

Abstract
This study was directed at exploring the structure-activity relationship for anandamide and certain of its analogues at the rat VR1 receptor in transfected cells and at investigating the relative extent to which anandamide interacts with CB1 and vanilloid receptors in the mouse vas deferens.
pKi values for displacement of [3H]-resiniferatoxin from membranes of rVR1 transfected CHO cells were significantly less for anandamide (5.78) than for its structural analogues N-(4-hydroxyphenyl)-arachidonylamide (AM404; 6.18) and N-(3-methoxy-4-hydroxy)benzyl-arachidonylamide (arvanil; 6.77).
pEC50 values for stimulating 45Ca2+ uptake into rVR1 transfected CHO cells were significantly less for anandamide (5.80) than for AM404 (6.32) or arvanil (9.29). Arvanil was also significantly more potent than capsaicin (pEC50=7.37), a compound with the same substituted benzyl polar head group as arvanil.
In the mouse vas deferens, resiniferatoxin was 218 times more potent than capsaicin as an inhibitor of electrically-evoked contractions. Both drugs were antagonized to a similar extent by capsazepine (pKB=6.93 and 7.18 respectively) but were not antagonized by SR141716A (1 μM). Anandamide was less susceptible than capsaicin to antagonism by capsazepine (pKB=6.02) and less susceptible to antagonism by SR141716A (pKB=8.66) than methanandamide (pKB=9.56). WIN55212 was antagonized by SR141716A (pKB=9.02) but not by capsazepine (10 μM).
In conclusion, anandamide and certain of its analogues have affinity and efficacy at the rat VR1 receptor. In the mouse vas deferens, which seems to express vanilloid and CB1 receptors, both receptor types appear to contribute to anandamide-induced inhibition of evoked contractions.
Keywords: Anandamide, vanilloid, cannabinoid, VR1 transfected cells, structure-activity, mouse vas deferens, AM404, arvanil, capsaicin, RTX
Introduction
There is mounting evidence that the endogenous cannabinoid, anandamide (Figure 1), can serve as an agonist at the vanilloid VR1 receptor as well as at cannabinoid receptors (Pertwee, 1999; 2000; 2001; Zygmunt et al., 1999; Smart et al., 2000). Thus, this fatty acid amide has been shown to mediate vasodilation that is inhibited by the vanilloid receptor antagonist, capsazepine, but not by the CB1-selective antagonist, SR141716A, and patch-clamp experiments in transfected cells have demonstrated that anandamide is an agonist at both rat and human VR1 receptors (Zygmunt et al., 1999; Smart et al., 2000). Certain analogues of anandamide have also been reported to activate vanilloid as well as cannabinoid receptors and experiments with these compounds suggest that the structural requirements for interaction with VR1 and CB1 receptors are quite different (Melck et al., 1999; Zygmunt et al., 1999; Smart et al., 2000). For example, it has been shown that methanandamide (Figure 1), which is more potent than anandamide as a CB1 receptor agonist (Pertwee, 1999; 2000), is considerably less potent than anandamide as a VR1 receptor agonist (Zygmunt et al., 1999; Smart et al., 2000). In addition, there is evidence that the anandamide uptake inhibitor, AM404 (Figure 1), activates rat VR1 receptors at concentrations lower than those at which it binds to CB1 receptors (Pertwee, 2000; 2001; Jerman et al., 2000; Zygmunt et al., 2000). On the other hand, non-eicosanoid CB1 receptor agonists such as CP55940 and (+)-WIN55212 do not appear to activate vanilloid VR1 receptors (Zygmunt et al., 1999; Smart et al., 2000).
Figure 1.
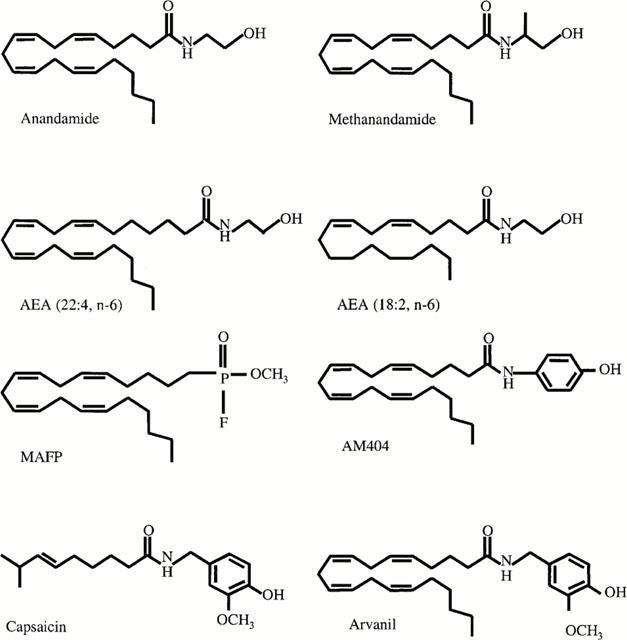
Structures of anandamide (arachidonyl ethanolamide), methanandamide, AEA (22 : 4, n-6) (docosatetraenyl ethanolamide), AEA (18 : 2, n-6) (linoleyl ethanolamide), MAFP (methyl arachidonyl fluorophosphonate), AM404 (4-hydroxyphenyl arachidonyl ethanolamide), capsaicin (3-methoxy-4-hydroxy benzyl-8-methyl-6-nonenamide) and arvanil (3-methoxy-4-hydroxy benzyl arachidonyl amide).
