

Abstract
The antimalarial drug halofantrine can prolong the QT interval and this may be enhanced by prior use of mefloquine. This possible interaction has been investigated by examining the effects of halofantrine and mefloquine alone and in combination.
In anaesthetized rabbits (n=6 per group), halofantrine given as bolus doses of 1, 3, 10, and 30 mg kg−1 at 25 min intervals dose-dependently prolonged the rate-corrected QT (QTc) interval from 313±12 ms pre-drug to 410±18 ms after the highest dose. Similar doses of mefloquine did not alter QTc intervals significantly. The highest dose of mefloquine (30 mg kg−1) caused cardiac contractile failure.
Pretreatment with 3 mg kg−1 mefloquine 25 min before the first dose of halofantrine potentiated the effects of all doses of halofantrine on QTc intervals.
The blood concentrations of halofantrine were two to six times higher in the group pretreated with mefloquine compared to the halofantrine alone group; e.g. 1.03±0.17 and 0.16±0.02 μM respectively after 1 mg kg−1 halofantrine. There was a significant correlation between blood halofantrine concentrations and QTc intervals (r=0.673). Even after making allowance for overestimation of the potency of halofantrine that may result from the hypokalaemia that is prevalent in anaesthetized rabbits, these effects occurred with concentrations of halofantrine that are found in clinical use.
These data indicate clearly that while mefloquine does not alter QTc intervals itself, it does enhance the effects of halofantrine by increasing the circulating concentration of halofantrine.
Keywords: Halofantrine, mefloquine; QT prolongation; QTc interval; cardiotoxicity; AV block; arrhythmias
Introduction
There have been several clinical reports of adverse cardiac effects of the antimalarial drug halofantrine, including QT prolongation, episodes of torsade de pointes, or sudden cardiac death (Castot et al., 1993; Karbwang et al., 1993; Monlun et al., 1993; 1995; Nosten et al., 1993; Toivonen et al., 1994). Animal studies have demonstrated that halofantrine prolongs the rate-corrected QT (QTc) interval in a dose-dependent manner and that there is a good correlation between blood halofantrine concentrations and QTc intervals (Batey et al., 1997). In contrast, there are conflicting reports about the effects of mefloquine on QTc intervals in clinical studies. It has been reported to either cause transient, mild prolongation of the QTc interval (Davis et al., 1996), or to have no effect on the QTc interval (Laothavorn et al., 1992). The conclusions of the former report have been challenged (Evans & Waller, 1997), and in animal studies, even when given at high doses which were sufficient to impair cardiac contractility, mefloquine did not alter QTc intervals (Coker et al., 2000a).
One of the early clinical reports of halofantrine-induced QTc prolongation indicated that its effects appeared to be greater in patients who had received mefloquine previously (Nosten et al., 1993). A patient who experienced recrudescence of malaria 21 days after mefloquine treatment was given high dose halofantrine, but suffered sudden cardiac death before receiving the final dose of halofantrine. In a subsequent prospective study, treatment with mefloquine during the previous month increased the slope of the halofantrine concentration-response curve resulting in greater QTc prolongation at any given blood concentration of halofantrine (Nosten et al., 1993). These results suggest that there may be an interaction between mefloquine and halofantrine which exacerbates QTc prolongation. We have therefore examined the effects of halofantrine and mefloquine, alone and in combination, on QTc intervals in an anaesthetized animal model.
Methods
Surgical preparation of rabbits
Female New Zealand White rabbits (2.3 – 3.8 kg) were sedated with Hypnorm (fentanyl citrate 0.315 mg ml−1, fluanisone 10 mg ml−1) 0.5 ml kg−1 i.m. Sodium pentobarbitone (Sagatal 60 mg ml−1 diluted 1 : 2 with saline) was injected slowly via a marginal ear vein until a depth of anaesthesia sufficient for surgery was achieved (∼15 mg kg−1). The trachea was cannulated and when necessary, rabbits were ventilated with room air at a rate of 38 cycles per min and a stroke volume of ∼7 ml kg−1. Cannulae were inserted into the descending vena cava via a femoral vein for drug administration and into the abdominal aorta via a femoral artery for blood pressure measurement. The right jugular vein was also isolated and prepared for cannulation. If necessary, additional sodium pentobarbitone was then administered to attain a greater depth of anaesthesia, and the chest was opened via a midline sternotomy. An incision was made in the pericardium to expose the anterior surface of the heart. A bipolar suction electrode was attached to the surface of the heart to permit recording of epicardial monophasic action potentials. Endocardial monophasic action potentials were recorded from the right ventricle with a bipolar electrode catheter inserted via the right jugular vein.
Data recording and measurement
A limb lead ECG was monitored from subcutaneous needle electrodes, and for each experiment, the lead which gave the best separation of the P wave from the T wave of the preceding complex was recorded. The ECG and the monophasic action potentials were recorded using Grass 7P6/7DA or 7P4/7DA amplifiers. The arterial cannula was connected to a Bell & Howell type 4 – 422 transducer linked to a Grass 7P122 amplifier. All the signals from the Grass amplifiers were sampled at 1000 Hz and fed into a Po-Ne-Mah data acquisition and analysis system (Linton Instrumentation, Diss, Norfolk, U.K.) running on an Opus 486/33 computer. Data were recorded directly on to the computer hard disk and transferred subsequently to compact disk (Hewlett-Packard SureStore CD Writer 6020) for archive purposes. Heart rate was calculated on line from either the blood pressure or ECG signal.
