

Abstract
Nerve injury often produces long-lasting spontaneous pain, hyperalgesia and allodynia that are refractory to treatment, being only partially relieved by clinical analgesics, and often insensitive to morphine. With the aim of assessing its therapeutic potential, we examined the effect of antisense oligonucleotide knockdown of spinal metabotropic glutamate receptor 1 (mGluR1) in neuropathic rats.
We chronically infused rats intrathecally with either vehicle, or 50 μg day−1 antisense or missense oligonucleotides beginning either 3 days prior to or 5 days after nerve injury. Cold, heat and mechanical sensitivity was assessed prior to any treatment and again every few days after nerve injury.
Here we show that knockdown of mGluR1 significantly reduces cold hyperalgesia, heat hyperalgesia and mechanical allodynia in the ipsilateral (injured) hindpaw of neuropathic rats.
Moreover, we show that morphine analgesia is reduced in neuropathic rats, but not in sham-operated rats, and that knockdown of mGluR1 restores the analgesic efficacy of morphine.
We also show that neuropathic rats are more sensitive to the excitatory effects of intrathecally injected N-methyl-D-aspartate (NMDA), and have elevated protein kinase C (PKC) activity in the spinal cord dorsal horn, two effects that are reversed by knockdown of mGluR1.
These results suggest that activity at mGluR1 contributes to neuropathic pain through interactions with spinal NMDA receptors and PKC, and that knockdown of mGluR1 may be a useful therapy for neuropathic pain in humans, both to alleviate pain directly, and as an adjunct to opioid analgesic treatment.
Keywords: Knockdown, antisense oligonucleotide, metabotropic glutamate receptor, neuropathy, hyperalgesia, allodynia, pain, opioid analgesia, NMDA sensitivity, nerve injury
Introduction
Neuropathic pain refers to pain due to injury or diseases of the peripheral or central nervous system (McQuay, 1997; Portenoy, 1992). Neuropathic pain is associated with severe, chronic sensory disturbances characterized by spontaneous pain, increased responsiveness to painful stimuli (hyperalgesia), and pain perceived in response to normally innocuous stimuli (allodynia). Prevalent symptoms in human patients include cold hyperalgesia, mechanical allodynia, and less commonly, heat hyperalgesia (McQuay, 1997; Portenoy, 1992). Approximately 4 million people in North America suffer from chronic neuropathic pain, and of these, no more than half achieve adequate pain control (Hansson, 1994). Moreover, neuropathic pain is particularly difficult to treat, often being only partially relieved by high doses of opioids in both humans (Cherny et al., 1994; MacDonald, 1991; McQuay et al., 1992) and rats (Ossipov et al., 1995a,1995b).
Recently, the excitatory amino acid glutamate has been shown to play a role in neuropathic pain. Glutamate release is enhanced in the spinal cord of neuropathic rats (Al-Ghoul et al., 1993). It has also been shown that antagonists at one type of glutamate receptor, the N-methyl-D-aspartate (NMDA) receptor, alleviate heat hyperalgesia and mechanical allodynia associated with nerve injury in rats (Chaplan et al., 1997; Mayer et al., 1995; Yamamoto & Yaksh, 1992). As well as NMDA receptors, glutamate also acts as a family of metabotropic glutamate receptors (mGluRs) that are directly coupled, via guanine nucleotide regulatory (G) proteins to intracellular second messengers. This family of mGluRs is classified into three groups based on sequence homology, signal transduction mechanisms and receptor pharmacology (Hayashi et al., 1994; Conn & Pin, 1997; Schoepp & Conn, 1993). Group I mGluRs, which include mGluR1 and mGluR5, are positively coupled to phosphatidylinositol (PI) hydrolysis, and activation of these receptors results in an increase in intracellular Ca2+ concentration and protein kinase C (PKC) activity. Group II (mGluR2 and mGluR3) and group III (mGluR4,6,7,8) mGluRs are negatively coupled to activation of adenylate cyclase and the production of cyclic adenosine 3′,5′-monophosphate (cyclic AMP). Activity at mGluRs modulates activity at NMDA receptors. Specifically, activation of group I mGluRs enhances NMDA receptor activity (Bleakman et al., 1992) via a PKC mediated mechanism (Chen & Huang, 1992; Harvey & Collingridge, 1993; Kelso et al., 1992; Kitamura et al., 1993; Raymond et al., 1994), while activation of group II and III mGluRs attenuates NMDA receptor activity (Martin et al., 1997).
Recent evidence suggests that mGluRs may contribute to neuropathic pain in rats. Inhibition of group I mGluRs with intrathecal (i.t.) injection of the relatively selective antagonist (S)-4-carboxyphenylglycine ((S)-4CPG) (Fisher et al., 1998) reduced cold hyperalgesia and mechanical allodynia in neuropathic rats. However, (S)-4CPG is a non-selective antagonist of both mGluR1 and mGluR5, as well as an agonist at group II mGluRs (Hayashi et al., 1994). Furthermore, although we have shown selective in vivo anti-hyperalgesic effects with i.t. administration of mGluR1 and mGluR5 selective antibodies (Fundytus et al., 1998b), a clear demonstration of treatment receptor-specific effectiveness could not be definitively established. Moreover, these previous studies did not address the specific role of mGluR1 in reduced opioid sensitivity after nerve injury. While the basis of the refractoriness of neuropathic pain to opioid therapy is not known, the parallel etiology of neuropathic pain and opioid tolerance/dependence may provide a clue (Mao et al., 1995a; Mayer et al., 1995). Thus, in addition to its hypothesized spinal effects on neuropathic pain, inhibition of group I mGluRs (Fundytus & Coderre, 1994; 1999a,1999b), as well as inhibition of PKC and release of intracellular Ca2+ (Fundytus & Coderre, 1996; 1999a,1999b), in the brain attenuates the severity of precipitated withdrawal symptoms in rats treated chronically with morphine. Once again, the data on opioid dependence was obtained using relatively non-selective antagonists, therefore a specific role of mGluR1 needs to be assessed.
Current technology allows for direct targeting of genes with antisense (AS) oligonucleotides, which are purported to selectively reduce protein synthesis of specific gene products, including receptor proteins. This method uses short oligonucleotides (i.e. base sequences) which are complementary to, and designed to hybridize to, a target mRNA transcript via Watson-Crick base pairing. Formation of an oligonucleotide-mRNA duplex leads to mRNA inactivation and therefore inhibition of protein synthesis. Antisense oligonucleotides are more specific than previously used antagonists, and unlike with the use of antibodies, a direct effect on receptor protein can be easily quantified using Western blot analysis. Recent evidence suggests that knockdown of spinal mGluR1 may reduce pain transmission in rats (Young et al., 1998).
In an effort to develop better therapies for neuropathic pain, as well as to increase opioid efficacy in neuropathic patients, we examined the effects of decreasing mGluR1 receptor number with mGluR1 AS oligonucleotide treatment in rats with a chronic constriction injury of one sciatic nerve. These results were initially presented at scientific meetings (Fundytus et al., 1997; 1998a; 1999a,1999b). Here we show that spinal knockdown of mGluR1 reduces cold and hot hyperalgesia and mechanical allodynia in neuropathic rats. We also show that knockdown of mGluR1 restores morphine sensitivity, and reduces NMDA sensitivity in neuropathic rats. In addition, we show that translocation of [3H]-PDBu binding sites associated with nerve injury is prevented by knockdown of mGluR1.
Methods
Subjects and surgery
The present studies employed male Long Evans rats weighing 275 – 300 g at the beginning of the experiment (Charles River, PQ). Rats were housed in groups of 3 – 4, with food and water available ad libitum, on a 12 : 12 h light : dark cycle (lights on at 0730). All surgical and testing procedures conformed to the ethical guidelines enforced by the Clinical Research Institute of Montreal, and the Canadian Council on Animal Care. Nerve injury was induced by placing a 2 mm length of PE90 polyethylene tubing around one sciatic nerve (Mosconi & Kruger, 1996). Sham surgery consisted of exposing the nerve, but not placing a cuff. Intrathecal (i.t.) catheters were inserted using a lumbar catheterization method (Storkson et al., 1996). The catheter was attached to an Alzet osmotic mini-pump (ALZA Model 2001) containing either artificial cerebrospinal fluid, antisense (AS) oligonucleotide solution, or missense (MS) oligonucleotide solution.
Oligonucleotides and drugs
We designed an antisense (AS: 5′-GAG CCG GAC CAT TGT GGC-3′) oligonucleotide complementary to base pairs 371-388 of the rat mGluR1 gene RATGPCR. A missense (MS: 5′-GAG CCG AGC ACT GTG TGC-3′) oligonucleotide was designed by taking the AS sequence and mismatching four base pair couples. Oligonucleotides were purchased from Medicorp Inc (Montreal, QC, Canada). We used unmodified, phosphodiester-bonded (PO) oligonucleotides, which, although quickly degraded in blood, have been shown to be stable, and non-toxic, in the CNS (Akhtar & Agrawal, 1997; Wahlestedt, 1994; Whitesell et al., 1993; Phillips & Gyurko, 1995; Yaida & Nowak, 1995). We chose to use PO oligonucleotides because modified oligonucleotides, particularly phosphorothioate-modified oligonucleotides, have been shown to be toxic in the CNS (Akhtar & Agrawal, 1997; Whitesell et al., 1993). The vehicle used to dissolve the oligonucleotides, and as the vehicle treatment, was artificial cerebrospinal fluid ((mM): NaCl 128.6, KCl 2.6, MgCl2 1.0, CaCl2 1.4, phosphate buffered to pH 7.4; ACSF). Vehicle, AS and MS were continuously infused i.t. for 7 days, via the catheter, in a volume of 1 μl h−1. The daily dose of AS and MS was 50 μg day−1. We chose this dose of oligonucleotide based on previous experiments utilizing antisense technology. Effective knockdown of receptors has been achieved with doses as low as 1 μg day−1, up to doses as high as 720 μg day−1 (Wahlestedt, 1994). This dose of AS and MS oligonucleotides was not found to produce any motoric or sedative side-effects, as examined using placing, righting and grasping reflexes. Morphine (Sabex, QC, Canada) was dissolved in 0.9% saline, and injected intrathecally (i.t.), via lumbar puncture between vertebrae L4 and L5, in doses of 3, 10 or 30 μg in 20 μl.
