

Abstract
The selective carbonic anhydrase inhibitor acetazolamide is known to increase blood flow in several organs. Acetazolamide directly dilates isolated resistance arteries associated with activation of calcium-activated potassium (KCa) channels. We examined both the presence and mechanism of the direct vascular action of acetazolamide in vivo in humans.
Forearm vasodilator responses of 30 healthy volunteers to infusion of placebo and increasing doses of acetazolamide (1-3-10 mg min−1 dl−1) into the brachial artery were recorded by venous occlusion plethysmography, before and after local administration of L-NMMA (0.2 mg min−1 dl−1, an inhibitor of NO-synthase, n=6), indomethacin (5.0 μg min−1 dl−1, an inhibitor of prostaglandin synthesis, n=6), glibenclamide (10 μg min−1 dl−1, an inhibitor of KATP channels, n=6), tetraethylammonium (0.1 mg min−1 dl−1, an inhibitor of KCa channels, n=6) or placebo (NaCl 0.9%, n=6). Lower dosages of acetazolamide did not affect vascular tone (n=6).
Acetazolamide infusions increased forearm blood flow from 2.41±0.17 to 2.99±0.18, 4.09±0.26 and 6.77±0.49 ml min−1 dl−1 in the infused forearm (P<0.001), with no significant changes in the non-infused forearm, blood pressure or heart rate. Acetazolamide-induced vasodilation was not inhibited by L-NMMA, indomethacin, or glibenclamide but was significantly attenuated by TEA (vasodilation: 23±6, 82±19, 241±38% versus 27±8, 44±22, 42±35%).
We conclude that acetazolamide exerts a direct vasodilator effect in vivo in humans mediated by vascular KCa channel activation. This makes acetazolamide the first drug known that specifically modulates this channel.
Keywords: Acetazolamide, carbonic anhydrase, vasodilation, potassium channels, human
Introduction
Acetazolamide has been used widely as a selective inhibitor of carbonic anhydrase (Maren, 1991). A number of studies has been published on vascular properties of acetazolamide, mostly concentrating on the cerebral (Wang et al., 1993; Krasney et al., 1973; Faraci et al., 1990) and retinal (Rassam et al., 1993) circulation. All studies used systemic administration of the drug, which limits the possibility to interpret the results as a direct interaction with the vascular wall or an indirect systemic metabolic or counter-regulating effect. However, we have recently found that acetazolamide exerts a direct vasodilator effect in isolated guinea-pig resistance arteries. This effect is associated with a change in smooth muscle cell pH and subsequent opening of calcium-activated potassium (KCa) channels, as acetazolamide-induced vasodilation is inhibited by charybdotoxin, a highly selective blocker of this channel (Pickkers et al., 1999). Opening of vascular potassium channels hyperpolarizes the cell membrane, leading to closure of voltage-operated calcium channels. A blunted calcium influx results in a decrease in cytoplasmic calcium levels and vasorelaxation (Pickkers et al 1995).
In the past few years there has been an increasing interest in the role of potassium channels in the cardiovascular system (Brayden & Nelson, 1992). The KCa channel is present in human vascular smooth muscle and the physiological importance of the channel in the regulation of arterial tone is emerging as it is now clear that myogenic tone (Nelson & Quayle, 1995) and flow-mediated vasodilation (Cooke et al., 1991) are regulated by the KCa channel in many tissues.
In view of the presence of the enzyme carbonic anhydrase in vascular smooth muscle cells (Geers & Gros, 1991), aim of the present study was to determine if acetazolamide induces vasodilation in vivo in humans due to a direct action on the vascular wall, and if so, whether the mechanism of action is similar as found in isolated vessels. Since systemic administration does not permit one to distinguish between direct and indirect vascular effect of a drug, we used the perfused forearm model to examine the direct effect and mechanism of action of acetazolamide on forearm vascular tone.
Methods
Subjects
After the approval of the Ethics Committee of our hospital and the written informed consent of each subject we studied a total of 42 (21 males) non-smoking, non-obese healthy volunteers, aged 18 – 33 years. Their demographic characteristics are summarized in Table 1. Volunteers who were taking prescription drugs (except for oral contraceptives) or acetylsalicylic acid or other non-steroidal anti-inflammatory drugs were excluded.
Table 1.
Demographic data for all participants
Procedures
The experiments were performed with subjects in the supine position in a quiet, temperature controlled room (23 – 24°C). Under local anaesthesia (xylocaine HCl 20 mg ml−1) a 20-gauge catheter (Angiomat, Deseret Medical, Becton Dickinson, Sandy UT, U.S.A.) was inserted into the left brachial artery connected with an arterial pressure monitoring line (Viggo Spectramed, 5269-129) to a Hewlett Packard 78353B monitor (GmbH, Böblingen, Germany). The arterial line was kept patent with saline infusion (3 ml h−1 with 2 U heparin ml−1 added) and was used for infusion of drugs and for blood pressure recordings. Mean arterial pressure (MAP) was determined by the electronically integrated area under the brachial arterial pulse-wave curve and averaged per FBF measurement.