For each experiment, data were retrieved from the computer before drug administration and 5, 10, 15, and 20 min after giving each dose of drug, and the ECG intervals were measured manually using on screen cursors, taking the mean of at least four ECG complexes at each time point measured. ECG intervals were only measured in beats that originated from the sino-atrial node. The PR interval was measured from the onset of the P wave to the onset of the R wave (i.e. Q), the QRS interval from the beginning of the Q wave to the end of the S wave, and the QT interval from Q to the end of the T wave. The QT interval was corrected for heart rate to give QTc using Bazett's formula expressed in ms as recommended by Molnar et al. (1995). Arterial blood gases, pH and K+ concentrations were measured using a Ciba-Corning 850 blood gas analyser (Chiron Diagnostics, Halstead, Essex, U.K.). After completion of the surgical procedures at least 20 min was allowed for stabilization during which ventilation volume was adjusted, if necessary, to maintain arterial PO2 >90 mmHg and PCO2 20 – 30 mmHg. Arterial blood samples were taken 20 min after administration of each dose of halofantrine and stored for later analysis of halofantrine concentrations by high performance liquid chromatography as described previously (Batey et al., 1997; Mberu et al., 1992).
Experimental protocol
Four groups of n=6 rabbits were studied; halofantrine, mefloquine, halofantrine plus mefloquine and control (vehicle). Both drugs were dissolved in dimethylacetamide (40%) /propylene glycol (60%) v v−1, at 50 mg ml−1. Immediately prior to administration of each dose, the appropriate volume of stock drug solution was diluted with 5% w v−1 D-glucose solution containing 10 u ml−1 heparin to give a total volume for administration of 1 ml kg−1. Drugs were given by slow intravenous injection (∼30 s) and flushed through the cannula with heparinized D-glucose solution. The halofantrine and mefloquine groups received doses of 1, 3, 10, and 30 mg kg−1 of drug at 25 min intervals, and the controls received appropriate volumes of vehicle at the same time points. In the group which received both drugs, a single dose of mefloquine (3 mg kg−1) was given 25 min before the first dose of halofantrine, after which the protocol was identical to that for the other groups.
Drugs
(±)-Halofantrine HCl (formula 9, batch no. 3D01HP; a 50 mg ml−1 solution) and (±)-mefloquine were gifts from SmithKline Beecham Pharmaceuticals and F Hoffman-La Roche, respectively. N,N-Dimethylacetamide and propylene glycol were purchased from Sigma (Poole, U.K.) and D-glucose from BDH (Poole, U.K.) Heparin sodium (mucous) injection was obtained from CP Pharmaceuticals (Wrexham, U.K.).
Statistics
Shapiro – Wilk tests revealed that some data may not be distributed normally. Friedman tests were used for comparisons within groups and Kruskal – Wallis tests for comparisons between groups. A probability value of P<0.05 was considered to be significant.
Results
Haemodynamics
The values for heart rate and mean arterial blood pressure measured throughout these experiments are illustrated in Figure 1. There were no significant differences between groups in heart rate or blood pressure at any time point during the course of these experiments. In all groups, however, mean arterial blood pressure tended to decline slightly as the experiments progressed. Injection of certain doses of drug caused transient within group alterations in blood pressure. In the mefloquine group administration of 3 mg kg−1 increased mean blood pressure from 48±3 to 52±2 mmHg (P<0.01, Friedman test). Similarly, in the group that received this dose of mefloquine before receiving increasing doses of halofantrine, mefloquine increased mean blood pressure from 52±2 to 59±2 mmHg (P<0.05, Friedman test). With higher doses of mefloquine, however, blood pressure declined (Figure 1b). The reduction in blood pressure following administration of 10 mg kg−1 mefloquine was transient in all but one rabbit, whereas 30 mg kg−1 mefloquine reduced blood pressure markedly in the remaining five rabbits leading to death within 5 min of administration. Only the highest dose of halofantrine reduced blood pressure transiently, from 46±3 to 35±2 mmHg in the halofantrine group, and from 48±6 to 33±5 mmHg in the group receiving both mefloquine and halofantrine (both P<0.001, Friedman test). No particular pattern of changes in heart rate was observed in the vehicle group, but heart rate did appear to decline with the higher doses of halofantrine, although none of these apparent changes reached statistical significance (Figure 1a).
Figure 1.
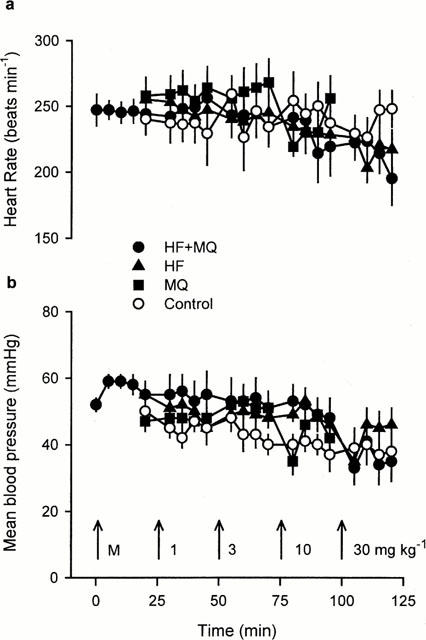
Heart rate (a) and mean arterial blood pressure (b) measured at various time points in the four groups of anaesthetized rabbits receiving vehicle (Control), mefloquine (MQ), halofantrine (HF) or halofantrine after 3 mg kg−1 mefloquine (HF+MQ). The arrows indicate the times at which bolus i.v. injections of 3 mg kg−1 mefloquine (M), or 1, 3, 10, and 30 mg kg−1 of mefloquine or halofantrine were given. Values are means with vertical bars indicating s.e.mean, n=6 per group.