Western blot analysis
Rats were quickly decapitated, and their spinal cords were pressure ejected and rapidly frozen. Spinal cords were stored at −70°C until analysis. The lumbar enlargement section of spinal cords were homogenized in Tris buffer containing protease inhibitors. Group I mGluRs are most likely found in the dorsal horn in lumbar spinal cord, with some expression in the intermediate grey matter and ventral horn (Hargett et al., 1998; Berthele et al., 1999; Alvarez et al., 2000). Concentration of protein in each sample was determined using the method of Bradford (1976). The concentration of protein in each sample fell on the linear portion of the curve. For separation, 20 μg of total protein was loaded onto the gel for electrophoresis. Proteins were separated by gel electrophoresis (SDS – PAGE; 7.5% polyacrylamide gel), and electrotransferred to PVDF membrane. The membrane was probed with an anti-rat mGluR1 or anti-rat mGluR5 antibody (primary antibody) raised in rabbits (Upstate Biotechnology, Lake Placid, NY, U.S.A.). These antibodies are raised against the C termini of the receptors, a region that is unique to each of these receptors, and specificity was verified with immunoblotting (Martin et al., 1992; Abe et al., 1992; Upstate Biotechnology). The primary antibody was later tagged with a peroxidase-conjugated anti-rabbit antibody (secondary antibody; Jackson Laboratories). The secondary antibody was detected by chemiluminescence (Boehringer Mannheim) and the membranes were apposed to Kodak Biomax MR film for 1 min. Band density was measured using Alpha Imager software. The mGluR1 is a protein of approximately 133 – 142 kD (Houamed et al., 1991; Martin et al., 1992; Masu et al., 1991) and the mGluR5 protein is approximately 128 kD (Abe et al., 1992). Note that separate gels were run for each analysis (mGluR1 in lumbar spinal cord at 7 days of oligonucleotide infusion; mGluR1 in thalamus/PAG at 7 days of oligonucleotide infusion; mGluR5 in lumbar spinal cord at 7 days of oligonucleotide infusion; mGluR1 in lumbar spinal cord 12 days after cessation of oligonucleotide infusion), therefore direct comparisons of raw density values between analyses cannot be made.
Spontaneous nociceptive behaviours
The ability of the selective group I mGluR agonist 3,5-dihydroxyphenylglycine (DHPG; Tocris Cookson, Ballwin, MO, U.S.A.) to induce spontaneous nociceptive behaviours (SNBs) (Fisher & Coderre, 1996a) was assessed in a separate group of rats. Intrathecal catheters were implanted as described above, and rats were infused with either ACSF, or 50 μg day−1 AS or MS oligonucleotides for 7 days. On the 7th day of treatment, rats were injected intrathecally via lumbar puncture with 50 nmol in 30 μl DHPG, and the time spent exhibiting SNBs over a 60 min observation period was recorded. SNBs included elevation of tail, licking of tail, elevation of hindpaws and licking of hindpaws. The time spent in each of these behaviours was combined into one ‘time spent exhibiting SNBs' score. Data were analysed by ANOVA followed by post-hoc Fisher's LSD t-tests on significant results.
The ability of intrathecal NMDA to induce pain behaviours was assessed in a separate group of nerve cuffed and sham-operated rats. NMDA (Sigma, Oakville, ON, Canada) was injected intrathecally (i.t.), via an i.t. catheter, in doses of 1.67 and 2.5 nmol in 20 μl. Rats were observed for a period of 8 min and the time spent exhibiting SNBs was recorded (time spent favouring paws, agitation, licking and biting paws).
Assessing neuropathic hyperalgesia and allodynia
In rats that were pre-treated with i.t. oligonucleotides, cold, hot and mechanical sensitivity were measured prior to any treatment (baseline) and again 4, 8, 12 and 16 days after nerve injury. In rats that were post-treated with i.t. oligonucleotides, cold, hot and mechanical sensitivity were measured prior to any treatment (baseline) and again 4 days after nerve injury (but before i.t. treatment), as well as 8, 12 and 18 days after nerve injury.
Cold sensitivity was measured by placing rats in a 1 cm deep 1°C cold water bath for 75 s, and counting the number of responses (lifting of hindpaw). Cold hyperalgesia was assessed by calculating the increase in number of responses, compared to baseline, from days 4 to 16 or 18 after nerve injury.
Mechanical sensitivity was measured by applying thin filaments (von Frey hairs) to the plantar surface of the hindpaw and determining the 50% response threshold (in grams) for paw withdrawal using the up-down method of filament presentation (Chaplan et al., 1994). Mechanical allodynia was assessed by calculating the per cent decrease in 50% response threshold (from baseline) on days 4 to 16 or 18 after nerve injury.
Heat sensitivity was measured by applying focussed radiant heat to the glass under the plantar surface of the hindpaw and measuring the latency for the rat to withdraw its paw (Hargreaves et al., 1988). A cut-off latency of 20 s was used to prevent tissue injury. Heat hyperalgesia was assessed by calculating the per cent decrease in latency (from baseline) on days 4 to 16 or 18 after nerve injury.
For tests of cold, mechanical and heat sensitivity, data from the pre-treatment schedule were analysed by repeated measures ANOVA with nerve injury condition and i.t. treatment as independent groups factors, and days post nerve injury as a repeated measures factor. Data from the post-treatment schedule were analysed by repeated measures ANOVA with i.t. treatment as the independent groups factor and days post nerve injury as the repeated measures factor. Significant results were further analysed with post-hoc Fisher's LSD t-tests.
Morphine dose-response curve
To assess the analgesic effect of intrathecal (i.t.) morphine sulphate, rats were tested by measuring their latency to withdraw their tail from 55°C water. A cut-off latency of 10 s was used to prevent tissue injury. Tail flick latencies were measured prior to any treatment (naïve rats before oligonucleotide infusion or nerve injury; BL1), and again 4 days after nerve injury (after 7 days of oligonucleotide infusion; BL2). In ACSF-treated neuropathic rats, BL2 (2.44±0.12) was slightly, but significantly higher than BL1 (2.01±0.22) (P<0.05), indicating that the rats were not hyperalgesic after sciatic nerve injury. There were no differences between BL1 and BL2 for any of the other treatment groups (ACSF sham BL1=2.01±0.15 and BL2=2.19±0.18; AS cuffed BL1=2.44 and BL2=2.46±0.16; MS cuffed BL1=1.90±0.17 and BL2=1.97±0.18). There were no differences between oligonucleotide-treated rats and ACSF-treated rats for either BL1 or BL2. These results indicate that rats with a sciatic nerve injury were not hyperalgesic in the tail, replicating the results of Ossipov et al. (1995a,1995b). Furthermore, because BL1 and BL2 were not different in oligonucleotide-treated rats versus vehicle-treated rats, this suggests that antisense treatment did not affect motor function and the ability to respond on this test. After the measurement of BL2, rats were injected intrathecally with morphine in a dose of either 3, 10 or 30 μg per 20 μl via lumbar puncture between vertebrae L4 and L5. Following morphine injection, tail flick latencies were measured every 15 min from 15 – 60 min post-morphine. Latency scores were converted to percent maximum possible effect scores [%MPE=((latency – BL2)/(cutoff – BL2))*100]. From the %MPE scores, we calculated the area under the curve (AUC) from 15 – 60 min post morphine injection (maximum AUC=300) to clarify the analgesic effect. We calculated the ED50 with 95% confidence intervals from the AUC scores, using a regression of the log dose response curve, with confidence intervals calculated according to the method of Goldstein (1964). We designed this experiment similarly to previously published studies of other investigators who showed that the tail is not hyperalgesic in sciatic nerve-injured rats, and who have demonstrated that NMDA antagonists restore morphine analgesic efficacy in neuropathic rats in a tail flick test (Ossipov et al., 1995b), as opposed to merely reversing hyperalgesia in the injured paw.
