

Abstract
Hypersensitivity to the drug sulfamethoxazole (SMX) is thought to be a consequence of bioactivation to the hydroxylamine metabolite (SMX-NHOH) and further oxidation to the ultimate reactive metabolite, nitroso-sulfamethoxazole (SMX-NO). SMX-NO covalently modifies self proteins which in turn might be recognized as neo-antigens by T-cells. The antioxidant glutathione (GSH) is known to protect cells from reactive metabolites by conjugation and subsequent dissociation to SMX-NHOH and/or SMX.
To study the reactivity of T-cells to SMX metabolites and their respective role in the generation of drug-specific T-cells, we analysed the effect of GSH on the response of PBMC to SMX and its metabolites SMX-NHOH and SMX-NO. Furthermore, we monitored the proliferative response of drug-specific T-cell clones in the presence or absence of GSH.
We found that addition of GSH to peripheral blood mononuclear cells had no effect on the SMX-specific response but enhanced the proliferation to SMX-metabolites. The response of SMX-NO-specific T-cell clones was abrogated when GSH was present during the covalent haptenation of antigen presenting cells (APC). Conversely, SMX-specific T-cell clones gained reactivity through the conversion of SMX-NO to the parent drug by GSH. While GSH had no effect on the initial activation of T-cell clones, it prevented covalent binding to APCs, reduced toxicity and thereby led to proliferation of drug-specific T-cells to non-reactive drug metabolites.
Our data support the concept that in allergic individuals T-cells recognize the non-covalently bound parent drug rather than APC covalently modified by SMX-NO.
Keywords: Non-peptide antigens; drug metabolism; sulfamethoxazole; reduced glutathione, drug hypersensitivity; T-lymphocytes; immunogenicity
Introduction
According to the classical hapten-carrier model of Landsteiner (Landsteiner & Jacoby, 1935) the sulfonamide SMX is thought to be too small to represent a complete antigen. It is not chemically reactive and thus requires metabolism to form a hapten-carrier complex. In vivo it is metabolized predominantly in the liver but also in the skin by N-acetyl transferase and N-glucuronyl transferase enzymes (Reilly et al., 2000). These biotransformations lead to the formation of non-toxic metabolites (Vree & Hekster, 1987). To a limited extent, SMX is N4-hydroxylated in a cytochrome P450- and/or myeloperoxidase-catalysed reaction to a hydroxylamine-metabolite (SMX-NHOH) which can be further oxidized to a nitroso compound (SMX-NO) (Cribb et al., 1990; 1995; Cribb & Spielberg, 1992). Figure 1 depicts these metabolic pathways of SMX. The nitroso compound can bind to thiol groups of proteins (Naisbitt et al., 1999), and is therefore able to covalently modify self-proteins, which in turn might be recognized as neo-antigens by the immune system (Meekins et al., 1994). In rats the in vivo administration of SMX-NO but not of SMX itself resulted in production of anti-SMX IgG antibodies (Gill et al., 1997). Thus, it is usually assumed that SMX gains its immunogenicity after oxidative metabolism.
Figure 1.
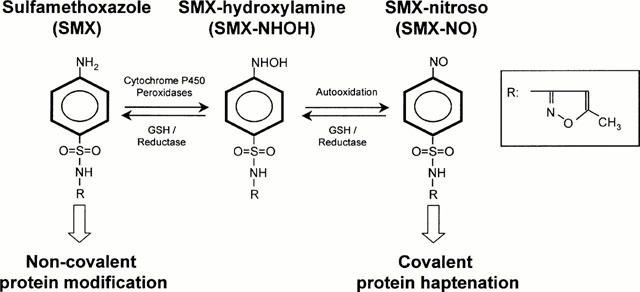
Scheme of SMX metabolism. SMX can be bioactivation to SMX-NHOH and further oxidation to SMX-NO. In turn, SMX metabolites can be reduced and detoxified by GSH. Reactive metabolite will bind preferentially in a covalent way to proteins, while the parent compound SMX associates non covalently.
Oxidation of SMX-NHOH to the nitroso intermediate is prevented by intracellular reduced sulfhydryl compounds such as GSH (Cribb et al., 1991). GSH is a tripeptide which reacts with SMX-NO to form an unstable semimercaptal. This conjugate rapidly rearranges to a stable sulfinamide metabolite or is cleaved to SMX-NHOH (Cribb et al., 1991). SMX-NHOH is less prone to haptenate proteins compared to SMX-NO and thereby less toxic (Naisbitt et al., 1999).