ECGs and monophasic action potentials
Intravenous administration of halofantrine prolonged the QT interval in a dose-dependent manner, whereas mefloquine did not alter the QT interval (Table 1). The effect of halofantrine on the QT interval was not influenced by any subtle changes in heart rate that may have occurred. Figure 2 illustrates clearly that halofantrine increased the rate-corrected QT (QTc) interval. It is also clear from Table 1 and Figure 2 that pretreatment with 3 mg kg−1 mefloquine markedly potentiated the halofantrine-induced QT and QTc prolongation. In contrast, mefloquine alone did not alter QT intervals and the vehicle was also devoid of activity. Mefloquine also failed to alter either the PR interval or the QRS interval (Table 1. After administration of the highest dose of halofantrine the PR interval increased significantly and the QRS interval tended to increase (Table 1). Similar effects of halofantrine on the QRS interval were seen in the group pretreated with mefloquine but in this group accurate measurements of the PR interval could not be obtained after administration of the higher doses of halofantrine because P waves were often masked by the extended T wave of the preceding ECG complex.
Table 1.
ECG intervals (PR, QRS and QT) measured before and 20 min after administration of each dose of drug

Figure 2.
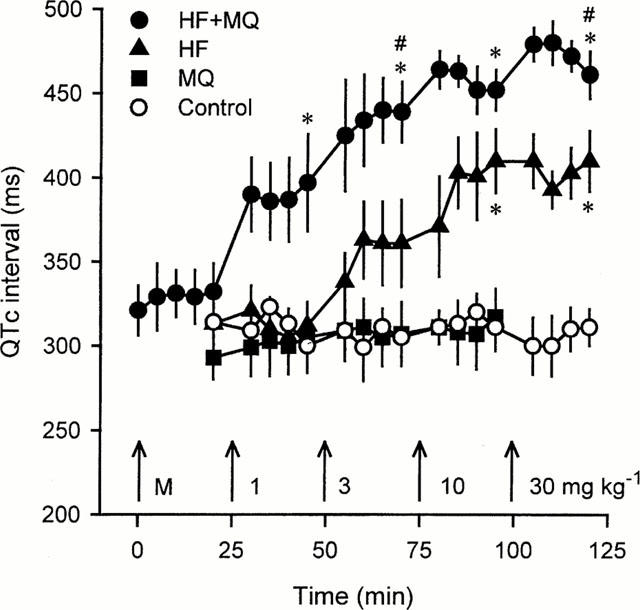
QTc intervals measured at various time points in the four groups of anaesthetized rabbits receiving vehicle (Control), mefloquine (MQ), halofantrine (HF) or halofantrine after 3 mg kg−1 mefloquine (HF+MQ). The arrows indicate the times at which bolus i.v. injections of 3 mg kg−1 mefloquine (M), or 1, 3, 10, and 30 mg kg−1 of mefloquine or halofantrine were given. Values are means with vertical bars indicating s.e.mean, n=6 per group. *P<0.05 compared with control group, #P<0.05 compared with halofantrine group at that time point, Kruskal-Wallis test.
Changes in ECG morphology and the shape of the monophasic action potential also occurred in the groups receiving halofantrine. The amplitude of T waves increased and ‘humps' indicative of delays in repolarisation appeared in phase 3 of the action potential (Figure 3). These effects were more obvious in the group which received halofantrine after mefloquine. Ventricular premature beats were observed in two of the rabbits in this group and AV block occurred in three of these animals after administration of the highest dose of halofantrine. In two cases Mobitz type I AV block was observed (see Figure 3 for an example and further description). Ventricular premature beats and AV block also occurred in one rabbit in the halofantrine group, but there were no cardiac rhythm disturbances in either the mefloquine or control groups.
Figure 3.
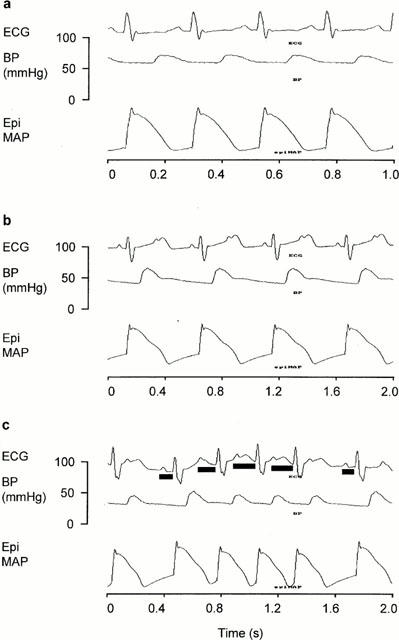
Lead II ECG, arterial blood pressure (BP) and epicardial monophasic action potential (EpiMAP) measured (a) before drug administration, (b) 19 min after administration of 10 mg kg−1 halofantrine and (c) 5 min after 30 mg kg−1 halofantrine in one rabbit from the group pretreated with 3 mg kg−1 mefloquine before receiving increasing doses of halofantrine. In (a) there is normal sinus rhythm but the P waves of each ECG complex are merged with the T wave of the preceding complex. In (b) there is 2 : 1 second degree atrio-ventricular (AV) block. Alternate P waves appear in the rising phase of the T wave of the preceding complex and the subsequent QRS complex is missing. (c) Illustrates second degree Mobitz type 1 (Wenkebach) AV block which is characterized by progressive lengthening of the PR interval until there is a missed QRS complex. The solid bars under the ECG complexes indicate the PR intervals. (b) and (c) also reveal halofantrine-induced increases in T wave amplitude and delays in repolarization during phase 3 of the epicardial monophasic action potentials.