[3H]PDBu binding
To determine amount of membrane-bound (i.e. activated) PKC, we utilized a [3H]-phorbol-12,13-dibutyrate ([3H]-PDBu) binding assay (Olds et al., 1989; Worley et al., 1986). We have previously used this method to determine pain-related increases in membrane-bound PKC in spinal cord dorsal horn (Yashpal et al., 1995). It has previously been shown that [3H]-PDBu binding is enhanced in spinal cord in rats with a sciatic nerve injury (Mao et al., 1992a,1992b; 1995b). Traditionally, it is assumed that PKC has to be transported to the membrane and bind DAG to be activated. Although this does not provide absolute quantification of PKC activity, it does provide some indication of the activation of PKC. Four days after nerve injury (7 days of oligonucleotide infusion), rats were decapitated, and spinal cords quickly removed by pressure ejection and frozen at −70°C. Serial transverse sections (20 μm) of lumbar spinal cord were cut at −18°C and thaw mounted onto gelatin coated slides. Slides were dried overnight under a vacuum, and stored at −70°C until autoradiographic processing. Sections were pre-incubated for 1 h at 4°C in buffer containing 50 mM Tris-HCl (pH 7.7), 100 mM NaCl and 1 mM CaCl2. Sections were then incubated in buffer containing 0.1% bovine serum albumin and 2.5 nM [3H]-PDBu for 1 h at 33°C. Non-specific binding was assessed by adding excess unlabeled PDBu (1 μM). Computer images were generated and binding density was analysed using the MCID image analysis system.
Treatment schedule
Pre-treatment
Three days prior to nerve injury, rats were implanted with i.t. catheters attached to mini osmotic pumps containing either vehicle, or 50 μg day−1 AS or MS. Nerve injury was induced 3 days later, and rats were tested 4, 8, 12 and 16 days after nerve injury for cold hyperalgesia, hot hyperalgesia and mechanical allodynia. Oligonucleotides were infused for a total of 7 days (from 3 days prior to until 4 days after nerve injury). This pre-treatment schedule was employed to determine if mGluR1 is involved in the development of hyperalgesia and/or allodynia associated with nerve injury. Western blot analysis, [3H]-PDBu binding, morphine analgesia and NMDA sensitivity were assessed in rats with this treatment schedule (pre-treatment).
Post-treatment
We used a separate group of rats to determine whether mGluR1 is involved in the maintenance of neuropathic pain, and whether AS oligonucleotide knockdown of mGluR1 could reverse hyperalgesia and/or allodynia associated with an established neuropathy. Briefly, nerve injury was induced by placing a polyethylene cuff around one sciatic nerve as described above. Because there was no effect of oligonucleotide treatment in sham-operated rats when it was given as a pre-treatment, and would be expected to have the greatest effect, we did not include a sham-operated group in the post-treatment test. Rats were confirmed to be neuropathic (i.e. display hot and cold hyperalgesia and mechanical allodynia as described below) 4 days after nerve injury. Five days after nerve injury, intrathecal catheters were implanted as described above, and rats were chronically infused with either ACSF, or 50 μg day−1 AS or MS oligonucleotides for 7 days (from 5 – 12 days post nerve injury). Hot, cold and mechanical sensitivity was measured again on days 8, 12 and 18 post nerve constriction (corresponding to days 3, 7 and 13 after beginning oligonucleotide infusion).
Results
Western blot analysis
As a first step, Western blot analysis was performed to determine whether AS oligonucleotide treatment inhibited production of mGluR1 protein in the lumbar spinal cord or the thalamus/periaqueductal grey region of rats 4 days after nerve constriction (7 days of oligonucleotide infusion). It has recently been shown that deafferentation of the olfactory neuroepitheium induces an up-regulation of mGluR1α in the olfactory bulb (Casabona et al., 1998). In the present study, we saw an increase in mGluR1 protein in ACSF-treated neuropathic rats compared to ACSF-treated sham-operated rats (+44%). In neuropathic rats, intrathecal AS oligonucleotide treatment decreased mGluR1 protein in lumbar spinal cord by 57%, as compared to ACSF treatment (Figure 1A). In sham-operated rats, intrathecal AS oligonucleotide treatment decreased mGluR1 protein by 38% compared to ACSF-treated rats. In contrast, compared to ACSF treatment, MS oligonucleotide treatment decreased mGluR1 protein by only 1% in neuropathic rats, and increased mGluR1 protein by 44% in sham-operated rats (Figure 1A). By 12 days after oligonucleotide infusion was stopped, mGluR1 protein had recovered in lumbar spinal cord of neuropathic AS-treated rats (per cent difference in binding density compared to ACSF: AS=+22±27%, and MS=−7±16%, t-test P>0.05), indicating that the treatment was reversible. In the thalamus/periaqueductal grey region of the brain, AS treatment induced some decrease in mGluR1 protein compared to ACSF treatment in both neuropathic (AS-treated −25% compared to ACSF-treated) and sham-operated (AS-treated −18% compared to ACSF-treated) rats, suggesting that some of the behavioural effects may be due to actions in the brain as well as in the spinal cord (Figure 1B). MS treatment induced a slight increase (+4%) in brain mGluR1 levels in neuropathic rats, and a slight decrease (−10%) in sham-operated rats (Figure 1B). The spinal levels of mGluR5 appeared to be slightly decreased in neuropathic rats (AS-treated −8% compared to ACSF-treated rats), and more pronouncedly decreased in sham-operated rats (AS-treated −26% compared to ACSF-treated) (Figure 1C). However, because the effect was so minimal in neuropathic rats compared to the effects on mGluR1 protein, it is unlikely that a decrease in mGluR5 contributed significantly to the behavioural effects seen in neuropathic rats. MS treatment induced an increase (54%) in neuropathic rats, and a decrease (19%) in sham-operated rats, in mGluR5 protein compared to vehicle treatment. These results show that AS treatment induced a consistent reduction in the amount of mGluR1 protein.
Figure 1.

Western blot analysis. Representative Western blots (from a single animal per group) and histogram summaries (n=3 per group) from Western blot analysis of lumbar spinal cords and the thalamus/periaqueductal region of brains taken from ACSF-, AS- and MS-treated rats after 7 days of oligonucleotide infusion, and 4 days after nerve constriction (induced by implantation of a cuff around one sciatic nerve) or sham-surgery. Depicted in the histograms is the mean of the binding density scores for each group (±s.e.m.). (A) Mean binding density of anti-rat mGluR1 IgG in lumbar spinal cord. AS treatment induced a 57% decrease of mGluR1 protein compared to vehicle treatment in neuropathic rats, and a 38% decrease in mGluR1 protein compared to vehicle treatment in sham-operated rats. MS treatment induced a 1% decrease in mGluR1 protein compared to vehicle treatment in neuropathic rats, and a 44% increase in mGluR1 protein compared to vehicle treatment in sham-operated rats. (B) Mean binding density of anti-rat mGluR1 IgG in thalamus/periaqueductal region of brain. AS treatment induced a 25% decrease in neuropathic rats, and an 18% decrease in mGluR1 protein in sham-operated rats. MS treatment induced a 4% increase in neuropathic rats, and a 10% decrease in sham-operated rats, compared to vehicle-treatment. (C) Mean binding density of anti-rat mGluR5 IgG in lumbar spinal cord. AS treatment induced a slight decrease in mGluR5 protein in neuropathic rats (8%), and a more pronounced decrease in sham-operated rats (26%). MS treatment induced an increase (54%) in neuropathic rats, and a decrease (19%) in sham-operated rats in mGluR5 protein compared to vehicle treatment.
DHPG-induced spontaneous nociceptive behaviours
To confirm that our in vivo mGluR1 AS treatment produced a functionally relevant block of mGluR1 in behaving animals, we assessed the effects of the mGluR1 AS treatment on nociception induced by intrathecal injection of the selective group I mGluR agonist DHPG. DHPG produced spontaneous nociceptive behaviours (SNBs), including elevating, licking and biting of the hindpaws and tail. These nociceptive behaviours were significantly reduced in AS-treated, but not MS-treated rats (Figure 2), confirming that the AS treatment is capable of reducing nociception induced by mGluR1 receptor activation.
Figure 2.

DHPG-induced spontaneous nociceptive behaviours. Mean time spent exhibiting nociceptive behaviour after i.t. injection of 50 nmol DHPG in ACSF – (n=8), AS- (n=5) and MS-treated (n=5) rats. *Significantly different from ACSF-treated (P<0.05); †significantly different from MS-treated (P<0.05).
Cold sensitivity in neuropathic rats
Cold sensitivity was tested by counting the number of lifts of the injured (ipsilateral) hindpaw when rats were placed in 1 cm deep water at 1°C for 75 s. Pre-treated ACSF- and MS-treated neuropathic rats exhibited a large increase from baseline in response frequency (Figure 3A) compared to sham-operated rats, on days 4 to 16 after nerve injury. Pre-treated AS-treated neuropathic rats showed a significantly lower increase in response frequency (Figure 3A) in the cold water test, compared to ACSF- and MS-treated rats, and were not significantly different from sham-operated rats. There were no differences between i.t. treatment groups in sham-operated rats. There were also no differences between treatment groups in the contralateral paw (data not shown). When i.t. oligonucleotides were given as a post-treatment, all nerve-injured (neuropathic) rats displayed cold hyperalgesia 4 days after nerve injury, prior to i.t. infusion, as indicated by a large increase in response frequency (Figure 3B) of the ipsilateral paw, compared to baseline. On days 8, 12 and 18 after nerve injury (3, 7 and 13 days after beginning i.t. drug infusion) ACSF- and MS-treated neuropathic rats still exhibited a large increase from baseline in response frequency (Figure 3B) of the ipsilateral paw. In contrast, the increase in response frequency (Figure 3B) of the ipsilateral paw was significantly reduced in AS-treated neuropathic rats after i.t. treatment (Figure 3B). There were no differences between groups in the contralateral paw (data not shown).