In this study we have used both radioligand binding and 45Ca2+ uptake assays to investigate the structural features of anandamide and of certain of its analogues that determine affinity and/or potency at rat VR1 receptors stably transfected into CHO cells. Included in this study are the endogenous ethanolamides: anandamide (22 : 4, n-6) (AEA 22 : 4); palmitylethanolamide (PEA), and mead acid ethanolamide (Martin et al., 1999; Priller et al., 1995). In addition we have investigated: methanandamide; anandamide (18 : 2, n-6) (AEA 18 : 2) (Martin et al., 1999); the anandamide transport inhibitor, AM404; the hybrid vanilloid/cannabinoid activator, ‘arvanil' (Melck et al., 1999) and the irreversible CB1 receptor antagonist, MAFP (Fernando & Pertwee, 1997).
We have also investigated the relative extent to which anandamide interacts with CB1 and vanilloid receptors in a tissue that appears to express both these receptor types naturally. This tissue is the mouse vas deferens, a preparation in which anandamide and other CB1 receptor agonists serve as potent inhibitors of electrically-evoked contractions (Pertwee, 1997; Rinaldi-Carmona et al., 1994). Results from previous experiments suggest that anandamide can act through pre-junctional CB1 receptors to produce its inhibitory effects on the mouse vas deferens (Rinaldi-Carmona et al., 1994). In the present investigation we have obtained evidence that this tissue expresses vanilloid receptors that can mediate inhibition of electrically-evoked contractions and that the inhibitory effect of anandamide on contractions of the mouse vas deferens is mediated by vanilloid receptors as well as by CB1 receptors. The extent to which vanilloid receptors contribute to the inhibitory effects on evoked contractions of the mouse vas deferens of two anandamide analogues, methanandamide and arvanil (Figure 1), and of the non-eicosanoid cannabinoid receptor agonist, (+)-WIN55212 (Pertwee, 1999), has also been investigated.
Methods
Drugs and chemicals
Anandamide (arachidonyl ethanolamide), AEA (22 : 4, n-6) (docosatetraenyl ethanolamide), AEA (18 : 2, n-6) (linoleyl ethanolamide), MAFP (methyl arachidonyl fluorophosphonate) and Mead acid ethanolamide, (eicosa-5Z,8Z,11Z-trienylethanolamide) were obtained from Biomol research laboratories. Arvanil (3-methoxy-4-hydroxy benzyl arachidonyl amide) was obtained from Alexis. AM404 (4-hydroxyphenyl arachidonyl ethanolamide), capsaicin, capsazepine, methanandamide, PEA, (palmitylethanolamide), resiniferatoxin (RTX) and (+)-WIN55212 ((R)-(+)-(2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo-(1,2,3-de)-1,4-benzoxazin-6-yl)-1-naphthalenyl-methanone mesylate) were obtained from Tocris and SR141716A (N-(piperidin-1-yl)-5-(4-chlorophenyl )-1-( 2,4-dichlorophenyl )- 4-methyl -1H- pyrazole-3-carboxamide hydrochloride) from Sanofi Recherche and from the National Institute on Drug Abuse. Bovine serum albumin (BSA), cell culture media, non-enzymatic cell dissociation solution, G418, l-glutamine, Krebs salts, penicillin/streptomycin, phenylmethylsulphonyl fluoride (PMSF), Tris buffer and Triton X-100 were all obtained from Sigma-Aldrich. [3H]-RTX (50 Ci mmol−1) and 45Ca2+ (5 – 50 mCi mg−1 calcium) were obtained from Amersham Pharmacia Biotech (U.K.). Rat VR1 transfected CHO cells were a gift from Novartis, London.
Cell culture
rVR1 transfected CHO cells were maintained in MEM Alpha minus media containing 2 mM L-glutamine supplemented with 10% hyclone fetal bovine serum, 350 μg ml−1 G418, 100 units ml−1 penicillin and 100 μg ml−1 streptomycin. Cells were maintained in 5% CO2 at 37°C and passed twice a week using non-enzymatic cell dissociation solution. For the radioligand binding assay, cells were removed from flasks by scraping and then frozen as a pellet at −20°C for up to one month. For 45Ca2+ uptake assay, cells were plated in 24-well plates at 5×105 cells ml−1.