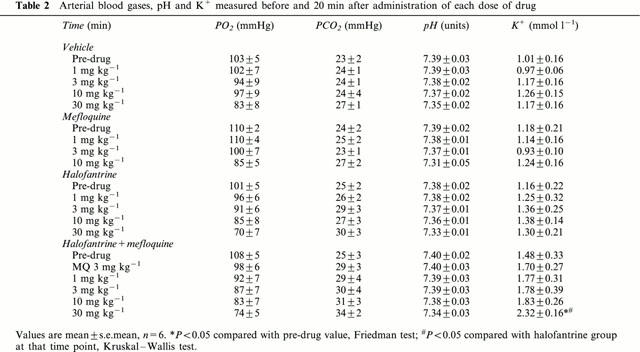
Blood gases, pH and K+
At the end of the experiments, blood K+ concentrations were higher in the group that received halofantrine and mefloquine (Table 2). No significant increases in K+ occurred in the other groups. As the experiments progressed PO2 tended to decline and PCO2 to increase. These effects were accompanied by a decrease in pH. The extent of these changes was similar in all the groups and there were no differences in PO2, PCO2 or pH among any of the groups at any time point (Table 2).
Table 2.
Arterial blood gases, pH and K+ measured before and 20 min after administration of each dose of drug
Halofantrine concentrations
The blood concentrations of halofantrine were measured 20 min after administration of each dose of halofantrine in the groups which were given this drug. Dose-dependent increases in blood halofantrine concentrations occurred in both groups (Figure 4). Figure 4 also illustrates clearly that the blood concentrations of halofantrine measured after each dose of halofantrine were approximately twice as high in the group pretreated with mefloquine compared to those receiving halofantrine alone. For example, after 30 mg kg−1 halofantrine the concentration of halofantrine was 13.5±1.6 μM (7230±870 ng ml−1) in the halofantrine group compared with 23.8±2.1 μM (12780±1140 ng ml−1) in the rabbits that were given mefloquine before halofantrine. Plotting the QTc interval measured 20 min after administration of halofantrine against the blood halofantrine concentration (on a log scale) revealed a significant correlation (r=0.673, P<0.0001) between these parameters (Figure 5).
Figure 4.
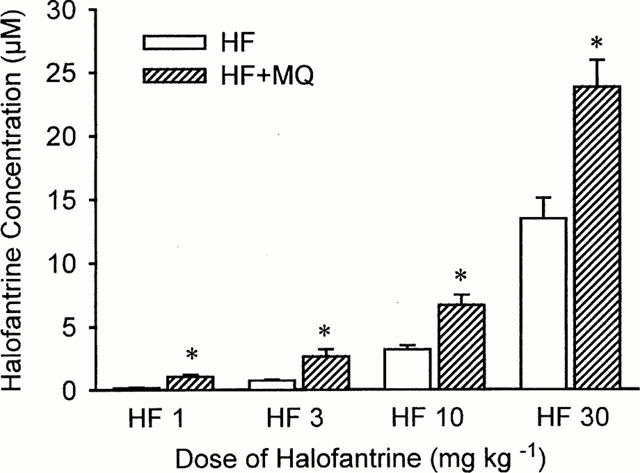
The concentrations of halofantrine measured by high performance liquid chromatography in blood samples taken 20 min after administration of each dose of halofantrine from animals receiving halofantrine alone (HF) or halofantrine after 3 mg kg−1 mefloquine (HF+MQ). Values are means with vertical bars indicating s.e.mean, n=6 per group. *P<0.05 compared halofantrine group, Kruskal – Wallis test.
Figure 5.
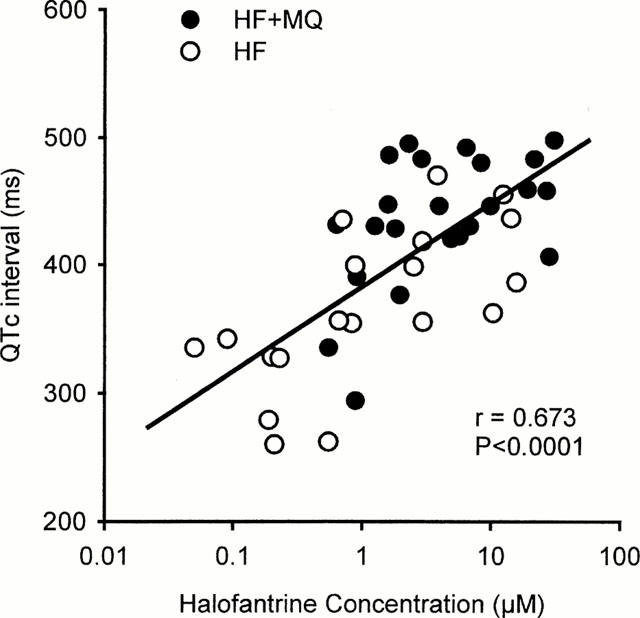
Correlation between the individual values for QTc intervals measured 20 min after administration of each dose of halofantrine and the blood concentrations of halofantrine in samples obtained at those time points from animals receiving halofantrine alone (HF) or halofantrine after 3 mg kg−1 mefloquine (HF+MQ).