Recent investigations have demonstrated that drugs such as lidocaine and SMX, which are considered to be chemically inert, can be recognized by drug-specific T-cell clones (Schnyder et al., 1997; Zanni et al., 1998a,1998b; von Greyerz et al., 1999). This recognition required the continuous presence of the drug, was MHC-restricted and very rapid. It could be best explained by an unstable and direct presentation of SMX or lidocaine without requirement for processing or drug metabolism. Surprisingly, and in contrast to what has been known from animal studies, CD4+ T-cells from patients allergic to SMX predominately respond to the inert parent compound rather than the covalently bound metabolites (Schnyder et al., 2000). Therefore, T-cell responses to the noncovalently presented SMX may contribute to the pathophysiology of drug-elicited allergy (Pichler & Yawalkar, 2000).
The objective of the present study was to analyse the effect of GSH on the toxicity of reactive SMX intermediates and to characterize the activation and proliferative response of T-cells from allergic individuals to SMX and its metabolites in the presence of GSH. Both peripheral blood mononuclear cells (PBMC) and SMX-specific T-cell clones responded more vigorously to SMX-NHOH and SMX-NO in the presence of GSH. This raises the possibility that detoxification of the reactive metabolites by reduction may promote drug hypersensitivity by increasing the plasma concentration of SMX at the site of toxicity (i.e., the skin).
Methods
Donor characteristics
The SMX hypersensitive individuals UNO and KG have been described previously (Zanni et al., 1998a; Schnyder et al., 2000). Donor UNO developed an erythematous exanthem after therapy with co-trimoxazole (=SMX-plus trimethoprim) 10 years ago and suffered later a generalized exanthem within 1 day of re-exposure to co-trimoxazole. The allergic donor KG, a young pharmacist, developed allergic symptoms (exanthem), after being treated for an infection with an unknown sulfonamide. Five years later, she developed a macular exanthem and dyspnea after working with sulfamethoxazole in the laboratory.
The patient RB developed Quincke's edema and acute difficulty in breathing 4 days after intake of co-trimoxazole. There followed no re-exposure to the drug. Three years later, the patient responded negative to various antibiotics, including co-trimoxazole, in scratch or patch tests. However, the lymphocyte transformation test (LTT) was positive for SMX (SI=8.7 at 1000 μM) and SMX-NO (SI=5.0 at 100 μM) but not for trimethoprim (SI=1.5 at 50 μg ml−1).
Culture media
The cell culture medium used was RPMI-1640 supplemented with 10% pooled, heat-inactivated human AB serum (Swiss Red Cross, Bern, Switzerland), 25 mM HEPES buffer, 2 mM L-glutamine (Seromed, Fakola, Basel, Switzerland), 25 μg ml−1 transferrin (Biotest, Dreieich, Germany), 100 μg ml−1 streptomycin and 100 u ml−1 penicillin. T-cell clones (TCC) were cultured using medium additionally enriched with 20 u ml−1 recombinant IL-2 (Dr A. Cerny, Inselspital, Bern, Switzerland). EBV-transformed B-lymphoblastoid cell lines (B-LCL) were grown in RPMI-1640 supplemented with 10% foetal calf serum (FCS, Gibco, Paisley, U.K.), 25 mM HEPES buffer, 100 μg ml−1 streptomycin and 100 u ml−1 penicillin.
Drugs and drug metabolites
The parent drug SMX was obtained from Hoffmann-La Roche (Basel, Switzerland) and stock solutions of 10 mg ml−1 freshly prepared before use in RPMI 1640 containing 5% (v v−1) of 1 N NaOH. SMX-NHOH and SMX-NO were synthesized as described by Naisbitt (Naisbitt et al., 1996) and were >95% pure as assessed by NMR and elemental analysis. Stock solutions (10 mM) were freshly prepared before use in a mixture of 80% RPMI 1640 and 20% DMSO. To facilitate dissolution, 1 N NaOH was added to the mixture to a final amount of 5% (v v−1).
Cytotoxicity assay
For evaluation of drug-toxicity, B-LCL (1×106 cells ml−1) were incubated in duplicate cultures at 37°C with indicated concentrations of SMX, SMX-NHOH or SMX-NO in the presence or absence of 1 mM GSH. After 2 h, cells were harvested, washed once with HBSS and viability was determined by trypan blue exclusion.