With the arm elevated just above heart level, blood flow was measured simultaneously in the infused and non-infused forearm three times per minute by electrocardiography-triggered venous occlusion plethysmography (Hokanson E20 rapid cuff inflator) using mercury-in-silastic strain gauges (Hokanson EC4, D.E. Hokanson, Washington, D.C., U.S.A.) as previously described (Whitney, 1953). To ensure that forearm blood flow (FBF) recordings were due predominantly to the forearm skeletal muscle resistance arteries, the hand circulation was occluded during all FBF-recordings by a wrist cuff inflated 100 mmHg above the systolic blood pressure (Lenders et al., 1991). FBF and drug administration (automatic syringe infusion pump, type STC-521, Terumo, Tokyo, Japan) were normalized to forearm volume (FAV) as measured with the water displacement method, and was expressed in ml min−1 dl−1.
Measurements were started at least 30 min after i.a. cannulation and 1 min after occlusion of the hand circulation. The FBF measurements during the last 2 min of each infusion period were taken as the response and used for further analysis.
Protocols
Low dose acetazolamide experiments
In six subjects, baseline forearm blood flow was measured for 5 min. Hereafter, increasing dosages of acetazolamide (0.1-0.3-1.0-3.0-10-30-100-300-1000 μg min−1 dl−1) were administered for 4 min each. Each measurement period lasted 8 to a maximum of 10 min and was alternated by a 5 min pause during which the wrist cuff was deflated to allow recovery of the hand circulation.
High dose acetazolamide experiments
The second series of experiments were conducted as described above, and intrabrachial infusion of acetazolamide at rates of 1-3-10 mg min−1 dl−1 were administered to 30 subjects. After termination of the last infusion period, extinction of the vasodilator blood flow response was measured after 10, 20 and 30 min. At t=45 min, the protocol was repeated after and during infusion of one of the antagonists or without a pharmacological intervention (NaCl 0.9%, to obtain time-control values) in five groups of six subjects each.
Time control experiments
After the first part of the experiment as descibed above, the second part of the experiment was started with local infusion of NaCl 0.9%. After obtaining baseline measurements for 5 min, the acetazolamide dose-response curve was repeated during concomitant NaCl 0.9% infusion.
Involvement of vascular prostaglandin- and nitric oxide-release in acetazolamide-induced vasodilation
Cerebral vasodilation after systemic administration of acetazolamide was inhibited by indomethacin (Wang et al., 1993), but not by NOLAG (Wang et al., 1992), an inhibitor of NO-synthesis. To study the role of vascular release of prostaglandins and nitric oxide in acetazolamide-induced vasodilation, we performed the following experiments: In the second and third subgroup of six subjects, the second part of the experiment was initiated by a 10 min intra-arterial infusion period of indomethacin (5.0 μg min−1 dl−1) or L-NMMA (0.2 mg min−1 dl−1) after which the acetazolamide dose-response curve was repeated during concomitant infusion of the blocker. Equipotent dosages of L-NMMA have been used before and have been shown to sufficiently attenuate vasodilation due to vascular NO synthesis (Smits et al., 1995). In a set of previous experiments adequate cyclo-oxygenase inhibition was confirmed by the absence of thromboxane B2 formation in vitro in blood drawn from an antecubital vein of the indomethacin-infused forearm, determined by a RIA using 3H-TxB2 as tracer (de Hoon and Pickkers, unpublished data).
Involvement of potassium channel activation in acetazolamide-induced vasodilation
It has been shown that in isolated resistance vessels, acetazolamide-induced vasodilation is inhibited by charybdotoxin, a selective blocker of KCa channels, but not by glibenclamide, a selective antagonist of KATP channels (Pickkers et al., 1999), indicating that acetazolamide-induced vasoactivity is mediated by KCa channel activation. Since charybdotoxin cannot be administered to man because of its toxicity, we used intraarterial administration of the potassium channel blocker tetra-ethyl ammonium (TEA) to investigate the role of KCa channel activation in the vascular effects of acetazolamide. TEA has been administered parenterally to man before, as a ganglion blocker in a total dose of 500 mg (i.v.) in patients suffering from peripheral vascular disease (Berry et al., 1946) and in normal subjects (Hoobler et al., 1949). TEA antagonizes different types of potassium channels with varying degrees of potency (Stanfield, 1983), but the compound has been shown to block single KCa channels of arterial smooth muscle and to be relatively selective for this channel at concentrations below 1 mM (Half-block, Ki ≈amp;200 μM) (Stanfield, 1983; Langton et al., 1991; Inone et al., 1985). In six subjects, we used intra-arterial administration of TEA at an infusion rate of 0.1 mg min−1 dl−1, calculated to lead to a local plasma concentration of approximately 0.5 mM l−1 (Bruning et al., 1998). In the second part of the experiment, after 10 min of TEA infusion, measurements of FBF and subsequent infusions of acetazolamide were started. TEA administration was continued during the acetazolamide infusions.