Radioligand binding experiments
Assays were performed in DMEM containing HEPES (25 mM) and BSA (0.25 mg ml−1). The total assay volume was 500 μl containing 20 μg of cell membranes. Binding was initiated by the addition of [3H]-RTX. Assays were carried out at 37°C for 1 h, before termination by addition of ice-cold wash buffer (50 mM Tris buffer, 1 mg ml−1 BSA, pH 7.4) and vacuum filtration using a 12-well sampling manifold (Brandel cell harvester) and Whatman GF/B filters that had been soaked in wash buffer at 4°C for at least 24 h. Each reaction was washed nine times with a 1.5 ml aliquot of wash buffer. The filters were oven-dried for 60 min and then placed in 5 ml of scintillation fluid. Radioactivity was quantified by liquid scintillation spectrometry. Specific binding was determined in the presence of 1 μM unlabelled RTX. Protein assays were performed using a Bio-Rad Dc Kit. In some experiments cell membranes were incubated for 15 min at 37°C with phenylmethylsulphonyl fluoride (PMSF) (50 or 200 μM) prior to the addition of cannabinoids. Unlabelled compounds were added in a volume of 50 μl after serial dilution using assay buffer from a 10 mM stock in ethanol (cannabinoids) or dimethylsulphoxide (DMSO) (RTX, capsaicin and capsazepine). [3H]-RTX was also added in a 50 μl volume following dilution in assay buffer. The concentration of [3H]-RTX used in displacement assays was 100 pM. Using this method we obtained specific binding of [3H]-RTX of 80.64±1.76% (n=21). Thus we did not think it necessary to include α1-acid glycoprotein (Szallasi et al., 1999) to reduce non-specific binding. For saturation experiments, concentrations of [3H]-RTX ranging from 0.01 to 1 nM were used. The KD value (0.117 nM) and Bmax (1223 fmol mg−1) for [3H]-RTX and the concentrations of competing ligands to produce 50% displacement of the radioligand (IC50) from specific binding sites were calculated using GraphPad Prism (GraphPad Software, San Diego). Dissociation constant (Ki) values were calculated using the equation of Cheng & Prusoff (1973).
45Ca2+ uptake experiments
Cells in 24-well plates were incubated for 20 min at 37°C with 100 μl of test compound and 1 μCi 45Ca2+ in a total volume of 1 ml of assay buffer (MEM containing 0.25 mg ml−1 BSA). All the concentrations of drug contained the same concentration of 0.1% vehicle, which was either ethanol (cannabinoids) or DMSO (RTX, capsaicin). A basal stimulation was measured in the presence of the appropriate vehicle equivalent. For some experiments the cells were incubated with PMSF (100 μM) for 15 min at 37°C prior to the addition of compounds. Following incubation, plates were placed on ice and washed three times with ice-cold assay buffer. Cells were incubated with assay buffer containing 0.5% Triton X-100 at 37°C for 20 min, before a 200 μl aliquot was removed for scintillation counting. The counts per minute above basal for each data point were expressed as a percentage of the response to 1 nM RTX and the concentration to produce 50% of maximum stimulation (EC50) of 45Ca2+ uptake was calculated using GraphPad Prism (GraphPad Software, San Diego).
Mouse vas deferens
Vasa deferentia were obtained from albino MF1 mice weighing 31 to 50 g. Each tissue was mounted in a 4 ml organ bath at an initial tension of 0.5 g. The baths contained Mg2+-free Krebs solution which was kept at 34°C and bubbled with 95% O2 and 5% CO2. The composition of the Krebs solution was (mM): NaCl 118.2, KCl 4.75, KH2PO4 1.19, NaHCO3 25.0, glucose 11.0 and CaCl2. 6H2O 2.54. Isometric contractions were evoked by stimulation with 0.5 s trains of three pulses of 110% maximal voltage (train frequency 0.1 Hz; pulse duration 0.5 ms) through a platinum electrode attached to the upper end and a stainless steel electrode attached to the lower end of each bath. Stimuli were generated by a Grass S48 stimulator, then amplified (Med-Lab channel attenuator) and divided to yield separate outputs to four organ baths (Med-Lab StimuSplitter). Contractions were monitored by computer (Apple Macintosh LCIII and Performa 475) using a data recording and analysis system (MacLab) that was linked via preamplifiers (Macbridge) to either UF1 transducers (Pioden Controls) or Model 1030 transducers (UFI, CA, U.S.A.). After placement in an organ bath, each tissue was subjected to a stimulation-free period of 10 min and then stimulated for 10 min. Tissues were then subjected to alternate periods of stimulation (5 min) and rest (10 min) until consistent twitch amplitudes were obtained. This equilibration procedure was followed by a stimulation-free period of 25 min. Tissues were then stimulated for 5 min after which the first agonist addition was made and stimulation continued until the end of the experiment. All agonist additions were made cumulatively without washout.