Figure 3.
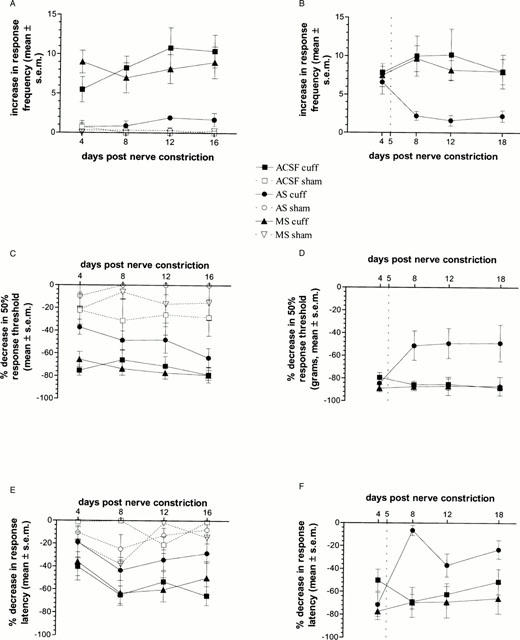
Nerve-injury induced hyperalgesia and allodynia. Change in response to cold, heat and mechanical stimulation on days 4, 8, 12 and 16 days (pre-treatment group) or 4, 8, 12 and 18 days (post-treatment group) after nerve constriction in neuropathic and sham-operated rats treated intrathecally with either ACSF (cuffed, n=12 pre-treatment, n=6 post-treatment; sham, n=4 pre-treatment), AS (cuffed, n=9 pre-treatment, n=5 post-treatment; sham, n=3 pre-treatment) or MS (cuffed, n=7 pre-treatment, n=6 post-treatment; sham, n=4 pre-treatment). (A) Cold water test in pre-treatment group: Mean increase in number of responses (lifting of hindpaw) when rats stood in water at 1°C. There was a significant interaction between neuropathic condition and i.t. treatment (F(2,33)=7.91, P<0.01). Post-hoc tests showed that AS-treated neuropathic rats had a significantly lower response frequency across test days compared to ACSF- or MS-treated neuropathic rats. (B) Cold water test in post-treatment group: Mean increase in number of responses after nerve injury, both prior to i.t. infusion (day 4), and after i.t. infusion (days 8, 12 and 18). All nerve injured rats showed a large increase in response frequency in the ipsilateral hindpaw, compared to baseline, on day 4 after nerve injury, prior to i.t. oligonucleotide infusion. The dotted line at day 5 indicates when the i.t. infusion of oligonucleotides began. There was a significant drug×day interaction (F(6,42)=2.30, P=0.05). Post-hoc Fisher's LSD t-tests showed that after i.t. infusion, AS-treated neuropathic rats showed a significantly lower increase in response frequency on days 8, 12 and 18 after nerve injury, compared to ACSF- and MS-treated rats regardless of test day. (C) von Frey hair test in pre-treatment group: Mean per cent decrease in 50% response threshold in grams. There were significant effects of neuropathic condition (F(1,33)=98.44, P<0.01), i.t. treatment (F(2,33)=9.23, P<0.01), and test day (F(3,99)=3.48, P<0.05). Post-hoc tests showed that neuropathic AS-treated rats had a significantly lower decrease in 50% response threshold than either ACSF- or MS-treated rats. (D) von Frey hair test in post-treatment group: Mean per cent decrease in 50% response threshold in grams. On day 4 after nerve injury, prior to i.t. drug infusion, all neuropathic rats showed a large decrease in 50% response threshold. The dotted line at day 5 indicates when the i.t. infusion of oligonucleotides began. There was a significant drug×day interaction (F(6,42)=3.35, P<0.05), and post-hoc tests showed that after i.t. infusion, neuropathic AS-treated rats had a significantly lower decrease in 50% response threshold than either ACSF- or MS-treated rats. (E) Radiant heat plantar test in pre-treatment group: Mean per cent decrease in response latency. There were significant effects of interaction between neuropathic condition and i.t. treatment (F(2,33)=3.26, P=0.05), and test day (F(3,99)=6.57, P<0.01). Post-hoc tests Fisher's LSD t-tests showed that AS-treated neuropathic rats had a significantly lower decrease in response latency than ACSF- or MS-treated neuropathic rats. (F) Radiant heat plantar test in post-treatment group: Mean per cent decrease in response latency. All nerve injured rats showed a large decrease in response latency, compared to baseline, on day 4 after nerve injury, prior to i.t. drug infusion. The dotted line at day 5 indicates when the i.t. infusion of oligonucleotides began. There was a significant drug×day interaction (F(6.42)=6.22, P<0.05), and post-hoc tests showed that after i.t. infusion, AS-treated neuropathic rats had a significantly lower decrease in response latency from days 8 to 18 after nerve injury.
Mechanical sensitivity in neuropathic rats
Mechanical sensitivity was assessed by determining the 50% response threshold, in grams, to stimulation of the plantar surface of the hindpaw with von Frey hairs. In the pre-treatment group, ACSF- and MS-treated neuropathic rats displayed a large decrease from baseline in 50% response threshold to von Frey hair stimulation of the ipsilateral hindpaw, as compared to sham-operated rats (Figure 3C). AS-treated neuropathic rats showed a significantly smaller decrease in response threshold, compared to ACSF- and MS-treated neuropathic rats, across test days, and were not different from sham-operated rats (Figure 3C). There were no differences between i.t. treatment groups in sham-operated rats (Figure 3C), or in the contralateral hindpaw (data not shown).
Similarly, in rats given post-nerve injury i.t. oligonucleotide infusion, all nerve injured rats displayed mechanical allodynia, as indicated by a large decrease in 50% response threshold in the ipsilateral hindpaw 4 days after nerve injury, prior to oligonucleotide infusion (Figure 3D). On days 8, 12 and 18 after nerve injury ACSF- and MS-treated neuropathic rats still exhibited a large decrease in 50% response threshold of the ipsilateral paw (Figure 3D). In contrast, the decrease in 50% response threshold in the ipsilateral hindpaw of AS-treated neuropathic rats was significantly lower than in ACSF- or MS-treated rats (Figure 3D) after i.t. oligonucleotide treatment. There were no differences between groups in the contralateral paw (data not shown).
Heat sensitivity in neuropathic rats
In the present study, heat sensitivity was measured by stimulating the plantar surface the hindpaw with a focused radiant heat source and measuring the latency of the rats to withdraw the paw. In the pre-treatment group, ACSF- and MS-treated neuropathic rats displayed a large decrease from baseline in withdrawal latency to radiant heat stimulation of the ipsilateral paw, as compared to sham-operated rats (Figure 3E). In AS-treated neuropathic rats this decrease was significantly less, compared to ACSF- and MS-treated neuropathic rats (Figure 3E). There were no differences between i.t. treatment groups in sham-operated rats (Figure 3E), or in the contralateral hindpaw (data not shown).
Similarly, in rats given post-nerve injury i.t. drug infusion, all nerve injured rats displayed heat hyperalgesia, as indicated by a large decrease in response latency in the ipsilateral hindpaw four days after nerve injury, prior to oligonucleotide infusion (Figure 3F). On days 8, 12 and 18 after nerve injury ACSF- and MS-treated neuropathic rats still exhibited a large decrease in response latency of the ipsilateral paw (Figure 3F). In contrast, the decrease in response latency in the ipsilateral hindpaw of AS-treated neuropathic rats was significantly lower than in ACSF- or MS-treated rats (Figure 3F) after i.t. treatment. There were no differences between groups in the contralateral paw (data not shown).
Morphine dose-response curve
Four days after injury, ACSF- and MS-treated neuropathic rats displayed a minimal analgesic response to intrathecal morphine in the tail withdrawal test (Figure 4) compared to sham-operated rats, which responded normally to intrathecal morphine. In contrast, AS-treated neuropathic rats displayed significant morphine analgesia, and were not different from sham-operated rats (Figure 4), indicating that AS treatment restored opioid sensitivity in neuropathic rats. Note that, as mentioned in the Methods section, the post-treatment baseline (prior to the i.t. injection of morphine) was not different from the pre-treatment baseline (prior to any i.t. infusion or nerve injury), therefore sciatic nerve injury did not cause hyperalgesia in the tail. Thus, our results in the present study indicate that AS treatment restored opioid sensitivity, rather than merely reversing hyperalgesia.
Figure 4.

Morphine dose-response curve. Morphine dose-response curve in neuropathic and sham-operated rats 4 days after nerve injury for ACSF- (n=15 each for cuffed and sham-operated), AS- (n=16, cuffed) and MS-treated (n=17, cuffed) rats. Prior to morphine injection, tail flick latencies were measured (BL2 as described in Methods). Following morphine injection, tail flick latencies were measured every 15 min from 15 – 60 min post-morphine. Latency scores were converted to percent maximum possible effect scores [%MPE=((latency −BL2)/(cutoff −BL2))*100]. From the %MPE scores, we calculated the area under the curve (AUC) from 15 – 60 min post morphine injection (maximum AUC=300) to determine the analgesic effect. The mean AUC for each treatment group at each morphine dose is illustrated in this figure. ACSF- and MS-treated neuropathic rats showed little analgesia after morphine was injected intrathecally (ED50=63.71 μg (21.38 μg-1950 mg) and 705.28 μg (95% C.I. not calculable) respectively). In contrast, AS-treated neuropathic rats showed a robust analgesic effect, and were not different from sham-operated rats (ED50=8.13 μg (5.25 – 12.02 μg) and 9.72 μg (4.68 – 20.89 μg) respectively).