T-cell proliferation assays
Proliferation of PBMC was measured using the lymphocyte transformation test (LTT) as previously described (Schnyder et al., 2000). In brief, PBMC from SMX-allergic donors UNO (Schnyder et al., 1997) and donor KG (Schnyder et al., 2000) were incubated in 96-well U-bottomed plates in triplicate cultures at a cell density of 1×106 ml−1. Cells were cultured in the continuous presence of different concentrations of SMX, SMX-NHOH, or SMX-NO. To some of the cultures GSH (Sigma, St. Louis, MO, U.S.A.) was added at 1 mM concentration. After 5 days, proliferation was determined by overnight incubation with 0.5 μCi of 3H-thymidine. Cells were harvested and incorporated radioactivity measured on a scintillation liquid-free β- counter (Trace 96, Inotech, Wohlen, Switzerland).
In order to measure the proliferation of T-cell clones (TCC), TCC (5×104 cells per well) were incubated in duplicate cultures in 96-well U-bottomed plates together with 5 – 10×104 B-LCL in 0.2 ml medium in the presence of indicated concentrations of antigen. To some of the cultures reduced GSH was added at a concentration of 1 mM. After 48 h, 0.5 μCi 3H-thymidine was added. Cells were harvested 12 h later and incorporated radioactivity determined as described above.
In some experiments, APC were haptenated by prepulse with SMX-NO as described (Schnyder et al., 2000). In brief, B-LCL (1×106 ml−1) were cultured in 0.5 ml medium containing 100 μM SMX-NO at 37°C in the presence or absence of 1 mM GSH. After 2 h, cells were washed twice with HBSS and irradiated (6000 Rad). Antigen-pulsed stimulator cells (1×104/well) were then added to 5×104 cells per well of the responding TCC. Proliferation was measured after 48 h as described.
All SMX-specific CD4+ TCC were generated from allergic donor UNO as described earlier (Schnyder et al., 1997; von Greyerz et al., 1999). TCC specific for covalently bound SMX were generated from SMX allergic donor RB using the same methods as described for UNO and KG. The cloning procedure resulted in 29 CD4+ TCC of which 24 (82.7%) were specific for SMX and five were specific for SMX-NO.
Determination of TCR downregulation
Cloned T-cells (2.5×104) were added to 5×104 autologous B-LCL and incubated in 0.2 ml of medium in U-bottomed plates in triplicates in the continuous presence of SMX, SMX-NHOH or SMX-NO. To some of the cultures 1 mM GSH was added. The plates were centrifuged for 2 min and incubated at 37°C. At various time points, cells were harvested, washed with PBS, containing 0.5 mM EDTA, and stained for 30 min at 4°C with FITC-labelled anti-CD3 (UCHT-1, Dako, Zug, Switzerland). The mean fluorescence intensity (MFI) of the FITC-fluorescence was measured on an Coulter XL flow cytometer using System II software (Coulter Corp., Hialeah, FL, U.S.A.) and the average MFI of triplicates calculated. Percentage of TCR downregulation was then calculated with the average MFI of TCC conjugated with APC without antigen taken as 100% value.
Statistical analysis
Values are expressed as the mean±standard deviation (s.d.). Since values were often to be non-normally distributed, the Mann-Whitney U-test was used for comparison of the two groups. A P-value below 0.05 was considered to be significant.
Results
Coincubation of reactive SMX metabolites with GSH reduces their cellular toxicity
The effect of GSH on the toxicity of SMX and its metabolites was addressed directly by enumerating trypan blue permissive cells after incubation for 2 h with different concentrations of drug (Figure 2). In agreement with previous studies (Rieder et al., 1995; Naisbitt et al., 1999) SMX-NO showed a far greater direct toxicity than SMX-NHOH, or SMX. B-LCL and TCC were similar in their sensitivity to the cytotoxic properties of the SMX metabolites (data not shown). This toxicity could be abolished by the coincubation with GSH. The reduction of toxicity was dependent on the GSH concentration and was accompanied by a loss in binding to the cell surface of B-LCL (Naisbitt et al., 1999 and data not shown).
Figure 2.
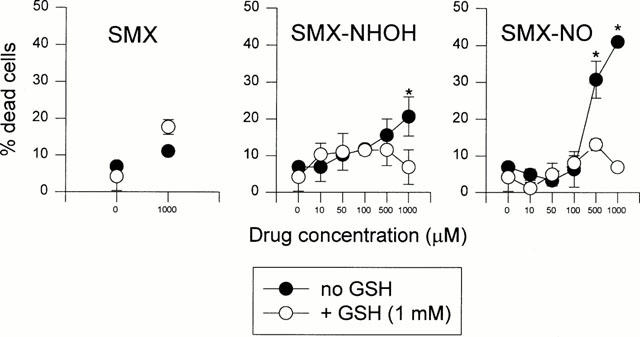
Reduced glutathione diminishes toxicity of SMX metabolites. B-LCL UNO were incubated at 37°C with indicated concentrations of SMX, SMX-NHOH or SMX-NO in the presence or absence of 1 mM GSH. After 2 h, cell viability was determined by trypan blue exclusion. The means of duplicate cultures±s.d. are shown. Statistical analysis was performed by comparing cell death at different concentrations of the compounds in the presence and absence of GSH (*P<0.05).