The onset of action of capsaicin was rapid, full development of responses to all effective concentrations used in this investigation occurring within 3 min. Inhibitory responses to capsaicin faded during continued exposure to capsaicin (data not shown). Consequently, in order to minimise the effects of desensitization, no more than three additions of capsaicin were made to any one tissue, later additions being made immediately after a full response to the previous addition had occurred (within 3 min). The same protocol was adopted for RTX. Additions of (+)-WIN55212, anandamide, methanandamide and arvanil were made at slightly greater intervals (5 min) as these agonists had a slower onset of action than capsaicin or RTX. Again, no more than three additions of agonist were made to any one tissue. In some experiments, capsazepine, SR141716A or DMSO was added 30 min before the first addition of an agonist. PMSF was sometimes also added at this time. This was added in all experiments with anandamide, methanandamide and arvanil because our cultured cell experiments with these agonists had also been conducted in the presence of PMSF. The dose of PMSF used yielded an organ bath concentration (20 μM) expected to protect anandamide from enzymic hydrolysis (Pertwee et al., 1995). All drugs were dissolved in DMSO. Drug additions were made in a volume of 10 μl. In control experiments, DMSO was added instead of agonist or antagonist.
Analysis of data
Values have been expressed as means and variability as s.e.mean or as 95% confidence limits. Mean values have been compared using Student's unpaired t-test or analysis of variance followed by Dunnett's test or the Newman-Keuls test. A P value <0.05 was considered to be significant. For in vitro experiments, values for EC50, IC50 and maximal effects (Emax) and the s.e.mean or 95% confidence limits of these values have been calculated by non-linear regression analysis using the equation for a sigmoid concentration-response curve (GraphPad Prism). In organ bath experiments, the degree of drug-induced inhibition of evoked contractions has been expressed in percentage terms. This was calculated by comparing the amplitude of the twitch response after each addition of a twitch inhibitor with its amplitude immediately before the first addition of the inhibitor. KB values of capsazepine or SR141716A for antagonism of agonists were calculated by substituting a single concentration ratio value into the equation (x−1)=B/KB, where x (the ‘concentration ratio') is the concentration of agonist that produced a particular size of effect in the presence of antagonist at a concentration, B, divided by the concentration of agonist that produced an identical effect in the absence of antagonist (Tallarida et al., 1979).
For data obtained in organ bath experiments, values of the concentration ratio and its 95% confidence limits were determined by symmetrical (2+2) dose parallel line assays (Colquhoun, 1971). This method was also used to determine the relative potency of RTX and capsaicin in the vas deferens and to establish whether 2-point log concentration-response plots deviated significantly from parallelism.
Results
Radioligand binding studies
Studies with PMSF
In the presence of the vehicle equivalent of PMSF, a concentration of 10 μM of anandamide only displaced 17.8±3.9% (n=4) of specific [3H]-RTX binding. However, in the presence of 50 and 200 μM PMSF, anandamide displaced the radioligand completely, with pKi values of 5.58±0.11 (n=4) and 5.78±0.06 (n=4) respectively (Figure 2a). The anandamide uptake inhibitor, AM404 had a significantly higher pKi value (Table 1) than anandamide in the presence of 200 μM PMSF (P<0.01, ANOVA followed by Dunnett's multiple comparison test). PMSF also enhances the affinity measurement for this compound. In the presence of the vehicle equivalent of PMSF the pKi value for AM404 (5.79±0.07, n=4) was significantly lower (P<0.01, unpaired t-test) than in PMSF treated cells (Figure 2b). The specific binding of the vanilloid agonist, capsaicin, was unaffected by PMSF, its pKi value being 5.70±0.18 in the presence of vehicle and 5.65±0.10 in the presence of the enzyme inhibitor (n=4).
Figure 2.
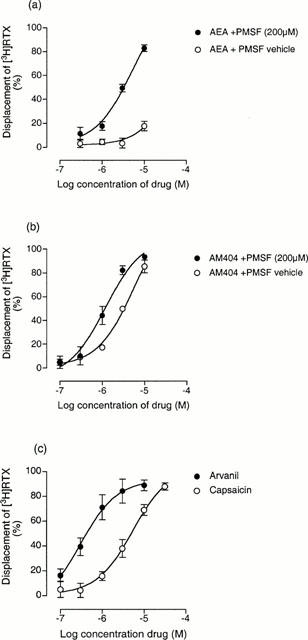
Displacement of [3H]-RTX (100 pM) by (a) anandamide (AEA) in the presence and absence of PMSF (b) AM404 in the presence and absence of PMSF and (c) arvanil and capsaicin in the presence of PMSF (200 μM). Each symbol represents the mean percent displacement±the s.e.mean (n=4 – 6).
Table 1.