Discussion
This is the first report of in vivo animal studies examining possible interactions between halofantrine and mefloquine affecting the ECG. The results demonstrate clearly that mefloquine potentiates halofantrine-induced QTc prolongation and suggest that this is a consequence of mefloquine either altering the disposition of halofantrine or reducing its metabolism. Pretreatment with mefloquine increased the blood concentrations of halofantrine 2 – 6 fold and there was a significant correlation between halofantrine concentrations and QTc intervals.
In general terms, these results confirm the clinical observation of greater effects of halofantrine on QTc intervals in patients who had previously received mefloquine (Nosten et al., 1993). There is, however, one difference between the clinical data and the present results. Nosten et al. (1993) showed that for any measured drug concentration, halofantrine caused a greater change in QTc intervals in the patients who had received mefloquine compared to those who received halofantrine alone. In the present study mefloquine not only increased the effect of halofantrine on QTc intervals but also increased the concentrations of halofantrine measured in blood after administration of the same dose of halofantrine. There is, however, a possible explanation for this apparent discrepancy. The clinical report (Nosten et al., 1993) plots the per cent change in QTc intervals against the total blood concentration of halofantrine and its metabolite desbutylhalofantrine. If mefloquine inhibited the metabolism of halofantrine to desbutylhalofantrine and the metabolite had less or no effect on QTc intervals this would reconcile the results of Nosten et al. (1993) with the present data. Although the total concentration of halofantrine and metabolite may not have changed in the former study, the proportion of halofantrine to metabolite may have increased.
Effects of mefloquine on metabolism and disposition of halofantrine
Cytochrome P450 3A4 (CYP 3A4) is the principal enzyme involved in the N-debutylation of halofantrine (Baune et al., 1999a). Recently, it has been shown that mefloquine can inhibit the metabolism of halofantrine by CYP 3A4 in vitro (Baune et al., 1999b), and the potent inhibitor of CYP 3A4, ketoconazole, also inhibited halofantrine metabolism in vitro (Halliday et al., 1995) and in vivo (Khoo et al., 1998). Thus mefloquine may have reduced the metabolism of halofantrine in the present experiments. Two clinical studies have suggested that after oral ingestion of halofantrine, QT prolongation is associated with plasma halofantrine concentrations but not with the concentrations of desbutylhalofantrine (Karbwang et al., 1993; Touze et al., 1996). However, i.v. administration of desbutylhalofantrine caused significant QTc prolongation in anaesthetized rabbits (Coker et al., 2000b). The concentrations of desbutylhalofantrine were not measured in the present study and it is therefore not possible to draw any firm conclusion about the contribution of this metabolite of halofantrine to the observed effects.
An in vitro study in isolated perfused rat liver demonstrated that mefloquine reduced the biliary clearance of halofantrine, but this may not reflect reduced metabolism as the biliary clearance of desbutylhalofantrine and bile production were also reduced (Leo et al., 1996). There is also evidence from in vivo studies that there is relatively little metabolism of halofantrine to desbutylhalofantrine after i.v. administration of halofantrine compared to oral administration (Humberstone et al., 1996; Krishna et al., 1993). Thus mefloquine-induced alterations in the metabolism of halofantrine may not be responsible for the increased blood halofantrine concentrations and prolongation of QTc intervals seen in the present study.
Mefloquine may have altered the disposition of halofantrine. Both drugs are highly lipophilic and it is known that halofantrine binds extensively to lipoproteins (Cenni et al., 1995; Humberstone et al., 1998). Mefloquine has also been reported to bind to lipoproteins (Desneves et al., 1996). If mefloquine limited the binding of halofantrine to lipoproteins this could have reduced the amount of halofantrine being removed from the circulation by the uptake of lipoproteins into tissues.
Pharmacodynamic effects of halofantrine and mefloquine
The results of the present study demonstrate that the effects of halofantrine and of mefloquine given alone to anaesthetized rabbits are very similar to those reported previously for each drug in anaesthetized guinea-pigs (Batey et al., 1997; Coker et al., 2000a). Mefloquine per se did not alter QTc intervals, or PR or QRS intervals. It did, however, have hypotensive effects which were severe enough to result in the death of one rabbit after 10 mg kg−1 and the remaining five within 5 min of administration of 30 mg kg−1 mefloquine. This result is remarkably similar to that seen in guinea-pigs, where additional in vitro studies indicated that mefloquine blocked L-type Ca2+ channels (Coker et al., 2000a). Thus it is likely that in these anaesthetized rabbits, as in the guinea-pigs, the higher doses of mefloquine caused cardiac contractile failure subsequent to Ca2+ channel blockade. This provides further evidence that the primary adverse cardiac effect of mefloquine is on contractile activity and not on the ECG. In contrast, halofantrine did not alter blood pressure significantly, but did cause marked dose-dependent QTc prolongation in both species. With the highest dose of halofantrine increases in PR interval and Mobitz type I AV block also occurred in both rabbits (present data) and guinea-pigs (Batey et al., 1997). Halofantrine has recently been reported to cause cardiotoxicity by blocking delayed rectifier K+ channels (Wesche et al., 2000), an action common to several drugs that prolong the QT interval. The differing actions of halofantrine and mefloquine on the heart suggest that a pharmacodynamic interaction between the two drugs is unlikely to account for the present observation of potentiation of halofantrine-induced QTc prolongation by mefloquine. Indeed, the Ca2+ channel blocking action of mefloquine may be expected to offset the QTc prolongation caused by halofantrine and reduce the potential of this drug combination to induce torsade de pointes in a manner similar to the combined K+ and Ca2+ channel blocking drug BRL-32872 (Bril et al., 1996)
Influence of blood K+ concentrations
The rise in blood K+ concentrations observed at the end of the experiments in the group receiving halofantrine and mefloquine, may be a consequence of very high concentrations of halofantrine causing haemolysis. In other studies in anaesthetized rabbits some haemolysis has been observed after administration of halofantrine (M.P. McIntosh, A.J. Batey & S.J. Coker, unpublished observations). Alternatively, the apparent decline in blood pressure with time during these experiments could have led to K+ release from underperfused skeletal muscle. However, blood pressure was similar in all the groups, and it was only in the rabbits with the highest halofantrine concentrations that blood K+ increased significantly at the end of the experiments.