Effect of GSH of T-cell proliferation in LTT
In order to test the influence of GSH on polyclonal T-cell responses, PBMC from SMX allergic donor UNO and donor KG were incubated with titrated amounts of SMX, SMX-NHOH, or SMX-NO. To some of the cultures reduced glutathione was added. Figure 3 shows the proliferation of the cultures after 5 days of incubation. PBMC from both donors respond in a concentration-dependent way to SMX (Figure 3, left, full circles). The proliferation to SMX metabolites was abolished at cytotoxic concentrations of the compounds. In accordance with previous studies, cells from donor UNO proliferated well to SMX-NO and SMX-NHOH at the non toxic concentration of 125 μM, the response of PBMC from donor KG was barely detectable (Schnyder et al., 2000).
Figure 3.
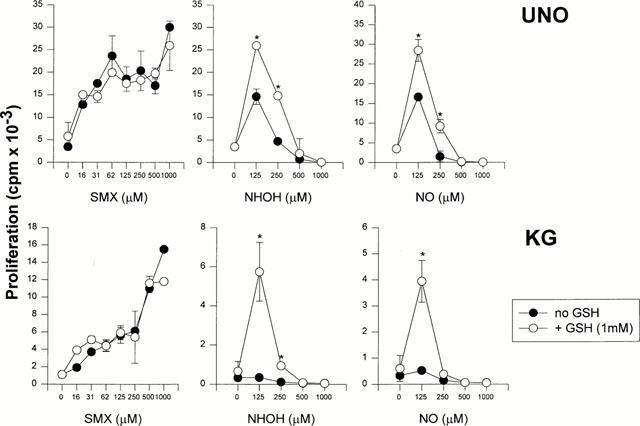
Reduced glutathione enhances proliferative T-cell responses to SMX metabolites. Freshly isolated PBMC from two allergic donors UNO (top row) and KG (bottom row) were incubated for 5 days in a standard LTT assay as described in Methods. Cells were either cultured in the continuous presence of titrated amounts of SMX, SMX-NHOH or SMX-NO. GSH was added to the cultures at indicated amounts for the whole time of the assay. Proliferation is shown as means of duplicate cultures±s.d. Statistical analysis was performed by comparing the proliferation at different concentrations of the compounds with GSH to that without added GSH (*P<0.05). Representative results from one out of three experiments are shown.
The presence of GSH in the cultures did not affect their response to SMX. In contrast, GSH significantly enhanced the proliferation of PBMC UNO to both SMX metabolites and induced responses at previously toxic metabolite concentrations (Figure 3, open symbols). Even more dramatic, GSH led to a good proliferation of PBMC KG to 125 μM of reactive SMX metabolites. Lower concentrations of GSH (0.5 and 0.25 mM) were also effective, albeit to a lesser extent, in enhancing the response of PBMC in the presence of SMX metabolites (data not shown).
Influence of GSH on proliferation of TCC specific for covalently bound SMX-NO or non covalently bound SMX
We have previously generated some TCC from the SMX-allergic patient RB that recognized exclusively SMX-NO modified APC. In order to demonstrate the biological effect of the conversion of SMX-NO by GSH, we pulsed autologous B-LCL with SMX-NO in the presence or absence of GSH. The APC were then added to the SMX-NO-specific CD4+ TCC NO28 and NO146. To some cultures GSH was added after the SMX-NO-pulse for the whole time of the assay. After 2 days of culture, T-cell proliferation was determined. Figure 4 shows that the presence of GSH during the SMX-NO pulse prevents the formation of haptenated cell surfaces and thereby leads to the elimination of the relevant antigen for SMX-NO-specific TCC. The addition of GSH after the pulse leads only to a weak (TCC NO28) or no decrease of specific T-cell proliferation.
Figure 4.