[3H]PDBu binding
Figure 5A is a histogram summary of [3H]-PDBu binding density from all the slides in each treatment group, while Figure 5B is a computer-generated image showing [3H]-PDBu binding in a single representative slide from each treatment group. Because mGluR1 is positively coupled to PI hydrolysis, and thus to activation of PKC, the involvement of PKC in neuropathic pain was examined by measuring the binding of [3H]-phorbol,12,13-dibutyrate ([3H]-PDBu) in lumbar spinal cord slices of rats 4 days after nerve constriction (7 days of oligonucleotide infusion). When activated, PKC has been shown to migrate to the cellular membrane, and [3H]-PDBu binds to the diacylglycerol site of PKC once it has been translocated to the membrane (Olds et al., 1989; Worley et al., 1986). [3H]-PDBu binding was significantly increased in lumbar spinal cord dorsal horn from ACSF- and MS-treated rats with nerve injury, as compared to sham-operated rats (Figure 5). In contrast, in lumbar spinal cord dorsal horn from AS-treated neuropathic rats, [3H]-PDBu binding was significantly lower compared to ACSF and MS-treated neuropathic rats, and was not different from binding seen in spinal cords from sham-operated rats (Figure 5). Although the representative slide from an AS-treated sham-operated rats appears to show reduced [3H]-PDBu binding compared to the slides from an ACSF- and MS-treated sham-operated rats, statistical analysis of all the data indicated no significant difference between the sham-operated groups.
Figure 5.

[3H]-PDBu binding. [3H]-PDBu binding autoradiography in lumbar spinal cord of neuropathic and sham-operated rats. (A) Histogram summary showing the density of [3H]-PDBu binding (nCi g−1) in ACSF-, AS- and MS-treated neuropathic and sham-operated rats (n=3 rats per treatment combination, with five slides from each rat, for a total of 15 slides per group). Planned comparisons showed that ACSF-treated sham-operated rats had a lower binding density than ACSF-treated neuropathic rats; AS-treated neuropathic rats had a lower binding density than either ACSF- or MS-treated neuropathic rats. *Significantly different from ACSF-treated neuropathic rats; # significantly different from MS-treated neuropathic rats. (B) Computer-generated image (MCID) of a single representative slide from each group showing sample binding of [3H]-PDBu in ACSF-, AS- and MS-treated neuropathic and sham-operated rats.
NMDA-induced spontaneous nociceptive behaviours
It was found that ACSF- and MS-treated rats with nerve injury displayed increased nociceptive behaviour in response to intrathecal injection of NMDA, compared to sham-operated rats (Figure 6). However, AS oligonucleotide treatment significantly reduced the nociceptive behaviours induced by intrathecal injection of NMDA in nerve injured rats (Figure 6).
Figure 6.

NMDA-induced spontaneous nociceptive behaviours. Mean time spent exhibiting nociceptive behaviour in neuropathic (n=4 – 6 per dose) and sham-operated (n=4 – 6 per dose) rats given increasing doses of i.t. NMDA; and in neuropathic rats treated with either AS (n=11) or MS (n=10), and given 2.5 nmol of NMDA. *Significantly different from ACSF-treated (P<0.05); †significantly different from MS-treated (P<0.05); #significantly different from sham-operated (P<0.05).
Discussion
In the present study, we first showed that intrathecal infusion of our AS oligonucleotide greatly decreased mGluR1 protein in lumbar spinal cord, and slightly decreased it in the brain. We also showed that by 12 days post-infusion, the amount of mGluR1 protein in AS-treated neuropathic rats had recovered to levels similar to ACSF-treated rats, indicating that the effect was reversible. As a functional correlate of a decrease in mGluR1 protein, we showed that AS-treated rats displayed significantly fewer DHPG-induced SNBs. Moreover, AS oligonucleotide treatment significantly decreased cold hyperalgesia, mechanical allodynia and heat hyperalgesia of the ipsilateral hindpaw of neuropathic rats. The attenuation of neuropathic pain in mGluR1 AS-treated rats was accompanied by a concomitant decrease in activated PKC, as indicated by a decrease in [3H]-PDBu binding. In relation to the possible common aetiology of neuropathic pain and opioid tolerance/dependence, it is important to point out here that inhibition of both mGluR1 and PKC activity in the brain has been shown to attenuate the precipitated morphine withdrawal syndrome (Fundytus & Coderre, 1994; 1996; 1999a,1999b). Furthermore, spinal PKCγ is translocated to the membrane in neuropathic rats (Mao et al., 1995a,1995b), inhibition of PKC in the spinal cord reduces morphine tolerance and neuropathic pain (Mao et al., 1992Mao et al., 1992; 1995b), and PKCγ knockout mice do not develop neuropathy (Malmberg et al., 1997). The activation of PKC may underlie the effects seen with inhibition of either mGluR1 or NMDA receptors.
Clinically, neuropathic pain is often difficult to treat, being only partially relieved by high doses of opioids (Cherny et al., 1994; MacDonald, 1991; McQuay et al., 1992). We demonstrated that in this model of neuropathy, ACSF- and MS-treated neuropathic rats were less sensitive to the analgesic effects of intrathecally administered morphine, while mGluR1 AS-treated neuropathic rats displayed a normal analgesic response to morphine. Thus, knockdown of mGluR1 at the spinal level prevented the development of morphine insensitivity in neuropathic rats.
Activation of group I mGluRs has been shown to enhance activity at NMDA receptors (Bleakman et al., 1992) via a PKC-mediated mechanism (Chen & Huang, 1992; Harvey & Collingridge, 1993; Kelso et al., 1992; Kitamura et al., 1993; Raymond et al., 1994), and NMDA receptors have been shown to contribute to nociception in animal models of persistent nociception (Chaplan et al., 1997; Mayer et al., 1995; Yamamoto & Yaksh, 1992). Therefore, we predicted, based on our results from [3H]-PDBu binding, that NMDA receptor activity would be enhanced in neuropathic rats, and that neuropathic rats would therefore be more sensitive to the excitatory effects of NMDA injected intrathecally. We also predicted that mGluR1 AS oligonucleotide treatment would attenuate this enhanced NMDA receptor activity, and thus NMDA sensitivity, in neuropathic rats. In experiments designed to test these predictions, we showed that neuropathic rats with nerve injury displayed increased nociceptive behaviours in response to intrathecal injection of NMDA, and that this effect was reversed by mGluR1 AS oligonucleotide treatment. Although it is possible that mGluR1 AS treatment induced a reduction in the number of NMDA receptors, this is highly unlikely. First, we used unmodified, phosphodiester bonded oligonucleotides, which have generally been shown not to have non-sequence-specific effects. Moreover, if mGluR1 AS treatment reduced the number of NMDA receptors, we would expect the response to i.t. NMDA to be blunted in the AS-treated rats compared to ACSF-treated sham-operated rats. However, the time spent exhibiting nociceptive behaviours was virtually identical for the two groups of rats. Thus, in this model of neuropathy, rats with nerve injury were confirmed to be more responsive to spinal administration of NMDA, and this could be attributed to mGluR1-associated mechanisms.
Although we saw profound effects in neuropathic rats, mGluR1 AS oligonucleotide treatment had no significant effect in sham-operated rats or in the contralateral paw of neuropathic rats, indicating that mGluR1 is involved in chronic neuropathic pain, but perhaps not to the same degree in the mediation of acute nociceptive stimuli. This is in agreement with a previous study where we showed that blockade of either mGluR1 or mGluR5 with selective antibodies alleviated cold hyperalgesia in neuropathic rats, but had no effect on response latency to focused radiant heat in naive rats, nor on formalin-induced pain scores (Fundytus et al., 1998b). However, this is in contrast to other investigators who found that antisense knockdown of mGluR1 increased response latency in a tail flick test in normal rats (Young et al., 1998). This discrepancy may be due to differences in the degree or exact location of mGluR1 depletion in various spinal neurons (depending on cannula placement, spread and penetration), or differences in characteristics of the phosphodiester bonded oligonucleotides used here and the phosphorothioate modified oligonucleotides used by Young et al. (1998).
Additional data suggests a role for group I mGluRs in acute (both reflexive and persistent) nociceptive responses. Intrathecal injection of the selective group I mGluR agonist DHPG induces spontaneous nociceptive behaviours (present study and Fisher & Coderre, 1996a). The relatively selective group I mGluR antagonist (S)-4CPG slightly attenuates formalin-induced pain scores (Fisher & Coderre, 1996b), and increases mechanical threshold and thermal latencies in normal and inflamed animals (while the non-selective mGluR antagonist L-AP3 does so only in inflamed animals) (Young et al., 1997). In addition, both L-AP3 and (S)-4CPG reduce dorsal horn neuronal responses following mustard oil stimulation (Young et al., 1994; 1995; 1997). It has also been shown in monkeys that the group I mGluR antagonists (S)-4CPG and L-AP3 inhibit dorsal horn neuronal responses to brief high intensity cutaneous stimuli, and reduce capsaicin-induced central sensitization of dorsal horn neurons (Neugebauer et al., 1999). However, as discussed earlier, (S)-4CPG has a dual action whereby it is an antagonist at group I mGluRs, but an agonist at group II mGluRs (Hayashi et al., 1994), and therefore it is unclear whether the group I antagonistic or group II agonistic actions, or some combination of both, are responsible for the reduction in nociceptive behaviours. In addition, the non-selective mGluR antagonist L-AP3 has actions at NMDA receptors as well as mGluRs (Birse et al., 1993). However, it has also been shown that antagonism of mGluRs does not attenuate the hyperalgesia associated with an incision injury in rats that mimics acute post-operative pain (Zahn & Brennan, 1998). Furthermore, Neugebauer et al. (1994) showed that antagonism of group I mGluRs decreases dorsal horn neuronal responses in rats with inflamed knee joint, but has no effect in normal animals. Thus, group I mGluRs may be involved in both acute nociception and chronic pain, and the degree to which these receptors are involved in each remains to be further clarified.