Interaction of anandamide and analogues with vanilloid receptors in CHO cells transfected with rVR1 receptors
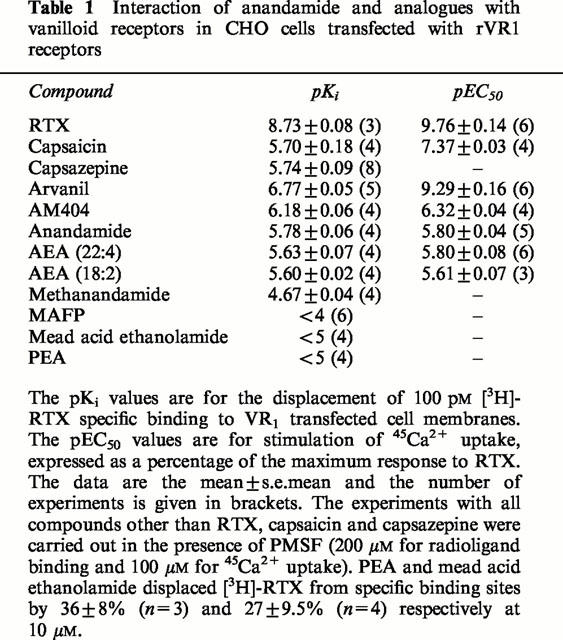
Competition studies with anandamide analogues
Having established the importance of the inclusion of PMSF, all the following studies were performed following pre-incubation of the cell membranes with 200 μM PMSF. The endogenous anandamide analogue AEA (22 : 4) and AEA (18 : 2) had pKi values of 5.63±0.07 and 5.60±0.02 respectively (Figure 1; Table 1) which are not significantly different (P>0.05, ANOVA followed by Newman-Keuls) from the value obtained for anandamide under the same conditions. The highest affinity anandamide analogue studied was the ‘hybrid' vanilloid/cannabinoid, arvanil with a pKi value of 6.77±0.05 (Figures 1 and 2c, Table 1). This value was significantly higher than the pKi value of either of the parent molecules, capsaicin and anandamide (P<0.001, ANOVA followed by Newman-Keuls). The anandamide analogue, methanandamide had a pKi value of 4.67±0.04 (n=4) which was significantly higher than that of anandamide (P<0.01, ANOVA followed by Newman-Keuls). The compound methyl arachidonyl fluorophosphonate (MAFP) (Figure 1), which is known to be a potent inhibitor of PLA2 and fatty acid amide hydrolase (FAAH) as well as a CB1 receptor antagonist (Pertwee, 1997) displaced specific [3H]-RTX binding by 16.9±4.74% (n=4) at a concentration of 100 μM. The putative endogenous cannabinoid ligands, palmitylethanolamide (PEA) and mead acid ethanolamide also displaced [3H]-RTX from specific binding sites, giving a maximum displacement of 36±8% (n=3) and 27±9.5% (n=4) respectively at 10 μM, as compared with 84±3.8% for anandamide at this concentration.
45Ca2+ uptake studies
As shown for the radioligand binding assay, the presence of PMSF was found to alter the potency of anandamide. In the absence of the enzyme inhibitor, a concentration of 10 μM anandamide only produced 14.2±3.43% (n=4) stimulation of 45Ca2+ uptake. In the presence of 100 μM PMSF the pEC50 value of anandamide was 5.80±0.04 (n=5). This was significantly greater (P<0.001, unpaired t-test; n=4) than the value of 5.00±0.05 obtained in the presence of 20 μM PMSF (Figure 3a). Consequently, all uptake experiments with anandamide analogues were carried out in the presence of 100 μM PMSF. The pEC50 values for stimulation of 45Ca2+ uptake by the endogenous anandamide analogues AEA (22 : 4) and AEA (18 : 2) were 5.80±0.08 and 5.61±0.07 respectively (Figure 3b). These were not significantly different from the pEC50 of anandamide (P>0.05, ANOVA followed by Dunnett's test). The anandamide uptake inhibitor AM404 had a pKi value of 6.32±0.04 in the presence of 100 μM PMSF (n=4), which was significantly higher than that of anandamide (P<0.01, ANOVA followed by Dunnett's test) (Figure 3b). The most potent compound in the stimulation of 45Ca2+ uptake was the hybrid vanilloid/cannabinoid arvanil, with a pEC50 value of 9.29±0.16 (n=6). This value was significantly higher than that of either anandamide or capsaicin (P<0.001, ANOVA followed by Newman-Keuls) (Figure 3c). An investigation of the VR1 desensitising characteristics of these compounds was not the purpose of this study. However, at concentrations above 10 nM, the concentration-response curve for calcium influx by arvanil appeared to reverse indicating desensitisation of the VR1 receptor (data not shown). None of the compounds tested produced significant 45Ca2+ uptake in untransfected CHO cells at a concentration of 10 μM (n=3; data not shown).
Figure 3.