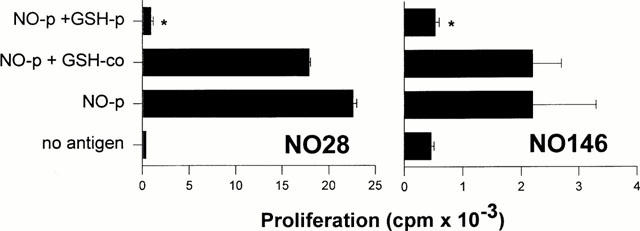
Response of SMX-NO specific TCC to SMX-NO in the presence of GSH. B-LCL were incubated for 2 h with 100 μM SMX-NO in the presence (NO-p+GSH p) or absence (NO-p) of 1 mM GSH. Controls were mock pulsed (no antigen). The APC were added to the CD4+ TCC NO28 and NO146 from allergic donor RB. To some cultures GSH was added after the SMX-NO-pulse for the whole time of the assay. After 2 days of culture, T-cell proliferation was determined as described in Methods. The means of duplicate cultures±s.d. are shown. Statistical analysis compares T-cell proliferation to SMX-NO pulsed APC in the presence of GSH to proliferation in the absence of GSH (*P<0.05).
Conversely, we investigated whether addition of GSH to SMX-NO would lead to the generation of the parent compound SMX. To do this, we used TCC derived from the allergic patient UNO which respond exclusively to non-covalently presented SMX. Figure 5 shows for the two representative TCC 8.15 and 9.5 that autologous B-LCL modified with SMX-NO in the presence or absence of GSH are not recognized by these T-cells. Controls were mock pulsed (no antigen). However, when both SMX-NO and GSH were left in the culture for the whole time of the assay, enough parent compound was generated to induce specific T-cell proliferation.
Figure 5.
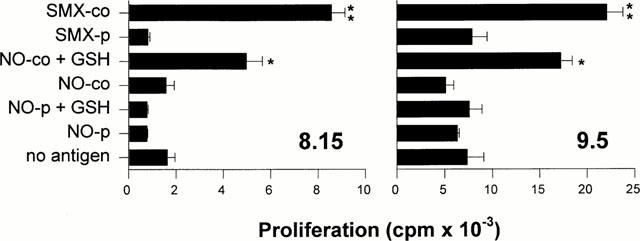
Response of SMX-specific TCC to SMX-NO in the presence of GSH. B-LCL were incubated for 2 h 100 μM SMX-NO in the presence (NO-p+GSH) or absence (NO-p) of 1 mM GSH. Controls were pulsed with 1000 μM SMX (SMX-p) or mock pulsed (no antigen). The APC were added to TCC 8.15 or 9.5. Alternatively, TCC and APC were incubated in the continuous presence of 100 μM SMX-NO (NO-co) or 1000 μM SMX (SMX-co). To some cultures GSH was added for the whole time of the assay (NO-co+GSH). After 2 days of culture, T-cell proliferation was determined as described in Methods. The means of duplicate cultures±s.d. are shown. Statistical analysis was performed by comparing the proliferation to continuously present SMX-NO in the presence and absence of GSH (*P<0.05) and the proliferation to continuously present SMX with SMX-pulsed APC (**P<0.05).
Effects of GSH on antigen dose-dependent proliferation of SMX-specific TCC
Next, we investigated the effect of GSH on the concentration-dependent proliferative response of CD4+ cell clones which react to noncovalently presented SMX and at least partly to its metabolites. None of the clones responds to SMX-NO-modified APC (Schnyder et al., 2000). Figure 6 shows the results for a representative selection from a total of seven clones tested. All clones responded well to SMX (left panel, closed symbols). However, individual T-cell clones proliferated differently well to SMX metabolites (center and right panel, closed symbols). While TCC 9.5 showed only a small response to SMX-NHOH or SMX-NO at 10 μM – a concentration which is usually not associated with cell toxicity – other clones such as TCC 9.3 and S23E also responded well to drug metabolites albeit with a lower proliferation. The addition of GSH had no effect on the SMX-specific proliferation but completely restored the response of clone 9.5 and remarkably enhanced the proliferation of clone S23E to the reduced drug metabolites (=SMX, empty symbols).
Figure 6.