At the cellular level, certain neurochemical, intracellular and molecular interactions can be hypothesized to explain the mechanisms by which mGluR1 may be involved in neuropathic pain and opioid sensitivity in neuropathic subjects. These mechanisms are similar to those proposed for interactions between other systems and the opioidergic system (e.g. NMDA and CCK) (Mao et al., 1995a; Ossipov et al., 2000). In this model, we assume that group I mGluRs, opioid receptors and NMDA receptors are co-localized in the same cells. Neuropathic injury has been shown to stimulate an increased release of glutamate (Al-Ghoul et al., 1993), and this would result in increased activation of both NMDA receptors and mGluRs. Increased activity of group I mGluRs (as well as NMDA receptors) would ultimately lead to an increase in the concentration of intracellular Ca2+ and activation of PKC, via increased PI hydrolysis. Activation of PKC has been shown to phosphorylate opioid receptors, and induce desensitization (Childers, 1991; Kramer & Simon, 1999; Fan et al., 1998; Narita et al., 1996; Wang et al., 1996), reducing the analgesic efficacy of opioid agonists. Moreover, PKC phosphorylates the ion channel associated with NMDA receptors (Chen & Huang, 1992; Lambert, 1993; Smart & Lambert, 1996; Swope et al., 1999), increasing NMDA receptor sensitivity and activity, and leading to enhanced influx of Ca2+. By decreasing the number of group I mGluRs, AS oligonucleotide treatment would decrease PI hydrolysis, and protein kinase C activation, attenuating the phosphorylation of opioid receptors and NMDA receptor channels. Excitation would be decreased, thus reducing pain transmission, and desensitization of opioid receptors would also be decreased, with a resultant increase in the efficacy of opioid analgesics. This discussion deals with only some of the mechanisms that may be involved, and elicits the question of whether all of these elements occur within one neuron. Further research is necessary to verify the mechanisms proposed here.
Thus, in the current study, we present data indicating a role for mGluR1 in neuropathic pain. Because knockdown of mGluR1 reverses nerve-injury induced insensitivity to morphine, as well as reducing neuropathic pain, we propose that knockdown of mGluR1 may be useful as therapy for neuropathic pain in the clinic. It may be used to alleviate pain directly, or as an adjunct to opioid analgesic therapy.
Acknowledgments
M.E. Fundytus was supported by an MRC/PMAC Post-doctoral Fellowship with J.L. Henry sponsored by the ASTRA Research Centre Montreal, and currently holds the Dr Ronald Melzack Pain Research Award sponsored by the Canadian Pain Society, the Canadian Anaesthesiologists Society, AstraZeneca Canada, and the Medical Research Council of Canada. This research was supported by the ASTRA Research Centre Montreal (now AstraZeneca Research & Development), and MRC Canada grants to J.L. Henry and T.J. Coderre. T.J. Coderre is an MRC Scientist. The authors wish to thank Dr Claes Wahlestedt and Dr Francois Denis for their valuable advice on antisense oligonucleotide technology, and Dr Robert Day for his valuable advice on Western blot analysis.
Abbreviations
- ACSF
artificial cerebrospinal fluid
- AS
antisense
- AUC
area under the curve
- DHPG
dihydroxyphenylglycine
- LSD
least significant difference
- mGluR
metabotropic glutamate receptor
- MS
missense
- NMDA
N-methyl-D-aspartate
- PAG
periaqueductal gray
- PDBu
phorbol-12,13-dibutyrate
- PI
phosphatidylinositol
- PKC
protein kinase C
- PO
phosphodiester bonded
- PS
phosphorothioate modified
- (S)-4CPG
(S)-4-carboxyphenylglycine
- SNB
spontaneous nociceptive behaviour
References
- ABE T., SUGIHARA H., NAWA H., SHIGEMOTO R., MIZUNO N., NAKANISHI S. Molecular characterization of a novel metabotropic glutamate receptor mGluR5 coupled to inositol phosphate/Ca2+ signal transduction. J. Biol. Chem. 1992;267:13361–13368. [PubMed] [Google Scholar]
- AKHTAR S., AGRAWAL S. In vivo studies with antisense oligonucleotides. Trends Pharmacol. Sci. 1997;18:12–18. doi: 10.1016/s0165-6147(96)01002-4. [DOI] [PubMed] [Google Scholar]
- AL-GHOUL W.M., LI VOLSI G., WEINBERG R.J., RUSTIONI A. Glutamate immunocytochemistry in the dorsal horn after injury or stimulation of the sciatic nerve of rats. Brain Res. Bull. 1993;30:453–459. doi: 10.1016/0361-9230(93)90278-j. [DOI] [PubMed] [Google Scholar]
- ALVAREZ F.I., VILLALBA R.M., CARR P.A., GRANDES P., SOMOHANO P.M. Differential distribution of metabotropic glutamate receptors 1a, 1b, and 5 in rat spinal cord. J. Comp. Neurol. 2000;422:464–487. doi: 10.1002/1096-9861(20000703)422:3<464::aid-cne11>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- BERTHELE A., BOXALL S.J., URBAN A., ANNESER J.M., ZIEGLGANSBERGER W., URBAN L., TOLLE T.R. Distribution and developmental changes in metabotropic glutamate receptor messenger RNA expression in the rat lumbar spinal cord. Brain. Res. Dev. Brain. Res. 1999;112:39–53. doi: 10.1016/s0165-3806(98)00156-4. [DOI] [PubMed] [Google Scholar]
- BIRSE E.F., EATON S.A., JANE D.E., JONES P.L., PORTER R.H., POOK P.C., SUNTER D.C., UDVARHELYI P.M., WHARTON B., ROBERTS P.J., SALT T.E., WATKINS J.C. Phenylglycine derivatives as new pharmacological tools for investigating the role of metabotropic glutamate receptors in the central nervous system. Neuroscience. 1993;52:481–488. doi: 10.1016/0306-4522(93)90400-a. [DOI] [PubMed] [Google Scholar]
- BLEAKMAN D., RUSIN K.I., CHARD P.S., GLAUM S.R., MILLER R.J. Metabotropic glutamate receptors potentiate ionotropic glutamate responses in the rat dorsal horn. Mol. Pharmacol. 1992;42:192–196. [PubMed] [Google Scholar]
- BRADFORD M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- CASABONA G., CATANIA M.V., STORTO M., FERRARIS N., PERROTEAU I., FASOLO A., NICOLLETTI F., BOVOLIN P. Deafferentation up-regulates expression of the mGlu1a metabotropic glutamate receptor protein in the olfactory bulb. Eur. J. Neurosci. 1998;10:771–776. doi: 10.1046/j.1460-9568.1998.00076.x. [DOI] [PubMed] [Google Scholar]
- CHAPLAN S.R., BACH F.W., POGREL J.W., CHUNG J.M., YAKSH T.L. Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Meth. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- CHAPLAN S.R., MALMBERG A.B., YAKSH T.L. Efficacy of spinal NMDA receptor antagonism in formalin hyperalgesia and nerve injury evoked allodynia. J. Pharmacol. Exp. Ther. 1997;280:829–838. [PubMed] [Google Scholar]
- CHEN L., HUANG L-Y.M. Protein kinase C reduces Mg2+ block of NMDA-receptor channels as a mechanism of modulation. Nature. 1992;356:521–523. doi: 10.1038/356521a0. [DOI] [PubMed] [Google Scholar]
- CHERNY N.I., THALER H.T., FRIEDLANDER-KLAR H., LAPIN J., FOLEY K.M., HOUDE R., PORTENOY R.K. Opioid responsiveness of cancer pain syndromes caused by neuropathic or nociceptive mechanisms: A combined analysis of controlled, single-dose studies. Neurology. 1994;44:857–861. doi: 10.1212/wnl.44.5.857. [DOI] [PubMed] [Google Scholar]
- CHILDERS S.R. Opioid receptor-coupled second messenger systems. Life Sci. 1991;48:1191–2003. doi: 10.1016/0024-3205(91)90154-4. [DOI] [PubMed] [Google Scholar]
- CONN P.J., PIN J.P. Pharmacology and function of metabotropic glutamate receptors. Ann. Rev. Pharmacol. Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- FAN G.H., ZHAO J., WU Y.L., LOU L.G., ZHANG Z., JING Q., MA L., PEI G. N-methyl-D-aspartate attenuates opioid receptor-mediated G protein activation and this process involves protein kinase C. Mol. Pharmacol. 1998;53:684–690. doi: 10.1124/mol.53.4.684. [DOI] [PubMed] [Google Scholar]
- FISHER K., CODERRE T.J. Comparison of nociceptive effects produced by intrathecal administration of mGluR agonists. NeuroReport. 1996a;7:2743–2747. doi: 10.1097/00001756-199611040-00067. [DOI] [PubMed] [Google Scholar]
- FISHER K., CODERRE T.J. The contribution of mGluRs to formalin-induced nociception. Pain. 1996b;68:255–263. doi: 10.1016/s0304-3959(96)03212-5. [DOI] [PubMed] [Google Scholar]
- FISHER K., FUNDYTUS M.E., CAHILL C.M., CODERRE T.J. Intrathecal administration of the mGluR compound, (S)-4CPG, attenuates hyperalgesia and allodynia associated with sciatic nerve constriction injury in rats. Pain. 1998;77:59–66. doi: 10.1016/S0304-3959(98)00082-7. [DOI] [PubMed] [Google Scholar]
- FUNDYTUS M.E., CODERRE T.J. Effect of activity at metabotropic, as well as ionotropic (NMDA), glutamate receptors on morphine dependence. Br. J. Pharmacol. 1994;113:1215–1220. doi: 10.1111/j.1476-5381.1994.tb17127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FUNDYTUS M.E., CODERRE T.J. Chronic inhibition of intracellular Ca2+ release or PKC activation significantly reduces the development of morphine dependence. Eur. J. Pharmacol. 1996;300:173–181. doi: 10.1016/0014-2999(95)00871-3. [DOI] [PubMed] [Google Scholar]
- FUNDYTUS M.E., CODERRE T.J. Opioid tolerance and dependence: A new model highlighting the role of metabotropic glutamate receptors. Pain Forum. 1999a;8:3–13. [Google Scholar]
- FUNDYTUS M.E., CODERRE T.J. mGluRs and opioid dependence: A further examination of the mechanisms. Pain Forum. 1999b;8:59–63. [Google Scholar]
- FUNDYTUS M.E., DRAY A., HENRY J.L., CODERRE T.J. An antisense oligonucleotide targeting mGluR1 restores opioid sensitivity in neuropathic rats. INRC ‘98 (International Narcotics Research Conference Abstract) 1998a. p. p. 42.