Two series of control experiments were conducted to exclude a non-specific inhibition of vasodilation by TEA and to address possible non-selective actions of TEA on KATP channels. Firstly, to investigate if TEA is a non-specific inhibitor of vasodilation we examined its effect on vasodilation induced by a drug that exerts its effect independent from vascular potassium channels: sodium nitroprusside. In a group of six subjects the increase of FBF to intrabrachial administration of SNP (0.2-2.0-6.0 μg min−1 dl−1) was assessed in the absence and presence of TEA (0.1 mg min−1 dl−1), in a fashion similar as described above for the acetazolamide experiments. Secondly, we have found that the vasodilation to ATP-dependent K+ channel opener diazoxide is also partly inhibited by TEA (Pickkers, unpublished data), indicating that TEA might also block vascular KATP channels in the perfused forearm. To confirm that the acetazolamide-induced vasodilation is solely mediated by the KCa channel, we used intra-arterial administration of the KATP selective blocker glibenclamide (10 μg min−1 dl−1) as a negative control for the acetazolamide-induced vasodilation. The dose of glibenclamide we used was three times higher than the dose that has been demonstrated to significantly attenuate the responses to the KATP channel opener diazoxide in the forearm vascular bed (Bijlstra et al., 1996).
Drugs and solutions
Acetazolamide (Diamox 500 mg ampoule, AHP Pharma), NGmonomethyl-L-arginine (L-NMMA)-acetate (Clinalpha), indomethacin (Indocid PDA 1 mg, Merck Sharp and Dome) and glibenclamide (2 mg vial, Hoechst) were diluted in NaCl 0.9% immediately prior to the experiments. The pH of the solution with the highest acetazolamide concentration was 9.05. For the other potassium channel antagonist experiments we used sterile tetra-ethyl ammonium (TEA). On the day of the experiment TEA was reconstituted from a sterile powder (Sigma), diluted in NaCl 0.9% to a concentration of 1 mg ml−1 and passed through a 0.22 μm Millipore (Milford, Massachusetts, U.S.A.) filter. Sodium nitroprusside (Nipride, 50 mg, Roche) was dissolved in 2 ml glucose 5%, further diluted in NaCl 0.9% just before the experiments and protected from light. Saline was used as placebo infusion. All drugs and placebo infusions were administered at the same infusion rate of 100 μl min−1 dl−1.
Data analysis, calculations and statistics
The effects of acetazolamide were analysed by comparing the haemodynamic variables at baseline and at the increasing dosages by one-way analysis of variance (ANOVA) with repeated measures. Post hoc comparisons between the different dosages were made by Scheffé-F-tests including Bonferroni correction. Paired Student t-test was used for the assessment of the effects of L-NMMA, indomethacin, TEA and glibenclamide on baseline parameters. In order to evaluate the effect of the intervention (NO-synthase blockade, prostaglandin synthesis blockade and potassium channel blockade) on the acetazolamide or sodium nitroprusside responses, two-way repeated measures ANOVA was performed on the changes from baseline. Since the mean arterial blood pressure was not affected by either drug infusion (see Results), FBF-changes were assumed to represent changes in forearm vascular tone, and forearm vascular resistance was not calculated (Benjamin et al., 1995). The mean FBF of the last 2 min recorded at each infusion rate was taken as the response. Changes in FBF were compared with values obtained during baseline measurements and expressed as absolute values or as percentage change in ratio of the infused and non-infused arm compared to the baseline ratio to correct for small baseline differences between the first and second part of the experiments with the various inhibitors. By using the ratio of the FBF measurements, the non-infused arm can be concerned as a contemporaneous control for the infused arm (Benjamin et al., 1995).
All data are expressed as means±s.e.mean of n experiments, unless indicated otherwise. A P-value <0.05 was taken as statistically significant.
Results
Baseline characteristics
Baseline characteristics of all studied groups are presented in Table 1.
Cardiovascular responses to local acetazolamide administration
In the first series of experiments, intrabrachial administration of acetazolamide 0.01 μg min−1 dl−1 to 1 mg min−1 dl−1 (n=6) did not change blood flow in the infused forearm (3.2±0.4 ml min−1 dl−1 at baseline to 3.0±0.4 ml min−1 dl−1 at the last dose, P=N.S.). There were also no changes in FBF in the contralateral arm (2.6±0.5 to 2.3±0.5 ml min−1 dl−1), nor in blood pressure and heart rate (data not shown).