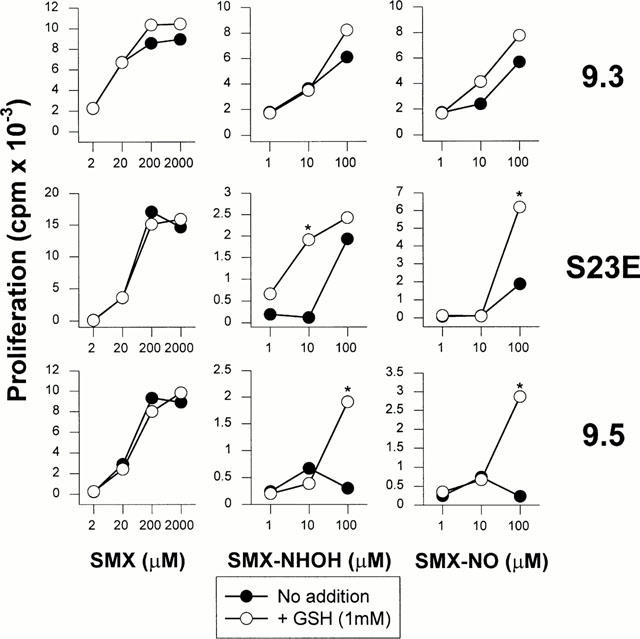
Response of SMX-specific TCC to SMX and SMX metabolites in the presence of GSH. CD4+ TCC from allergic donor UNO were incubated with autologous APC in the continuous presence of titrated amounts of SMX, SMX-NHOH or SMX-NO. GSH (1 mM) was added to some of the cultures for the whole time of the assay. After 2 days of culture, T-cell proliferation was determined as described in Methods. Proliferation is shown as means of duplicate cultures. Representative data are shown for one out of two experiments performed. s.d. was for all values below 5%. Statistical analysis compares T-cell proliferation to SMX and SMX metabolites in the presence of GSH to proliferation in the absence of GSH (*P<0.05).
Effect of GSH on TCR downregulation of TCC
Finally, we addressed the question whether the lack of response of T-cell clone 9.5 to SMX-NOH and SMX-NO was due to antigen toxicity or distinct specificities. Therefore, we tested the recognition of SMX, SMX-NHOH and SMX-NO by the T-cell receptor of clone 9.5. The rapid down-regulation of TCR surface expression was used as a read-out system for specific antigen recognition. As shown in Figure 7, clone 9.5 recognized all three antigens equally well. TCR down-regulation was dependent on the antigen concentration and independent of the presence of GSH.
Figure 7.
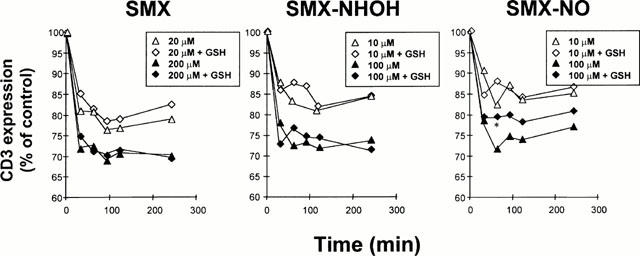
TCR downregulation of SMX-specific TCC in the presence of GSH. The CD4+ TCC 9.5 was incubated in the continuous presence of SMX, SMX-NHOH or SMX-NO. SMX and metabolite were used at indicated concentrations. GSH was added at 1 mM to part of the cultures for the whole time of the assay while controls were left without GSH addition. TCR downregulation was measured at the indicated time points and is given as the percentage of CD3 expression relative to the control values without drugs. s.d. was for all values below 5%. Statistical analysis compares TCR downregulation induced by SMX and SMX metabolites in the presence of GSH to downregulation in the absence of GSH (*P<0.05).
Discussion
Due to their electrophilic nature, the oxidative SMX metabolites SMX-NO and, to a lesser extent, SMX-NHOH are able to covalently modify APCs. The ability of SMX metabolites to bind APCs is concentration-dependent and correlated with their ability to induce cell death. The addition of GSH transforms the reactive metabolite back to the parent compound and thereby prevents binding and reduces toxicity. While in animal models only immune reactivity to the covalently bound drug was observed (Gill et al., 1997), T-cell response in humans has been found to be directed to the non covalently bound parent drug (Schnyder et al., 2000).
If the target structure for T-cells in SMX-allergy was the covalently bound SMX-NO (or SMX-NHOH) the presence of high levels of GSH would be expected to abrogate T-cell recognition. However, we observed that PBMC from allergic individuals responded more vigorously to SMX-NHOH and SMX-NO in the presence of GSH. In some cases, reactivities that were hardly measurable before GSH addition became clearly apparent. An unspecific effect of GSH on T-cell proliferation could be ruled out since GSH had no influence on the response to SMX.
When we analysed TCC generated from drug-allergic patients, clones specific for the covalently bound drug failed to respond to their antigen in the presence of GSH. Importantly, GSH has to be present during the haptenation of APC by SMX-NO. When GSH was added after haptenation it was not able to reverse the process of covalent modification.
On the other hand and as expected, SMX-specific T-cell clones did not respond to SMX-NO modified APC. The presence of GSH during the SMX-NO pulse did not lead to T-cell proliferation as the parent compound, SMX, generated during that time was washed away after the pulse. However, when SMX-NO and GSH were coincubated with TCC and APC a clear proliferative response was induced. This indicates that GSH coincubated with SMX-NO generates sufficient amounts of parent compound to activate SMX-specific T-cells. On the other hand, GSH in coincubation is not able to fully prevent covalent modification and thus abrogate the response of SMX-NO-specific TCC.