- FUNDYTUS M.E., FISHER K., DRAY A., HENRY J.L., CODERRE T.J. Antisense oligonucleotides targeting group I mGluRs attenuate nerve constriction-induced hyperalgesia and allodynia. Soc. Neurosci. Abstr. 1997;23:1013. [Google Scholar]
- FUNDYTUS M.E., FISHER K., DRAY A., HENRY J.L., CODERRE T.J. In vivo antinociceptive activity of anti-rat mGluR1 and anti-rat mGluR5 antibodies in rats. NeuroReport. 1998b;9:731–735. doi: 10.1097/00001756-199803090-00031. [DOI] [PubMed] [Google Scholar]
- FUNDYTUS M.E., HENRY J.L., DRAY A., CODERRE T.J. Antisense knockdown of mGluR1 reverses hyperalgesia and allodynia associated with an established neuropathic injury in rats. Proc. 9th World Cong. Pain. 1999a;16:343–349. [Google Scholar]
- FUNDYTUS M.E., YASHPAL K., DRAY A., CODERRE T.J., HENRY J.L. Antisense oligonucleotide knockdown of mGluR1 reverses enhanced NMDA sensitivity in neuropathic rats. Soc. Neurosci. Abstr. 1999b;25:449. [Google Scholar]
- GOLDSTEIN A. Biostatistics: An Introductory Text. New York: The MacMillan Company; 1964. [Google Scholar]
- HANSSON P.Neurogenic Pain: Diagnosis and treatment Pain Clinical Updates 1994IIIssue 3, December [Google Scholar]
- HARGETT G.L., COGGESHALL R.E., CARLTON S.M. Metabotropic glutamate receptors are differentially distributed in primary afferent terminals in dorsal horn. Soc. Neurosci. Abstr. 1998;24:1869. [Google Scholar]
- HARGREAVES K., DUBNER R., BROWN F., FLORES C., JORES J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- HARVEY J., COLLINGRIDGE G.L. Signal transduction pathways involved in the acute potentiation of NMDA responses by 1S,3R-ACPD in rat hippocampal slices. Br. J. Pharmacol. 1993;109:1085–1090. doi: 10.1111/j.1476-5381.1993.tb13733.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAYASHI Y., SEKIYAMA N., NAKANISHI S., JANE D.E., SUNTER D.C,. , BIRSE E.F., UDVARHELYI P.M., WATKINS J.C. Analysis of agonist and antagonist activities of phenylglycine derivatives for different cloned metabotropic glutamate receptor subtypes. J. Neurosci. 1994;14:3370–3377. doi: 10.1523/JNEUROSCI.14-05-03370.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOUAMED K.M., KUIJPER J.L., GILBERT T.L., HALDEMAN B.A., O'HARA P.J., MULVIHILL E.R., ALMERS W., HAGEN F.S. Cloning, expression, and gene structure of a G protein-coupled glutamate receptor from rat brain. Science. 1991;252:1318–1321. doi: 10.1126/science.1656524. [DOI] [PubMed] [Google Scholar]
- KELSO S.R., NELSON T.E., LEONARD J.P. Protein kinase C-mediated enhancement of NMDA currents by metabotropic glutamate receptors in Xenopus oocytes. J. Physiol. 1992;449:705–718. doi: 10.1113/jphysiol.1992.sp019110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KITAMURA Y., MIYAZAKI A., YAMANAKA Y., NOMURA Y. Stimulatory effects of protein kinase C and calmodulin kinase II on N-methyl-D-aspartate receptor/channels in the postsynaptic density of rat brain. J. Neurochem. 1993;61:100–109. doi: 10.1111/j.1471-4159.1993.tb03542.x. [DOI] [PubMed] [Google Scholar]
- KRAMER H.K., SIMON E.J. Role of protein kinase C (PKC) in agonist-induced mu-opioid receptor down-regulation: II. Activation and involvement of the alpha, epsilon and zeta isoforms of PKC. J. Neurochem. 1999;72:594–604. doi: 10.1046/j.1471-4159.1999.0720594.x. [DOI] [PubMed] [Google Scholar]
- LAMBERT D.G. Signal transduction: G proteins and second messengers. Br. J. Anaesth. 1993;71:86–95. doi: 10.1093/bja/71.1.86. [DOI] [PubMed] [Google Scholar]
- MACDONALD N. Opiate-resistant pain: a therapeutic dilemma. Recent Results in Cancer Res. 1991;121:24–35. doi: 10.1007/978-3-642-84138-5_4. [DOI] [PubMed] [Google Scholar]
- MALMBERG A.B., CHEN C.C., TONEGAWA S., BASBAUM A.I. Preserved acute pain and reduced neuropathic pain in mice lacking PKC gamma. Science. 1997;278:279–283. doi: 10.1126/science.278.5336.279. [DOI] [PubMed] [Google Scholar]
- MAO J., PRICE D.D., HAYES R.L., LU J., MAYER D.J. Intrathecal GM1ganglioside and local nerve anesthesia reduce nociceptive behaviours in rats with experimental peripheral mononeuropathy. Brain Res. 1992a;584:28–53. doi: 10.1016/0006-8993(92)90874-9. [DOI] [PubMed] [Google Scholar]
- MAO J., PRICE D.D., MAYER D.J., HAYES R.L. Pain-related increases in spinal cord membrane-bound protein kinase C following peripheral nerve injury. Brain Res. 1992b;588:144–149. doi: 10.1016/0006-8993(92)91354-h. [DOI] [PubMed] [Google Scholar]
- MAO J., PRICE D.D., MAYER D.J. Mechanisms of hyperalgesia and morphine tolerance: a current view of their possible interactions. Pain. 1995a;62:259–274. doi: 10.1016/0304-3959(95)00073-2. [DOI] [PubMed] [Google Scholar]
- MAO J., PRICE D.D., PHILLIPS L.L., LU J., MAYER D.J. Increases in protein kinase C gamma immunoreactivity in the spinal cord dorsal horn of rats with painful mononeuropathy. Neurosci Lett. 1995b;198:75–78. doi: 10.1016/0304-3940(95)11975-3. [DOI] [PubMed] [Google Scholar]
- MARTIN G., NIE Z., SIGGINS G.R. Metabotropic glutamate receptors regulate N-methyl-D-aspartate-mediated synaptic transmission in nucleus accumbens. J. Neurophysiol. 1997;78:3028–3038. doi: 10.1152/jn.1997.78.6.3028. [DOI] [PubMed] [Google Scholar]
- MARTIN L.J., BLACKSTONE C.D., HUGANIR R.L., PRICE D.L. Cellular localization of a metabotropic glutamate receptor in rat brain. Neuron. 1992;9:259–270. doi: 10.1016/0896-6273(92)90165-a. [DOI] [PubMed] [Google Scholar]
- MASU M., TANABE Y., TSUCHIDA K., SHIGEMOTO R., NAKANISHI S. Sequence and expression of a metabotropic glutamate receptor. Nature. 1991;349:760–765. doi: 10.1038/349760a0. [DOI] [PubMed] [Google Scholar]
- MAYER D.J., MAO J., PRICE D.D. The association of neuropathic pain, morphine tolerance and dependence, and the translocation of protein kinase C. NIDA Res. Mongr. 1995;147:269–298. [PubMed] [Google Scholar]
- MCQUAY H.J.Opioid use in chronic pain Acta. Anesth. Scand. 199741175–183.(1 Pt 2) [DOI] [PubMed] [Google Scholar]
- MCQUAY H.J., JADAD A.R., CARROLL D., FAURA C., GLYNN C.J., MOORE R.A., LIU Y. Opioid sensitivity of chronic pain: a patient-controlled analgesia method. Anesthesia. 1992;47:757–767. doi: 10.1111/j.1365-2044.1992.tb03253.x. [DOI] [PubMed] [Google Scholar]
- MOSCONI T., KRUGER L. Fixed-diameter polyethylene cuffs applied to the rat sciatic nerve induce a painful neuropathy: ultrastructural morphometric analysis of axonal alterations. Pain. 1996;64:37–57. doi: 10.1016/0304-3959(95)00077-1. [DOI] [PubMed] [Google Scholar]