To assess the vascular response to graded infusion of higher acetazolamide concentrations, the data of the first dose-response curve for all experiments with the higher acetazolamide dosages were pooled (n=30). Table 2 summarizes the results of this analysis. High dose acetazolamide infusions induced a dose dependent increase in FBF from 2.4±0.2 at baseline to 3.0±0.2, 4.1±0.3 and 6.8±0.5 ml min−1 dl−1, which was significantly different from baseline for all three dosages. The dose-dependency was supported by significant differences between dosages (Scheffé-F-tests: P<0.05). After cessation of the last infusion of acetazolamide, FBF decreased (from 6.8±0.5 ml min−1 dl−1 at the last acetazolamide infusion) to 2.3±0.2, 1.8±0.2 and 1.9±0.2 at t=10, 20 and 30 min, respectively. Thus, within 20 to 30 min the FBF was identical to the baseline level, indicating total extinction of the vasodilator response. As shown in Table 2, there were no changes in blood flow in the non-infused forearm (from 2.2±0.2 at baseline to 2.1±0.2 ml min−1 dl−1 during the last dose) or changes in blood pressure or heart rate during acetazolamide infusion, arguing against an acute systemic vascular effect of the dosages used.
Table 2.
The results of graded intra-arterial acetazolamide infusion on haemodynamic parameters

Time control experiments
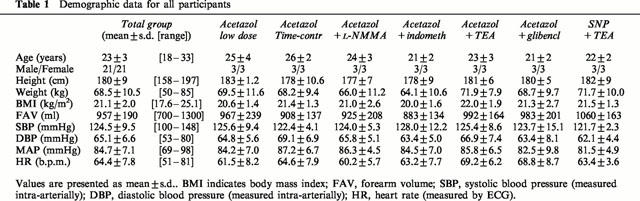
Before conducting experiments where the acetazolamide dose-response curve was repeated after and in the presence of one of the inhibitors, time control experiments were carried out to investigate the repeatability of the vasodilator response. In the subset of experiments where the acetazolamide infusions were repeated after 45 min using NaCl 0.9% as the pharmacological intervention, acetazolamide led to a maximum vasodilation of 228±56% at the first and 286±49% at the second dose response-curve. There was no significant difference between the percentage vasodilation between this first and second series of measurements (Figure 1, right panel), indicating good repeatability of the dose-response curve. During both acetazolamide experiments, there were no significant changes in contralateral FBF, blood pressure or heart rate.
Figure 1.
Percentage increase in the ratio of the infused and non-infused arms during graded acetazolamide infusion, both in the absence (•/solid line) as well as during concomitant infusion (○/dashed line) of TEA (left panel) or placebo (right panel) is presented as mean±s.e.mean. The P-value refers to the statistical difference between conditions for these responses as analysed by repeated measures ANOVA.
Involvement of vascular autocoid release in acetazolamide-induced vasodilation-Nitric oxide
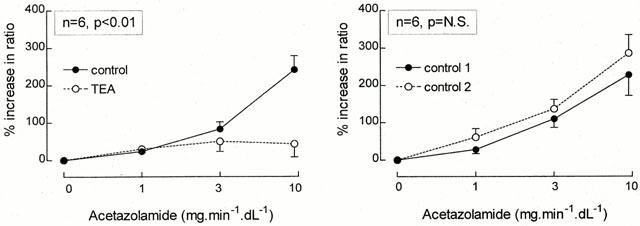
Figure 2 (left panel) illustrates the percentage vasodilation to the three increasing acetazolamide dosages in the absence and presence of L-NMMA infusions. In this group acetazolamide increased the ratio of FBF at a maximum of 219±38% in the absence of L-NMMA. Administration of L-NMMA reduced FBF from 2.0±0.5 to 1.2±0.1 ml min−1 dl−1 (paired t-test, P<0.05) with no significant change in the contralateral FBF (from 1.7±0.4 to 1.6±0.4 ml min−1 dl−1), blood pressure (123±2/66±2 to 123±2/69±2 mmHg) or heart rate (61±3 to 60±2 b.p.m.). Compared to this new baseline, the maximum percentage vasodilation induced by acetazolamide was 253±21%, not significantly different when compared with the first dose response curve.
Figure 2.
Percentage increase in the ratio of the infused and non-infused arms during graded acetazolamide infusion, both in the absence (•/solid line) as well as during concomitant infusion (○/dashed line) of L-NMMA (left panel) or indomethacin (right panel) is presented as mean±s.e.mean. The P-value refers to the statistical difference between conditions for these responses as analysed by repeated measures ANOVA.
Prostaglandins
The right panel of Figure 2 demonstrates the interaction of acetazolamide and indomethacin. Maximum vasodilation to acetazolamide was 219±27% in this group. Indomethacin did not change FBF (1.8±0.3 to 1.8±0.3 ml min−1 dl−1) or other hemodynamic parameters significantly, and the vasodilator response to acetazolamide was also unchanged (maximum: 199±34%, P=N.S.).
Involvement of potassium channel activation in acetazolamide-induced vasodilation
Figure 1 (left panel) demonstrates the effect of TEA on the forearm vasodilator response to acetazolamide. Baseline FBF after 10 min of TEA administration was 1.9±0.4, compared to 1.9±0.4 ml min−1 dl−1 at the start of the experiment (P=N.S.). Acetazolamide-induced vasodilation averaged 23±6, 82±19 and 241±38% in the absence of TEA and 27±8, 44±22, and 42±35% in the presence of TEA (P<0.02). The concomitant infusion of acetazolamide and TEA elicited no significant changes in contralateral FBF (from 1.7±0.3 at baseline to 1.3±0.2 ml min−1 dl−1 at the highest acetazolamide dose), blood pressure (from 125±3/71±3 to 128±4/77±4 mmHg) or heart rate (from 65±3 to 68±4 b.p.m.).