We had previously seen that SMX-specific TCC can crossreact with non-covalently bound SMX-NHOH and SMX-NO (Schnyder et al., 2000). When we analysed the response of TCC to SMX-metabolites in detail, we found that SMX-specific T-cell clones showed differential responses to coincubated SMX-NHOH or SMX-NO (Figure 6). Some clones responded well to both SMX-NO and SMX-NHOH, others responded better to SMX-NHOH than SMX-NO and another group hardly proliferated to any metabolite. These differences could reflect a range of susceptibilities to the toxic effects of SMX-NO or SMX-NHOH. TCC clones like 9.3 or S23E may for example have elevated levels of intracellular GSH. However, all clones showed comparable amounts of intracellular GSH (data not shown). Alternatively, the different responses to SMX-NHOH and SMX-NO may reflect distinct specificities of T-cell clones. Clones S23E or 9.3 would then recognize soluble SMX as well as soluble metabolites, while clone 9.5 is specific only for SMX. Both possibilities are not mutually exclusive. When we assessed the early activation events, namely TCR downregulation of TCC 9.5 upon antigen stimulation it was evident that the clone could respond to the SMX metabolites as well as to the parent compound itself. Thus, differential proliferative responses of T-cells do not necessarily correspond to distinct crossreactivity patterns. The most relevant finding in the analysis of TCC was, however, that in the presence of GSH all TCC tested showed a considerable increase or recovery of their proliferation. Considering the results of TCR downregulation after both SMX and SMX metabolite stimulation, TCC 9.5 presents an example of a T-cell susceptible to SMX-NO-mediated toxicity which can be rescued by detoxification via GSH.
Taken together, our results demonstrate the importance of labile, non-covalent drug presentation in drug hypersensitivity. Furthermore, it is tempting to speculate that under certain circumstances protection from oxidative stress could be detrimental in cases where non reactive drugs and their non reactive metabolites contribute to the generation and/or reactivation of drug-specific T-cells and thereby promote allergic reactions to drugs.
Acknowledgments
We are grateful for the generosity of blood donors UNO, KG and RB. We thank Dr B. Lauterburg and E. Junker, Clinical Pharmacology, Inselspital Bern, for determination of intracellular GSH. In addition, we thank Dr R. Torricelli for helpful cooperation and I. Strasser for excellent technical assistance. This work was supported by grant No. 97.0431 of the Federal Office of Education and Science (EU-Research Program BIOMED) and a grant of the International Program of Animal Alternatives 1998 by Procter & Gamble (both to W.J. Pichler). D.J. Naisbitt holds a Wellcome Trust Research Career Development Fellowship and K.B. Park is a Wellcome Principal Fellow.
Abbreviations
- APC
antigen presenting cell
- B-LCL
B-lymphoblastoid cell line
- LTT
Lymphocyte transformation test
- PBMC
Peripheral blood mononuclear cells
- SMX
Sulfamethoxazole
- SMX-NHOH
SMX-hydroxylamine
- SMX-NO
Nitroso-SMX
- TCC
T-cell clone
References
- CRIBB A.E., MILLER M., LEEDER J.S., HILL J., SPIELBERG S.P. Reactions of the nitroso and hydroxylamine metabolites of sulfamethoxazole with reduced glutathione. Drug Metab. Dispos. 1991;19:900–906. [PubMed] [Google Scholar]
- CRIBB A.E., MILLER M., TESORO A., SPIELBERG S.P. Peroxidase-dependent oxidation of sulfonamides by monocytes and neutrophils from humans and dogs. Mol. Pharmacol. 1990;38:744–751. [PubMed] [Google Scholar]
- CRIBB A.E., SPIELBERG S.P. Sulfamethoxazole is metabolized to the hydroxylamine in humans. Clin. Pharmacol. Ther. 1992;51:522–526. doi: 10.1038/clpt.1992.57. [DOI] [PubMed] [Google Scholar]