The very low baseline K+ concentrations in these experiments were a consequence of pentobarbitone anaesthesia and ventilation status. Blood K+ concentrations of approximately 2.5 mmol l−1 have been reported previously in pentobarbitone-anaesthetized rabbits (Barrett et al., 1997; Barrett & Walker, 1998). Recently we have measured blood K+ in conscious rabbits and then after pentobarbitone anaesthesia and surgical preparation. The K+ values were 5.26±0.05 and 1.30±0.02 mmol l−1 respectively (A. Farkas & S.J. Coker, unpublished observations). In two other groups of pentobarbitone-anaesthetized, open-chest rabbits the baseline blood gas and K+ values were PO2=97±2 mmHg, PCO2=25±1 mmHg, pH=7.44±0.01, K+=1.16±0.07 mmol l−1 (n=36) and PO2=95±1 mmHg, PCO2=35±1 mmHg, pH=7.43±0.01, K+ 2.1±0.09 mmol l−1 (n=40) (A. Farkas & S.J. Coker, unpublished observations). The PCO2 and K+ values in the first of these groups are very similar to those in the present study (see Table 2) whereas in the second group, when the PCO2 values were higher, the K+ values were also higher and closer to those reported by Barrett et al. (1997) and Barrett & Walker (1998). There were strong correlations between PCO2 and K+ values; r=0.809 and r=0.811 respectively. These data illustrate the influence of blood PCO2 on K+ concentrations and emphasise the need for detailed reporting of blood gas and K+ values.
Since hypokalaemia can enhance blockade of current passing through delayed rectifier K+ channels (IKr) (Yang & Roden, 1996), it is possible that the low K+ values have exaggerated the effects of halofantrine on QT prolongation. Yang and Roden's (1996) in vitro data indicate that the potency of IKr blockade by dofetilide was increased approximately 4 fold at 1 mmol l−1 extracellular K+ compared to the potency at 4 mmol l−1 extracellular K+, whereas the potency of quinidine was increased 2.5 fold under similar conditions. Assuming that a similar relationship holds for blockade of IKr by halofantrine, approximately two to four times higher concentrations of halofantrine may be required in normokalaemic man to produce the extent of QTc prolongation seen here in anaesthetized rabbits. It is interesting to note, however, that the enhancement of halofantrine-induced QTc prolongation by mefloquine was still evident at the end of the experiments when the K+ concentrations had increased in this group.
Clinical relevance
In clinical studies on halofantrine in patients with malaria, blood or plasma concentrations of halofantrine ranging from approximately 550 – 1750 ng ml−1 (1.03 – 3.27 μM) have been reported after oral (Karbwang et al., 1993; Nosten et al., 1993; Touze et al., 1996) or i.v. (Krishna et al., 1993) administration. The doses of 3 and 10 mg kg−1 halofantrine used in the present study resulted in blood concentrations of 392±24 and 1708±166 ng ml−1 (0.73±0.12 and 3.18±0.76 μM) respectively, which cover the range reported in clinical studies. In the rabbits pretreated with mefloquine, a plasma concentration of halofantrine of 1414±314 ng ml−1 (2.63±0.58 μM) was achieved after administration of the 3 mg kg−1 dose. Thus, even after allowing for any overestimation of the potency of halofantrine resulting from hypokalaemia, as discussed above, the marked potentiation of halofantrine-induced QTc prolongation by mefloquine occurred within the range of concentrations of halofantrine achieved in clinical use.
Attempts were made to measure mefloquine concentrations in the present studies, but due to small sample volumes, reliable results could not be obtained. Our previous in vitro experiments with mefloquine indicated that substantial effects on Ca2+ current in single ventricular myocytes occurred with concentrations of mefloquine only two to five times higher than those found routinely in clinical studies (Coker et al., 2000a). Since the dose of 3 mg kg−1 mefloquine used in the interaction group was three to 10 times lower than the dose causing cardiac contractile failure (which we believe to be consequence of Ca2+ channel blockade), it is likely that this 3 mg kg−1 dose is clinically relevant. However, further studies in which mefloquine concentrations were measured would be necessary to confirm this. In view of the extremely long half life of mefloquine, it is quite possible that it would persist at concentrations high enough to affect the actions of halofantrine given some time later, thus providing an explanation for the clinical observations of Nosten et al. (1993).
Conclusions
These studies have revealed a significant interaction between mefloquine and halofantrine, resulting in potentiation of halofantrine-induced prolongation of the QTc interval. The finding of increased circulating concentrations of halofantrine in the rabbits pretreated with mefloquine, the strong correlation between halofantrine concentrations and QTc intervals, and the qualitative differences in the effects of the two drugs on the heart, suggest a pharmacokinetic rather than a pharmacodynamic interaction. Whether the primary effect of mefloquine is on the distribution or on the metabolism of halofantrine remains to be determined.