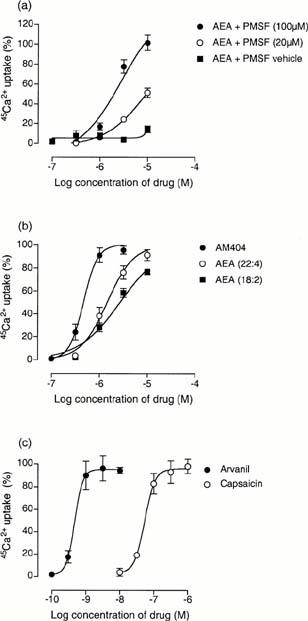
The stimulation of 45Ca2+ uptake by (a) anandamide (AEA) in the presence and absence of PMSF (b) AM404, AEA (22 : 4) and AEA (18 : 2) in the presence of 100 μM PMSF and (c) arvanil and capsaicin in the presence of 100 μM PMSF. The per cent stimulation is calculated from an RTX maximum for each experiment. Each symbol represents the mean per cent stimulation±the s.e.mean (n=4 – 6).
Mouse vas deferens
Effects of RTX and capsaicin on electrically-evoked contractions of the mouse vas deferens
Capsaicin was found to inhibit electrically-evoked contractions of the vas deferens in a concentration-related manner (Figure 4). Electrically-evoked contractions of the vas deferens were also inhibited by RTX. Mean inhibitory responses to 1 and 100 pM RTX were 16.4±4.0 and 42.3±4.9% respectively (n=6). Corresponding inhibitory responses to 0.1 and 10 nM capsaicin were 23.7±5.3 and 63.1±5.8% respectively (n=6). The relative positions of the pair of 2-point log concentration-response plots constructed from these data indicate the mean relative potency of RTX and capsaicin to be 218.3 and the 95% confidence limits of this value to be 52.4 and 728.4 (n=6). The slopes of these two plots were not significantly different. At 10 μM, the vanilloid receptor antagonist, capsazepine, readily antagonized both capsaicin and RTX (Figure 4 and Table 2). Capsazepine was equally effective against both agonists. Thus the parallel dextral shifts in 2-point log concentration-response plots of capsaicin and RTX that this antagonist produced were not significantly different from each other (Table 2). Neither capsaicin nor RTX was antagonized by 1 μM SR141716A (Table 3).
Figure 4.
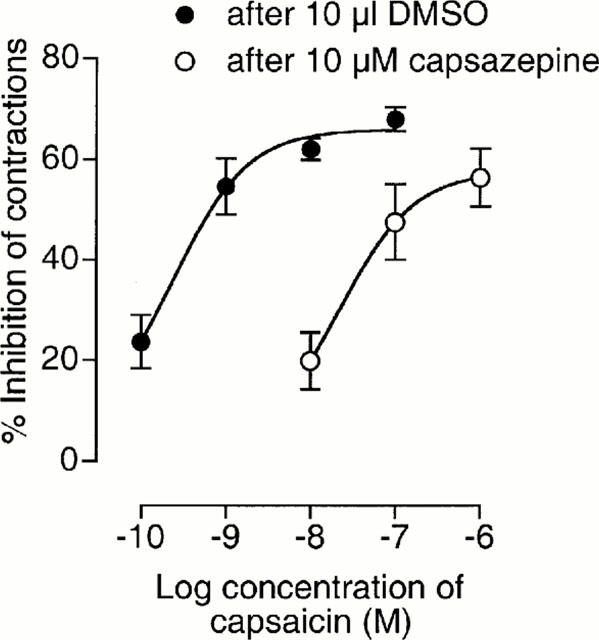
Mean concentration-response curves for capsaicin constructed in the presence of capsazepine or DMSO. Each symbol represents the mean value±s.e.mean for inhibition of electrically-evoked contractions of the mouse isolated vas deferens expressed as a percentage of the amplitude of the twitch response measured immediately before the first addition of capsaicin to the organ bath (n=6). Capsazepine and DMSO (10 μl) were added 30 min before the first addition of capsaicin. To minimize the effects of desensitization, responses to no more than two concentrations of capsaicin were measured in any one tissue. When 10 μl DMSO was added instead of capsaicin to tissues pretreated with 10 μl DMSO, mean twitch amplitude was not significantly affected by the first addition of DMSO and decreased by 12.4±3.9% after the second DMSO addition (n=6). pEC50 and Emax values of capsaicin were 9.61±0.36 and 66.0±3.2% respectively in the presence of DMSO and 7.64±0.78 and 57.6±7.6% respectively in the presence of capsazepine (GraphPad Prism).
Table 2.