- CRIBB A.E., SPIELBERG S.P., GRIFFIN G.P. N4-hydroxylation of sulfamethoxazole by cytochrome p450 of the cytochrome p4502C subfamily and reduction of sulfamethoxazole hydroxylamine in human and rat liver microsomes. Drug Metab. Dispos. 1995;23:406–414. [PubMed] [Google Scholar]
- GILL H.J., HOUGH S.J., NAISBITT D.J., MAGGS J.L., KITTERINGHAM N.R., PIRMOHAMED M., PARK K.B. The relationship between the disposition an immunogenicity of sulfamethoxazole in the rat. J. Pharmacol. Exp. Ther. 1997;282:795–801. [PubMed] [Google Scholar]
- LANDSTEINER K., JACOBY J. Studies on the sensitization of animals with simple chemical compounds. J. Exp. Med. 1935;61:643–656. doi: 10.1084/jem.61.5.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MEEKINS C.V., SULLIVAN T.J., GRUCHALLA R.S. Immunochemical analysis of sulfonamide drug allergy: Identification of sulfamethoxazole-substituted human serum proteins. J. Allergy Clin. Immunol. 1994;94:1017–1024. doi: 10.1016/0091-6749(94)90120-1. [DOI] [PubMed] [Google Scholar]
- NAISBITT D.J., HOUGH S.J., GILL H.J., PIRMOHAMED M., KITTERINGHAM N.R., PARK B.K. Cellular disposition of sulfamethoxazole and its metabolites: implications for hypersensitivity. Br. J. Pharmacol. 1999;126:1393–1407. doi: 10.1038/sj.bjp.0702453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAISBITT D.J., O'NEILL P.M., PIRMOHAMED M., PARK B.K. Synthesis and reactions of nitroso sulfamethoxazole with biological nucleophiles: Implications for immune mediated toxicity. Bioorg. Med. Chem. Lett. 1996;6:1511–1516. [Google Scholar]
- PICHLER W.J., YAWALKAR N. Pathophysiology of drug-induced exanthems. Int. Arch. Allergy Immunol. 2000;12:166–170. doi: 10.1159/000053750. [DOI] [PubMed] [Google Scholar]
- REILLY T.P., LASH L.H., DOLL M.A., HEIN D.W., WOSTER P.M., SVENSSON C.G. A role for bioactivation and covalent binding within epidermal keratinocytes in sulfonamide-induced cutaneous drug reactions. J. Invest. Dermatol. 2000;114:1164–1173. doi: 10.1046/j.1523-1747.2000.00985.x. [DOI] [PubMed] [Google Scholar]
- RIEDER M.J., KRAUSE R., BIRD I.A. Time-course of toxicity of reactive sulfonamide metabolites. Toxicology. 1995;95:141–146. doi: 10.1016/0300-483x(94)02900-f. [DOI] [PubMed] [Google Scholar]
- SCHNYDER B., BURKHART C., SCHNYDER-FRUTIG K., VON GREYERZ S., NAISBITT D.J., PIRMOHAMED M., PARK K.B., PICHLER W.J. Recognition of sulfamethoxazole and its reactive metabolites by drug-specific CD4+ T-cells from allergic individuals. J. Immunol. 2000;164:6647–6654. doi: 10.4049/jimmunol.164.12.6647. [DOI] [PubMed] [Google Scholar]
- SCHNYDER B., MAURI-HELLWEG D., ZANNI M., BETTENS F., PICHLER W.J. Direct, MHC-dependent presentation of the drug sulfamethoxazole to human αβ T cell clones. J. Clin. Invest. 1997;100:136–141. doi: 10.1172/JCI119505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHNYDER B., ZANNI M.P., PICHLER W.J. Drug-induced hypersensitivity syndrome. A review and presentation of two cases. Schweiz. Med. Wochenschr. 1997;127:355–359. [PubMed] [Google Scholar]
- VON GREYERZ S., ZANNI M.P., FRUTIG K., SCHNYDER B., BURKHART C., PICHLER W.J. Interaction of sulfonamide derivatives with the TCR of sulfamethoxazole-specific human αβ+T cell clones. J. Immunol. 1999;162:595–602. [PubMed] [Google Scholar]
- VREE T.B., HEKSTER Y.A. Clinical pharmacokinetics of sulfonamides and their metabolites. Antibiot. Chemother. 1987;37:1–214. [PubMed] [Google Scholar]
- ZANNI M.P., VON GREYERZ S., SCHNYDER B., BRANDER K.A., FRUTIG K., HARI Y., VALITUTTI S., PICHLER W.J. HLA-restricted, processing- and metabolism-independent pathway of drug recognition by human alpha beta T lymphocytes. J. Clin. Invest. 1998a;102:1591–1598. doi: 10.1172/JCI3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZANNI M.P., VON GREYERZ S., SCHNYDER B., WENDLAND T., PICHLER W.J. Allele-unrestricted presentation of lidocaine by HLA-DR molecules to specific αβ+T cell clones. Int. Immunol. 1998b;10:507–515. doi: 10.1093/intimm/10.4.507. [DOI] [PubMed] [Google Scholar]