- NARITA M., MIZOGUCHI H., KAMPINE J.P., TSENG L.F. Role of protein kinase C in desensitization of spinal delta-opioid-mediated antinociception in the mouse. Br. J. Pharmacol. 1996;118:1829–1835. doi: 10.1111/j.1476-5381.1996.tb15610.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NEUGEBAUER V., CHEN P.S., WILLIS W.D. Role of metabotropic glutamate receptor subtype mGluR1 in brief nociception and central sensitization of primate STT cells. J. Neurophysiol. 1999;82:272–282. doi: 10.1152/jn.1999.82.1.272. [DOI] [PubMed] [Google Scholar]
- NEUGEBAUER V., LUCKE T., SCHAIBLE H.G. Requirement of metabotropic glutamate receptors for the generation of inflammation-evoked hyperexcitability in rat spinal cord neurons. Eur. J. Neurosci. 1994;6:1179–1186. doi: 10.1111/j.1460-9568.1994.tb00616.x. [DOI] [PubMed] [Google Scholar]
- OLDS J.L., ANDERSON M.L., MCPHIE D.L., STATEN L.D., ALKON D.L. Imaging of memory-specific changes in the distribution of protein kinase C in the hippocampus. Science. 1989;245:866–869. doi: 10.1126/science.2772638. [DOI] [PubMed] [Google Scholar]
- OSSIPOV M.H., LAI J., MALAN T.P., JR, PORRECA F. Spinal and supraspinal mechanisms of neuropathic pain. Ann. NY. Acad. Sci. 2000;909:12–24. doi: 10.1111/j.1749-6632.2000.tb06673.x. [DOI] [PubMed] [Google Scholar]
- OSSIPOV M.H., LOPEZ Y., NICHOLS M.L., BIAN D., PORRECA F. Inhibition by spinal morphine of the tail-flick response is attenuated in rats with nerve ligation injury. Neurosci. Lett. 1995a;199:83–86. doi: 10.1016/0304-3940(95)12026-z. [DOI] [PubMed] [Google Scholar]
- OSSIPOV M.H., LOPEZ Y., NICHOLS M.L., BIAN D., PORRECA F. The loss of antinociceptive efficacy of spinal morphine in rats with nerve ligation injury is prevented by reducing spinal afferent drive. Neurosci. Lett. 1995b;199:87–90. doi: 10.1016/0304-3940(95)12022-v. [DOI] [PubMed] [Google Scholar]
- PHILLIPS M.I., GYURKO R. In vivo applications of antisense oligonucleotides for peptide research. Regul. Pept. 1995;59:131–141. doi: 10.1016/0167-0115(95)00104-j. [DOI] [PubMed] [Google Scholar]
- PORTENOY R.K. Cancer pain: pathophysiology and syndromes. Lancet. 1992;339:1026–1031. doi: 10.1016/0140-6736(92)90545-e. [DOI] [PubMed] [Google Scholar]
- RAYMOND L.A., TINGLEY W.G., BLACKSTONE C.D., ROCHE K.W., HUGANIR R.L. Glutamate receptor modulation by protein phosphorylation. J. Physiol. Paris. 1994;88:181–192. doi: 10.1016/0928-4257(94)90004-3. [DOI] [PubMed] [Google Scholar]
- SCHOEPP D.D., CONN P.J. Metabotropic glutamate receptors in brain function and pathology. Trends. Pharmacol. Sci. 1993;14:13–25. doi: 10.1016/0165-6147(93)90107-u. [DOI] [PubMed] [Google Scholar]
- SMART D., LAMBERT D.G. The stimulatory effects of opioids and their possible role in the development of tolerance. Trends Pharmacol. Sci. 1996;17:264–269. doi: 10.1016/0165-6147(96)10023-7. [DOI] [PubMed] [Google Scholar]
- STORKSON R.V., KJORSVIK A., TJOLSEN A., HOLE K. Lumbar catheterization of the spinal subarachnoid space in the rat. J. Neurosci. Meth. 1996;65:167–172. doi: 10.1016/0165-0270(95)00164-6. [DOI] [PubMed] [Google Scholar]
- SWOPE S.L., MOSS S.I., RAYMOND L.A., HUGANIR R.L. Regulation of ligand-gated ion channels by protein phosphorylation. Adv. Second Messenger Phosphoprotein Res. 1999;33:49–78. doi: 10.1016/s1040-7952(99)80005-6. [DOI] [PubMed] [Google Scholar]
- WAHLESTEDT C. Antisense oligonucleotide strategies in neuropharmacology. Trends Pharmacol. Sci. 1994;15:42–46. doi: 10.1016/0165-6147(94)90107-4. [DOI] [PubMed] [Google Scholar]
- WANG L., MEDINA V.M., RIVERA M, GINTZLER A.R. Relevance of phosphorylation state to opioid responsiveness in opiate naive and tolerant/dependent tissue. Brain Res. 1996;723:61–69. doi: 10.1016/0006-8993(96)00217-x. [DOI] [PubMed] [Google Scholar]
- WHITESELL L., GESELOWITZ D., CHAVANY C., FAHMY B., WALBRIDGE S., ALGER J.R., NECKERS L.M. Stability, clearance and disposition of intraventricularly administered oligonucleotides: Implications for therapeutic application within the central nervous system. Proc. Natl. Acad. Sci. U.S.A. 1993;90:4665–4669. doi: 10.1073/pnas.90.10.4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WORLEY P.F., BARABAN J.M., SNYDER S.H. Heterogeneous localization of protein kinase C in rat brain: autoradiographic analysis of phorbol ester receptor binding. J. Neurosci. 1986;6:199–207. doi: 10.1523/JNEUROSCI.06-01-00199.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAIDA Y., NOWAK T.S., JR Distribution of phosphodiester and phosphorothioate oligonucleotides in rat brain after intraventricular and intrahippocampal administration determined by in situ hybridization. Regul. Pept. 1995;59:193–199. doi: 10.1016/0167-0115(95)00100-p. [DOI] [PubMed] [Google Scholar]
- YAMAMOTO T., YAKSH T.L. Spinal pharmacology of thermal hyperesthesia induced by constriction injury of sciatic nerve. Pain. 1992;49:121–128. doi: 10.1016/0304-3959(92)90198-K. [DOI] [PubMed] [Google Scholar]
- YASHPAL K., PITCHER G.M., PARENT A., QUIRION R., CODERRE T.J. Noxious thermal and chemical stimulation induce increases in 3H-phorbol 12,13-dibutyrate binding in spinal cord dorsal horn as well as persistent pain and hyperalgesia, which is reduced by inhibition of protein kinase C. J. Neurosci. 1995;15:3263–3272. doi: 10.1523/JNEUROSCI.15-05-03263.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YOUNG M.R., BLACKBURN-MUNRO G., DICKINSON T., JOHNSON M.J., ANDERSON H., NAKALEMBE I., FLEETWOOD-WALKER S.M. Antisense ablation of type I metabotropic glutamate receptor mGluR1 inhibits spinal nociceptive transmission. J. Neurosci. 1998;18:10180–10188. doi: 10.1523/JNEUROSCI.18-23-10180.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YOUNG M.R., FLEETWOOD-WALKER S.M., DICKINSON T., BLACKBURN-MUNRO G., SPARROW H., BIRCH P.J., BOUNTRA C. Behavioural and electrophysiological evidence supporting a role for metabotropic glutamate receptors in the mediation of nociceptive inputs to the rat spinal cord. Brain Res. 1997;777:161–169. [PubMed] [Google Scholar]
- YOUNG M.R., FLEETWOOD-WALKER S.M., MITCHELL R., DICKINSON T. The involvement of metabotropic glutamate receptors and their intracellular signaling pathways in sustained nociceptive transmission in rat dorsal horn neurons. Neuropharmacology. 1995;34:1033–1041. doi: 10.1016/0028-3908(95)00071-d. [DOI] [PubMed] [Google Scholar]
- YOUNG M.R., FLEETWOOD-WALKER S.M., MITCHELL R., MUNRO F.E. Evidence for a role of metabotropic glutamate receptors in sustained nociceptive inputs to rat dorsal horn neurons. Neuropharmacology. 1994;33:141–144. doi: 10.1016/0028-3908(94)90109-0. [DOI] [PubMed] [Google Scholar]
- ZAHN P.K., BRENNAN T.J. Intrathecal metabotropic glutamate receptor antagonists do not decrease mechanical hyperalgesia in a rat model of postoperative pain. Anesth. Analg. 1998;87:1354–1359. [PubMed] [Google Scholar]