Acknowledgments
This work was supported by a grant from SmithKline Beecham Pharmaceuticals Ltd. and we thank Dr R.J. Horton, International Medical Division, SmithKline Beecham Pharmaceuticals Ltd., Brentford for his assistance in organising this and arranging the supply of halofantrine. We also thank F. Hoffman-La Roche Ltd., Basel, Switzerland for providing mefloquine.
Abbreviations
- AV
atrioventricular
- CYP
cytochrome P450
- ECG
electrocardiogram
- IKr
current through delayed rectifier potassium channels
- QTc
rate-corrected QT interval
References
- BARRETT T.D., MCLEOD B.A., WALKER M.J.A. A model of myocardial ischemia for the simultaneous assessment of electrophysiological changes and arrhythmias in intact rabbits. J. Pharmacol. Toxicol. Methods. 1997;37:27–36. doi: 10.1016/s1056-8719(96)00145-1. [DOI] [PubMed] [Google Scholar]
- BARRETT T.D., WALKER M.J.A. Glibenclamide does not prevent action potential shortening induced by ischemia in anesthetized rabbits but reduces ischemia-induced arrhythmias. J. Mol. Cell. Cardiol. 1998;30:999–1008. doi: 10.1006/jmcc.1998.0664. [DOI] [PubMed] [Google Scholar]
- BATEY A.J., LIGHTBOWN I.D., LAMBERT J.P., EDWARDS G., COKER S.J. Comparison of the acute cardiotoxicity of the antimalarial drug halofantrine in vitro and in vivo in anaesthetized guinea-pigs. Br. J. Pharmacol. 1997;122:563–569. doi: 10.1038/sj.bjp.0701402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAUNE B., FLINOIS J., FURLAN V.F.G., TABURET A., BECQUEMONT L., FARINOTTI R. Halofantrine metabolism in microsomes in man: major role of CYP 3A4 and CYP 3A5. J. Pharm. Pharmacol. 1999a;51:419–426. doi: 10.1211/0022357991772628. [DOI] [PubMed] [Google Scholar]
- BAUNE B., FURLAN V., TABURET A., FARINOTTI R. Effect of selected antimalarial drugs and inhibitors of cytochrome P-450 3A4 on halofantrine metabolism by human liver microsomes. Drug Metab. Dispos. 1999b;27:565–568. [PubMed] [Google Scholar]
- BRIL A., GOUT B., BONHOMME M., LANDAIS L., FAIVRE J.-F., LINEE P., POYSER R., RUFFOLO R. Combined potassium and calcium channel blocking activities as a basis for antiarrhythmic efficacy with low proarrhythmic risk: experimental profile of BRL-32872. J. Pharmacol. Exp. Ther. 1996;276:637–646. [PubMed] [Google Scholar]
- CASTOT A., RAPOPORT P., LECOZ P. Prolonged QT interval with halofantrine. Lancet. 1993;341:1541–1541. [PubMed] [Google Scholar]
- CENNI B., MEYER J., BRANDT R., BETSCHART B. The antimalarial drug halofantrine is bound mainly to low and high density lipoproteins in human serum. Br. J. Clin. Pharmacol. 1995;39:519–526. doi: 10.1111/j.1365-2125.1995.tb04489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COKER S.J., BATEY A.J., LIGHTBOWN I.D., DIAZ M.E., EISNER D.A. Effects of mefloquine on cardiac contractility and electrical activity in vivo, in isolated cardiac preparations, and in single ventricular myocytes. Br. J. Pharmacol. 2000a;129:323–330. doi: 10.1038/sj.bjp.0703060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COKER S., MCINTOSH M., BATEY A., EDWARDS G., PORTER C., CHARMAN W. QTc prolongation by desbutylhalofantrine, the metabolite of the antimalarial drug halofantrine. Proc. Australasian Soc. Clin. Exp. Pharmacol. Toxicol. 2000b;7:44. [Google Scholar]
- DAVIS T.M.E., DEMBO L.G., KAYE-EDDIE S.A., HEWITT B.J., HISLOP R.G., BATTY K.T. Neurological, cardiovascular and metabolic effects of mefloquine in healthy volunteers: A double-blind, placebo-controlled trial. Br. J. Clin. Pharmacol. 1996;42:415–421. doi: 10.1046/j.1365-2125.1996.04745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DESNEVES J., THORN G., BERMAN A., GALATIS D., LAGRECA N., SINDING J., FOLEY M., DEADY L., COWMAN A., TILLEY L. Photoaffinitiy labeling of mefloquine-binding proteins in human serum, uninfected erythrocytes and Plasmodium falciparum-infected erythrocytes. Mol. Biochem. Parisit. 1996;82:181–194. doi: 10.1016/0166-6851(96)02732-6. [DOI] [PubMed] [Google Scholar]
- EVANS S., WALLER P. Neurological, cardiovascular and metabolic effects of mefloquine in healthy volunteers: a double-blind, placebo-controlled trial (Letter to the Editors) Br. J. Clin. Pharmacol. 1997;43:665. [PubMed] [Google Scholar]