Effect of 10 μM capsazepine on inhibition of electrically-evoked contractions of the mouse vas deferens induced by certain cannabinoid and vanilloid receptor agonists
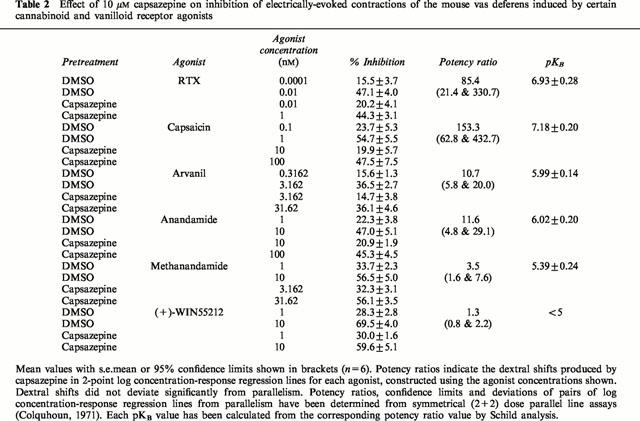
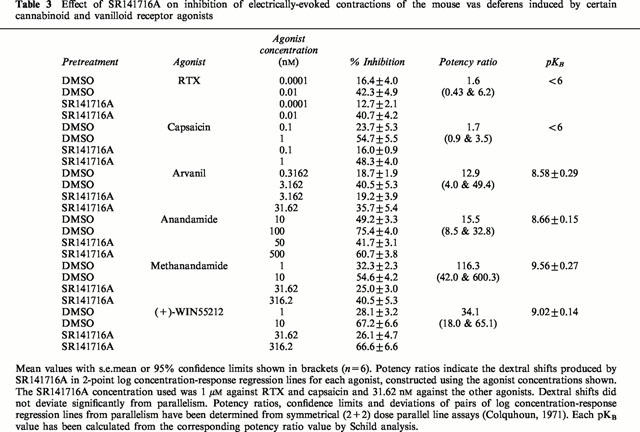
Table 3.
Effect of SR141716A on inhibition of electrically-evoked contractions of the mouse vas deferens induced by certain cannabinoid and vanilloid receptor agonists
Effects of capsazepine and SR141716A on inhibition of electrically-evoked contractions of the mouse vas deferens induced by anandamide, methanandamide, (+)-WIN55212 and arvanil
SR141716A (31.62 nM) produced significant parallel dextral shifts in 2-point log concentration-response plots of anandamide, methanandamide and (+)-WIN55212 (Table 3). The dextral shift in the log concentration-response plot of methanandamide was significantly greater than the dextral shift produced by SR141716A in the log concentration-response plot of anandamide (Table 3). At 10 μM, capsazepine induced dextral shifts in 2-point log concentration-response plots of anandamide and methanandamide that were not significantly different from each other but were significantly less than the dextral shift induced by 10 μM capsazepine in the log concentration-response plot of capsaicin (Table 2). (+)-WIN55212 was not antagonized by 10 μM capsazepine (Table 2). Arvanil was also found to inhibit electrically-evoked contractions of the vas deferens. It showed the same susceptibility to antagonism by 31.62 nM SR141716A and 10 μM capsazepine as anandamide (Tables 2 and 3).
Discussion
This study measures for the first time the affinity of the endogenous cannabinoid, anandamide, for rat vanilloid VR1 receptors using a radioligand binding assay in rVR1 transfected CHO cells. It thus provides further evidence for the interaction of this compound with the rat vanilloid VR1 receptor, confirming the findings of Zygmunt et al. (1999) at native rat VR1 receptors. As has been previously demonstrated for CB1 receptor assays (Pertwee et al., 1995; Pertwee, 1997), the inclusion of the FAAH inhibitor, PMSF, was found to significantly enhance the affinity of anandamide. In the presence of 200 μM PMSF, the Ki value of anandamide at rVR1 receptors is 1.66 μM. This value is considerably higher than the Ki value for this compound at the CB1 receptor, which has been shown to be between 37 and 116 nM (Pertwee, 1997). However, it would appear that the affinity of anandamide for rVR1 receptors is similar to that of the established vanilloid receptor agonist, capsaicin (Ki=1.98 μM). We found that the endogenous cannabinoid AEA (22 : 4), in which the arachidonyl chain of anandamide has been lengthened by two carbon atoms (Figure 1), had similar affinity (Ki=2.35 μM) for the rVR1 receptor as anandamide. It has been demonstrated that the structural difference between AEA (22 : 4) and anandamide does not significantly affect binding affinity for the CB1 receptor (Martin et al., 1999). Shortening the arachidonyl chain and reducing the number of double bonds to two as in the anandamide analogue, AEA (18 : 2) also has little impact on affinity for rVR1 receptors (Ki=2.50 μM). This is in contrast to the structure-activity relationship at the CB1 receptor where three or four double bonds have been shown to be optimal for binding and AEA (18 : 2) has been shown to have a significantly lower CB1 affinity than anandamide (Martin et al., 1999).