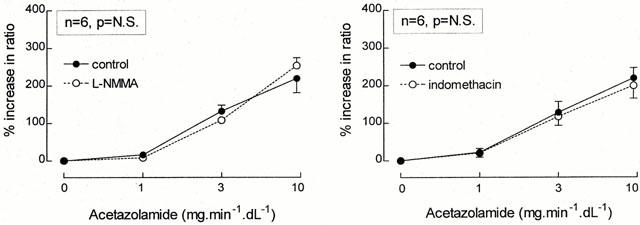
The intra-arterial infusion of TEA did not change the forearm vasodilator response to sodium nitroprusside (Figure 3, right panel). In the absence of TEA, the percentage increase in ratio of the infused and non-infused arms for the three increasing dosages SNP averaged 75±11, 366±38 and 740±78%, while these values were 78±23, 485±66 and 991±190% during concomitant TEA administration (P=N.S.). No significant effects on systemic haemodynamics were observed during the combined infusion of SNP and TEA. (The contralateral FBF was 2.1±0.3 before and 2.0±0.4 during the highest SNP dose.)
Figure 3.
Percentage increase in the ratio of the infused and non-infused arms during graded acetazolamide infusion (left panel), both in the absence (•/solid line) as well as during concomitant infusion of glibenclamide (○/dashed line) and during graded sodium nitroprusside infusion (right panel), both in the absence (•/solid line) as well as during concomitant infusion of TEA (○/dashed line) are presented as mean±s.e.mean. The P-value refers to the statistical difference between conditions for these responses as analysed by repeated measures ANOVA.
As demonstrated in Figure 3 (left panel), the vasodilator response to acetazolamide was not attenuated by local glibenclamide infusion. In this last series of experiments, maximum acetazolamide-induced vasodialtion were 209±33 and 292±44%, in the absence and presence of glibenclamide, respectively (P=N.S.). Local glibenclamide infusion itself did not change FBF in either arm (infused arm: from 1.8±0.3 to 2.0±0.4 ml min−1 dl−1; non-infused arm: from 2.2±0.3 to 2.4±0.5 ml min−1 dl−1), blood pressure (from 125±6/67±4 to 126±6/68±3 mmHg) or heart rate (from 63±3 to 65±3 b.p.m.).
Discussion
Our study is the first to demonstrate that acetazolamide exerts a directs vascular effect in vivo. We showed that local administration of acetazolamide induces rapid vasodilation in the human forearm, without affecting blood pressure, heart rate and blood flow in the non-infused arm. This indicates a direct local vascular effect of the drug, independent of its renal effects and possible actions on other organs. Although an interaction between acetazolamide and prostaglandin and nitric oxide production has been found in different tissues (Puscas & Coltau, 1995; Gutiérrez-Cabano, 1994), acetazolamide-induced vasodilation appeared to be independent of these autocoids in the human forearm, since local indomethacin and L-NMMA administration had no inhibitory effect on the acetazolamide-induced vasodilation. Consequently, our data support the conclusion that vascular autocoid release does not attribute to the vasodilatory response to intra-arterial acetazolamide infusion in man. In contrast, vasodilation was almost totally abolished by the potassium channel blocker TEA.
Role of potassium channel activation
From data obtained in experiments in isolated arteries (Pickkers et al., 1999) we postulated that activation of vascular KCa channels may represent the mechanism of vascular action of acetazolamide in vivo. In the absence of a highly selective KCa channel inhibitor that can be administered to man, we used the less selective blocker TEA. Tetraethylammonium has been administered parenterally to humans before (Berry et al., 1946; Hoobler et al., 1949). The vascular effects of KCa channel inhibition in isolated arteries is variable, as some studies reported an increase in basal tone for instance in cerebral arteries (Brayden & Nelson, 1992), but others found no change for example in human and guinea-pig resistance arteries (Pickkers & Hughes, 1995; Calder et al., 1994). Recently, it was published that TEA decreased coronary blood flow in ischemic heart, but not in non-ischemic hearts in open-chest dogs (Node et al., 1997), indicating that TEA is a vasoconstrictor only when the KCa channel is activated. Systemic administration in humans increases peripheral blood flow due to ganglion blockade (Berry et al., 1946; Hoobler et al., 1949), but in our experiments, intra-arterial administration (distal from ganglia) of TEA had no significant effect on baseline FBF.
