [PubMed] [Google Scholar]
- THOMAS S., VIRGINIO C., NORTH R.A., SURPRENANT A. The antagonist trinitrophenyl-ATP reveals co-existence of distinct P2X receptor channels in rat nodose neurones. J. Physiol. 1998;509:411–417. doi: 10.1111/j.1469-7793.1998.411bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TJOSEN A., BERGE O.G., HUNSKAAR S., ROSLAND J.H., HOLE K. The formalin test: an evaluation of the method. Pain. 1992;51:5–17. doi: 10.1016/0304-3959(92)90003-T. [DOI] [PubMed] [Google Scholar]
- TSUDA M., UENO S., INOUE K. In vivo pathway of thermal hyperalgesia by intrathecal administration of α,β-methyleneATP in mouse spinal cord: Involvement of glutamate-NMDA receptor system. Br. J. Pharmacol. 1999a;127:449–456. doi: 10.1038/sj.bjp.0702582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TSUDA M., UENO S., INOUE K. Evidence for the involvement of spinal endogenous AATP and P2X receptors in nociceptive responses caused by formalin and capsaicin in mice. Br. J. Pharmacol. 1999b;128:1497–1504. doi: 10.1038/sj.bjp.0702960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TSUDA M., KOIZUMI S., KITA A., SHIGEMOTO Y., UENO S., INOUE K. Mechanical allodynia caused by intraplantar injection of P2X receptor agonist in rats: Involvement of heteromeric P2X2/3 receptor signaling in capsaicin-insensitive primary afferent neurons. J. Neurosci. 2000;20:RC90. doi: 10.1523/JNEUROSCI.20-15-j0007.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VIRGINIO C., ROBERTSON G., SUPRENANT A., NORTH R.A. Trinitrophenyl-substituted nucleotides are potent antagonists selective for P2X1, P2X3 and heteromeric P2X2/3 receptors. Mol. Pharmacol. 1998;53:969–973. [PubMed] [Google Scholar]
- VULCHANOVA L., RIEDL M.S., SHUSTER S.J., BUELL G., SUPRENANT A., NORTH R.A., ELDE R. Immunohistochemical study of the P2X2 and P2X3 receptor subunits in rat and monkey sensory neurons and their central terminals. Neuropharmacology. 1997;36:1229–1242. doi: 10.1016/s0028-3908(97)00126-3. [DOI] [PubMed] [Google Scholar]
- VULCHANOVA L., RIEDL M.S., SHUSTER S.J., STONE L.S., HARGREAVES K.M., BUELL G., SUPRENANT A., NORTH R.A., ELDE R. P2X3 is expressed by DRG neurons that terminate in inner lamina II. Eur. J. Neurosci. 1998;10:3470–3478. doi: 10.1046/j.1460-9568.1998.00355.x. [DOI] [PubMed] [Google Scholar]