- HALLIDAY R., JONES B., SMITH D., KITTERINGHAM N., PARK B. An investigation of the interaction between halofantrine, CYP2D6 and CYP3A4: studies with human liver microsomes and heterologous enzyme expression systems. Br. J. Clin. Pharmacol. 1995;40:369–378. doi: 10.1111/j.1365-2125.1995.tb04559.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUMBERSTONE A.J., PORTER C.J.H., CHARMAN W.N. A physicochemical basis for the effect of food on the absolute oral bioavailability of halofantrine. J. Pharmaceut. Sci. 1996;85:525–529. doi: 10.1021/js950472p. [DOI] [PubMed] [Google Scholar]
- HUMBERSTONE A.J., PORTER C.J.H., EDWARDS G.A., CHARMAN W.N. Association of halofantrine with postprandially derived plasma lipoproteins decreases its clearance relative to administration in the fasted state. J. Pharmaceut. Sci. 1998;87:936–942. doi: 10.1021/js9704846. [DOI] [PubMed] [Google Scholar]
- KARBWANG J., BANGCHANG K.N., BUNNAG D., HARINASUTA T., LAOTHAVORN P. Cardiac effect of halofantrine. Lancet. 1993;342:501–501. doi: 10.1016/0140-6736(93)91631-u. [DOI] [PubMed] [Google Scholar]
- KHOO S.M., PORTER C.J.H., EDWARDS G.A., CHARMAN W.N. Metabolism of halofantrine to its equipotent metabolite, desbutylhalofantrine, is decreased when orally administered with ketoconazole. J. Pharmaceut. Sci. 1998;87:1538–1541. doi: 10.1021/js980185w. [DOI] [PubMed] [Google Scholar]
- KRISHNA S., TER KUILE F., SUPANARANOND W., PUKRITTAYAKAMEE S., TEJAISAVADHARM P., KYLE D., WHITE N.J. Pharmacokinetics, efficacy and toxicity of parenteral halofantrine in uncomplicated malaria. Br. J. Clin. Pharmacol. 1993;36:585–591. doi: 10.1111/j.1365-2125.1993.tb00419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAOTHAVORN P., KARBWANG J., NA BANGCHANG K., BUNNAG D., HARINASUTA T. Effect of mefloquine on electrocardiographic changes in uncomplicated falciparum malaria patients. Southeast Asean J. Trop. Med. Public Health. 1992;23:51–54. [PubMed] [Google Scholar]
- LEO K., WESCHE D., MARINO M., BREWER T. Mefloquine effect on disposition of halofantrine in the isolated perfused rat liver. J. Pharm. Pharmacol. 1996;48:723–728. doi: 10.1111/j.2042-7158.1996.tb03959.x. [DOI] [PubMed] [Google Scholar]
- MBERU E., MUHIA D., WATKINS W. Measurement of halofantrine and its major metabolite desbutylhalofantrine in plasma and blood by high-performance liquid chromatography: a new methodology. J. Chromato. 1992;581:156–160. doi: 10.1016/0378-4347(92)80461-x. [DOI] [PubMed] [Google Scholar]
- MOLNAR J., WEISS J., ROSENTHAL J. The missing second: what is the correct unit for the Bazett corrected QT interval. Am. J. Cardiol. 1995;75:537–538. doi: 10.1016/s0002-9149(99)80603-1. [DOI] [PubMed] [Google Scholar]
- MONLUN E., LEMETAYER P., SZWANDT S., NEAU D., LONGYBOURSIER M., HORTON J., LEBRAS M. Cardiac complications of halofantrine - a prospective-study of 20 patients. Transac. Royal Soc. Trop. Med. Hyg. 1995;89:430–433. doi: 10.1016/0035-9203(95)90041-1. [DOI] [PubMed] [Google Scholar]
- MONLUN E., PILLET O., COCHARD J.F., GARRIGUES J.C.F., LEBRAS M. Prolonged QT interval with halofantrine. Lancet. 1993;341:1541–1542. [PubMed] [Google Scholar]
- NOSTEN F., TER KUILE F.O., LUXEMBURGER C., WOODROW C., KYLE D.E., CHONGSUPHAJAISIDDHI T., WHITE N.J. Cardiac effects of antimalarial treatment with halofantrine. Lancet. 1993;341:1054–1056. doi: 10.1016/0140-6736(93)92412-m. [DOI] [PubMed] [Google Scholar]
- TOIVONEN L., VIITASALO M., SIIKAMAKI H., RAATIKKA M., POHJOLASINTONEN S. Provocation of ventricular-tachycardia by antimalarial drug halofantrine in congenital long QT syndrome. Clin. Cardiol. 1994;17:403–404. doi: 10.1002/clc.4960170711. [DOI] [PubMed] [Google Scholar]
- TOUZE J.E., BERNARD J., KEUNDJIAN A., IMBERT P., VIGUIER A., CHAUDET H., DOURY J.C. Electrocardiographic changes and halofantrine plasma level during acute falciparum malaria. Am. J. Trop. Med. Hyg. 1996;54:225–228. doi: 10.4269/ajtmh.1996.54.225. [DOI] [PubMed] [Google Scholar]
- WESCHE D., SCHUSTER B., WANG W.-X., WOOSLEY R. Mechanism of cardiotoxicity of halofantrine. Clin. Pharmacol. Therapeut. 2000;67:521–529. doi: 10.1067/mcp.2000.106127. [DOI] [PubMed] [Google Scholar]
- YANG T., RODEN D.M. Extracellular potassium modulation of drug block of IKr. Implications for torsade de pointes and reverse use-dependence. Circulation. 1996;93:907–411. doi: 10.1161/01.cir.93.3.407. [DOI] [PubMed] [Google Scholar]