Despite its lack of effect on baseline FBF, TEA almost totally abolished acetazolamide-induced vasodilation in the human forearm. Control experiments showed that acetazolamide-induced vasodilation was not inhibited by the KATP channel antagonist glibenclamide. Studies with nitroprusside excluded non-specific inhibitory effects of TEA. From these results we conclude that acetazolamide-induced vasodilation in the human forearm is mediated by KCa channel opening. Up to now, no other agent that specifically opens the KCa channel is known. NS-004, a putative opener of the KCa channel, has recently been shown not to be specific nor selective for the KCa channel (Sargant et al., 1993). Thus acetazolamide could be the first specific KCa channel opener. Clearly, electrophysiologic studies in isolated vascular smooth muscle cells need to be performed to define the mechanism of acetazolamide-induced modulation of KCa channels.
Besides the possibilty of a direct interaction with the KCa channel, it is also possible that acetazolamide acts via an indirect effect, e.g. through a second messenger or through a metabolic effect. Inhibition of carbonic anhydrase reduces the capacity of the red blood cells to carry CO2, thereby increasing tissue pCO2 (Bickler et al., 1988). The autoregulatory response to local hypercapnia is vasodilation (Kontos et al., 1967; Daugherty et al., 1967), in order to increase tissue blood flow, washing out the excess of CO2. From our studies, we cannot exclude that a build-up of tissue CO2 is responsible for K-channel activation (Taki et al., 1993). Interestingly, it has been reported that increasing pCO2 hyperpolarizes the plasma membrane of isolated small arteries (Jensen et al., 1993), and we have found that CO2 induces vasodilation and that this response is inhibited by blockers of KCa channels (Pickkers & Hughes, 1996). The role of K-channel activation in the interaction between pCO2 and vascular tone has been insufficiently studied and requires more attention.
Also, in in vitro experiments, we have demonstrated that the vasorelaxant effect of several carbonic anhydrase inhibitors and thiazide diuretics is triggered by a rise in intracellular pH due to the carbonic anhydrase-inhibiting activity of the drug, leading to KCa channel activation and relaxation (Pickkers et al., 1999). These observations make it plausible that acetazolamide activates the KCa channel through an indirect metabolic effect and not via a direct interaction with the channel.
Although the action of acetazolamide appears to be similar to previous findings in isolated resistance arteries (Pickkers et al., 1999), the calculated acetazolamide concentration needed to exert a comparable effect in vivo is much higher. Besides the possible barrier function of the endothelium in our experiments (in the in vitro experiments the drug is administered from the outside of the vessel directly onto the vascular smooth muscle cells), the difference in species and vascular beds could play an important role. It is known that carbonic anhydrase activity varies between species and from organ to organ (Effros & Weissman 1979; Muhleisen & Kreye, 1985, O'Brasky & Crandall, 1980). Isolated vessels were obtained from mesenteric or subcutaneous fat tissue, whereas forearm blood flow predominantly reflects forearm muscle perfusion. Other differences between in vivo and in vitro condition may also be important. We have recently found that, independent of the route of administration (from the outside of the vessel or via the luminal space) vasodilation induced by KCa channel activation is less in isotonic pressurized vessels than in unpressurized vessels in a conventional wire myograph (Ruijtenbeek & Hughes, 1997). At present, the origin of this disparity is not clear, but it could also explain the difference between the in vitro and in vivo results.
In conclusion, we report that intra-arterial infusion of acetazolamide directly dilates resistance arteries in the human forearm, and that this vasodilation is not dependent on local prostaglandin or NO release, but mediated by vascular KCa channels. This novel mechanism of action of acetazolamide deserves further attention, since no other drug is known that specifically modulates the vascular KCa channel.
Acknowledgments
This study was supported by the Dutch Heart Foundation, grant no. 94.059
Abbreviations
- FAV
forearm volume
- FBF
forearm blood flow
- KCa
Calcium-activated potassium channel
- KATP
ATP-dependent potassium channel
- L-NMMA
L-NG-monomethyl-arginine
- NO
Nitric oxide
- pHi
intracellular pH
- TEA
tetraethylammonium
References
- BENJAMIN N., CALVER A., COLLIER J., ROBINSON B., VALLANCE P., WEBB D. Measuring forearm blood flow and interpreting the responses to drugs and mediators. Hypertension. 1995;25:918–923. doi: 10.1161/01.hyp.25.5.918. [DOI] [PubMed] [Google Scholar]
- BERRY R.L., CAMPBELL K.N., LYONS R.H., MOE G.K., SUTLER M.R. The use of tetraethylammonium in peripheral vascular disease and causalgic states. Surgery. 1946;20:525–535. [PubMed] [Google Scholar]
- BICKLER P.E., LITT L., BANVILLE D.L., SEVERINGHAUS J.W. Effects of acetazolamide on cerebral acid-base balance. J. Appl. Physiol. 1988;65:422–427. doi: 10.1152/jappl.1988.65.1.422. [DOI] [PubMed] [Google Scholar]
- BIJLSTRA P.J., LUTTERMAN J.A., RUSSEL F.G.M., THIEN T., SMITS P. Interaction of sulphonylurea derivatives with vascular ATP-sensitive potassium channels in humans. Diabetologia. 1996;39(9):1083–1090. doi: 10.1007/BF00400658. [DOI] [PubMed] [Google Scholar]
- BRAYDEN J.E., NELSON M.T. Regulation of arterial tone by activation of calcium-dependent potassium channels. Science. 1992;256:532–535. doi: 10.1126/science.1373909. [DOI] [PubMed] [Google Scholar]
- BRUNING T.A., KEMME M.J., CHANG P.C., MUIZERT Y., VAN ZWIETEN P.A. Calculation of plasma concentrations of intraarterially infused compounds in forearm plethysmography. Cardiovasc Res. 1998;37:210–215. doi: 10.1016/s0008-6363(97)00218-6. [DOI] [PubMed] [Google Scholar]
- CALDER J.A., SCHACHTER M., SEVER P.S. Potassium channel opening properties of thiazide diuretics in isolated guinea-pig resistance arteries. J. Cardiovasc. Pharmacol. 1994;24:158–164. doi: 10.1097/00005344-199407000-00024. [DOI] [PubMed] [Google Scholar]
- COOKE J.P., ROSSITCH E.R., ANDON N.A., LOSCALZO J., DZAU V.J. Flow activates an endothelial potassium channel to release an endogenous nitrovasodilator. J. Clin. Invest. 1991;88:1663–1671. doi: 10.1172/JCI115481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAUGHERTY R.M., Jr, SCOTT J.B., DABNEY J.M., HADDY F.J. Local effects of O2 and CO2 on limb, renal, and coronary vascular resistances. Am. J. Physiol. 1967;213:1102–1110. doi: 10.1152/ajplegacy.1967.213.5.1102. [DOI] [PubMed] [Google Scholar]
- EFFROS R.M., WEISSMAN M.L. Carbonic anhydrase activity of the cat hind leg. J. Appl. Physiol. 1979;47:1090–1098. doi: 10.1152/jappl.1979.47.5.1090. [DOI] [PubMed] [Google Scholar]
- FARACI F.M., MAYHAN W.G., HEISTAD D.D. Vascular effects of acetazolamide on the choroid plexus. J. Pharmacol. Exp. Ther. 1990;254:23–27. [PubMed] [Google Scholar]
- GEERS C., GROS G. Muscle carbonic anhydrases. Function in muscle contraction and in the homeostasis of muscle pH and pCO2. New York: Plenum Press; 1991. The carbonic anhydrases; pp. 227–240. [Google Scholar]
- GUTIÉRREZ-CABANO C.A. Gastric Mucosal protection by acetazolamide in rats. Roles of prostaglandins, sulfhydryls, and gastric motility. Act. Gastroent. Latinoamer. 1994;24:89–97. [PubMed] [Google Scholar]
- HOOBLER S.W., MALTON S.D., BALLANTINE H.T., COHEN S., NELIGH R.B., PEET M., LYONS R.H. Studies on vasomotor tone. I. The effect of tetraethylammonium ion on the peripheral blood flow of normal subjects. J. Clin. Invest. 1949;28:638–647. doi: 10.1172/JCI102113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- INONE R., KITAMURA K., KURIYAMA H. Two Ca-dependent K-channels classified by the application of tetraethylammonium distribute to smooth muscle membranes of the rabbit portal vein. Eur. J. Physiol. 1985;405:173–179. doi: 10.1007/BF00582557. [DOI] [PubMed] [Google Scholar]
- JENSEN P.E., HUGHES A.D., BOONEN H.C.M., AALKJÆR C. Force, membrane potential, and [Ca2+]i during activation of rat mesenteric small arteries with norepinephrine, potassium, aluminum fluoride, and phorbol ester. Effects of changes in pHi. Circ. Res. 1993;73:314–324. doi: 10.1161/01.res.73.2.314. [DOI] [PubMed] [Google Scholar]
- KONTOS H.A., RICHARDSON D.W., PATTERSON J.L., JR Effects of hypercapnia on human forearm blood vessels. Am. J. Physiol. 1967;212:1070–1080. doi: 10.1152/ajplegacy.1967.212.5.1070. [DOI] [PubMed] [Google Scholar]
- KRASNEY J.A., LEVIZKY M.G., DAVIES D.G. Cardiovascular effects of intracisternal injections of acetazolamide. Cardiovasc. Res. 1973;7:684–686. doi: 10.1093/cvr/7.5.684. [DOI] [PubMed] [Google Scholar]
- LANGTON P.D., NELSON M.T., HUANG Y., STANDEN N.B. Block of calcium-activated potassium channels in mammalian arterial myocytes by tetraethylammonium ions. Am. J. Physiol. 1991;260:H927–H934. doi: 10.1152/ajpheart.1991.260.3.H927. [DOI] [PubMed] [Google Scholar]
- LENDERS J., JANSEN G.J., SMITS P., THIEN T. The role of the wrist cuff in forearm plethysmography. Clin. Sci. 1991;80:413–417. doi: 10.1042/cs0800413. [DOI] [PubMed] [Google Scholar]
- MAREN T.H. Direct measurements of the rate constants of sulfonamides with carbonic anhydrase. J. Pharmacol. Exp. Ther. 1991;41:419–426. [PubMed] [Google Scholar]
- MUHLEISEN M., KREYE V.A. Lack of soluble carbonic anhydrase in aortic smooth muscle of the rabbit. Eur. J. Physiol. 1985;405:234–236. doi: 10.1007/BF00582566. [DOI] [PubMed] [Google Scholar]
- NELSON M.T., QUAYLE J.M. Physiological roles and properties of potassium channels in arterial smooth muscle. Am. J. Physiol. 1995;268:C799–C822. doi: 10.1152/ajpcell.1995.268.4.C799. [DOI] [PubMed] [Google Scholar]
- NODE K., KITAKAZE M., KOSAKA H., MINAMINO T., HORI M. Bradykinin mediation of Ca2+-activated K+ channels regulates coronary blood flow in ischemic myocardium. Circulation. 1997;95:1560–1567. doi: 10.1161/01.cir.95.6.1560. [DOI] [PubMed] [Google Scholar]
- O'BRASKY J.E., CRANDALL E.D. Organ and species differences in tissue vascular carbonic anhydrase activity. J. Appl. Physiol. 1980;49:211–217. doi: 10.1152/jappl.1980.49.2.211. [DOI] [PubMed] [Google Scholar]
- PICKKERS P., GARCHA R.S., SCHACHTER M., SMITS P., HUGHES A.D. Inhibition of carbonic anhydrase accounts for the direct vascular effects of thiazide diuretics. Hypertension. 1999;33:1043–1048. doi: 10.1161/01.hyp.33.4.1043. [DOI] [PubMed] [Google Scholar]
- PICKKERS P., HUGHES A.D. Relaxation and decrease in [Ca2+]i by hydrochlorothiazide in guinea-pig isolated mesenteric arteries. Br. J. Pharmacol. 1995;114:703–707. doi: 10.1111/j.1476-5381.1995.tb17195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PICKKERS P., HUGHES A.D. Blockade of Ca2+-activated potassium channels inhibits relaxation in response to carbonic anhydrase inhibitors or reduction in pCO2 in guinea-pig isolated mesenteric resistance arteries (Abstr.) J. Vasc. Res. 1996;32:179. [Google Scholar]
- PUSCAS I., COLTAU M. Inhibition of carbonic anhydrase by nitric oxide. Arzneim-Forsch/Drug Res. 1995;45:846–848. [PubMed] [Google Scholar]
- RASSAM S.M., PATEL V., KOHNER E.M. The effect of acetazolamide on the retinal circulation. Eye. 1993;7:697–702. doi: 10.1038/eye.1993.159. [DOI] [PubMed] [Google Scholar]
- RUIJTENBEEK K., HUGHES A.D. Action of hydrochlorothiazide on guinea-pig isolated pressurized mesenteric arteries. (Abstr.) Hypertension. 1997;30:1003. [Google Scholar]
- SARGANT C.A., GROVER G.J., ANTONACCIO M.J., MCCULLOUGH J.R. The cardioprotective, vasorelaxant and electrophysiological profile of the large conductance calcium-activated potassium channel opener NS-004. J. Pharmacol. Exp. Ther. 1993;266:1422–1429. [PubMed] [Google Scholar]
- SMITS P., WILLIAMS S.B., LIPSON D.E., BANITT P., RONGEN G.A., CREAGER M.A. Endothelial release of nitric oxide contributes to the vasodilator effect of adenosine in humans. Circulation. 1995;92:2135–2141. doi: 10.1161/01.cir.92.8.2135. [DOI] [PubMed] [Google Scholar]
- STANFIELD P.R. Tetraethylammonium ions and the potassium permeability of excitable cells. Rev. Physiol. Biochem. Pharmacol. 1983;97:1–67. doi: 10.1007/BFb0035345. [DOI] [PubMed] [Google Scholar]
- TAKI K., HIRAHARA K., TOTOKI T., TAKAHASHI N. Retention of carbon dioxide in tissue following carbonic anhydrase inhibition in dogs. Clin Ther. 1993;15:884–889. [PubMed] [Google Scholar]
- WANG Q., PAULSON O.B., LASSEN N.A. Effect of nitric oxide blockade by NG-nitro-l-arginine on cerebral blood flow response to changes in carbon dioxide tension. J. Cereb. Blood. Flow. Metab. 1992;12:947–953. doi: 10.1038/jcbfm.1992.131. [DOI] [PubMed] [Google Scholar]
- WANG Q., PAULSON O.B., LASSEN N.A. Indomethacin abolishes cerebral blood flow increase in response to acetazolamideinduced extracellular acidosis: a mechanism for its effect on hypercapnia. J. Cereb. Blood. Flow. Metab. 1993;13:724–727. doi: 10.1038/jcbfm.1993.92. [DOI] [PubMed] [Google Scholar]
- WHITNEY R.J. The measurement of volume changes in human limbs. J. Physiol. Camb. 1953;121:1–21. doi: 10.1113/jphysiol.1953.sp004926. [DOI] [PMC free article] [PubMed] [Google Scholar]