This investigation also involved the study of anandamide analogues with an altered ethanolamido head group. We found that the anandamide uptake inhibitor AM404, which contains a 4-hydroxyphenyl head group, had a Ki value for the rVR1 receptor of 661 nM. Thus, the affinity is 4 fold higher than the affinity of capsaicin for this receptor. The highest affinity analogue investigated was arvanil, a ‘hybrid' activator of cannabinoid and vanilloid receptors (Melck et al., 1999). The affinity of this compound for the rVR1 receptor (Ki=170 nM) was 12 times higher than that of capsaicin. This compound contains the same 3-methoxy-4-hydroxybenzyl head group as capsaicin and has been shown to both inhibit anandamide uptake and bind to the CB1 receptor (Ki=250 – 520 nM) (Melck et al., 1999).
In line with previous studies demonstrating that methanandamide is less potent than anandamide at the VR1 receptor, we have found that the Ki value of this fatty acid amide (11 μM) is significantly higher at these receptors than that of anandamide (Zygmunt et al., 1999; Smart et al., 2000). The fluorophosphonate substitution found in MAFP appears to have greatly reduced the affinity for VR1 receptors. This is in contrast to the affinity of this compound for the CB1 receptor, which is higher than that of anandamide (Pertwee, 1997). The endogenous ethanolamide compounds, PEA and mead acid ethanolamide, appear to have affinity for the rVR1 receptor, although lower than that of anandamide.
In these studies anandamide, AEA (22 : 4) and AEA (18 : 2) appear to be full agonists in functional 45Ca2+ uptake studies with CHO cells stably transfected with rVR1 receptors (EC50 values: 1.57, 1.58 and 2.49 μM respectively). These findings compare with the data of Smart et al. (2000). Using HEK239 cells stably transfected with the hVR1 receptors and a FLIPR-based calcium assay, they obtained an EC50 value of 1.15 μM for anandamide. There are, however, some methodological differences that are worth noting. Smart et al. (2000) carried out their experiments at 25°C in the absence of PMSF. We used a 45Ca2+ uptake assay at 37°C, and found that the potency of anandamide was markedly attenuated in the absence of PMSF. There is emerging evidence that capsaicin binds to the cytosolic domain of the receptor and that an endogenous capsaicin-like substance may be present in the cell (Jung et al., 1999). Thus it is tempting to suggest that the PMSF is particularly important in the VR1 receptor assay carried out at 37°C, due to the rapid inactivation of anandamide by endogenous intracellular FAAH (Pertwee, 1997). In assays carried out at lower temperatures (e.g. Smart et al., 2000) the level of hydrolysis of anandamide may be reduced, rendering inclusion of PMSF unnecessary. In addition, because of inter-tissue differences in the activity of FAAH (Pertwee, 1997), it may not always be necessary to include PMSF.
It has recently been demonstrated that lipoxygenase products of arachidonic acid are vanilloid receptor agonists (Hwang et al., 2000). In the absence of PMSF, tissues that contain FAAH will hydrolyse anandamide to ethanolamide and arachidonic acid. Thus, it may be postulated that anandamide can activate the vanilloid receptor indirectly via arachidonic acid metabolites. However, the data presented here suggest that, under our experimental conditions, the action of anandamide at the VR1 receptor is a direct action, in that the compound is more potent when breakdown is inhibited by the inclusion of the FAAH inhibitor, PMSF. The importance of the inhibition of FAAH is supported by our finding that even the potency of AM404, a relatively poor substrate for the enzyme (Lang et al., 1999), was significantly enhanced by the inclusion of PMSF, although to a lesser extent than that of anandamide. In the presence of PMSF the EC50 of AM404 reported in this study (476 nM) compares with that of 109 nM obtained at 25°C in experiments with rVR1 transfected HEK239 cells using a FLIPR-based assay (Jerman et al., 2000).
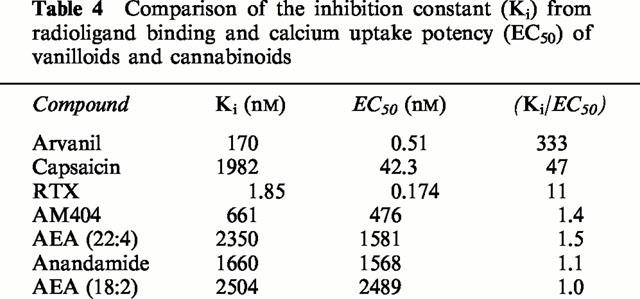
Agonist potency is a function of both affinity and efficacy, such that it depends on the quotient intrinsic efficacy/disassociation constant (Kenakin, 1997). The ratio of Ki (the inhibition constant) to EC50 value is indicative of the relative intrinsic efficacy of a compound. The Ki /EC50 ratios from this study (Table 4) would suggest that anandamide, AEA (22 : 4) and AEA (18 : 2) are relatively low affinity/low efficacy VR1 agonists. AM404 appears to have higher affinity but again displays relatively low efficacy. This is in contrast to the profile of capsaicin, which has the characteristics of a low affinity/high efficacy compound. In arvanil the arachidonyl side chain of anandamide has been added to the capsaicin ring system, a change that appears to have produced an increase in both affinity and efficacy at the VR1 receptor. It is notable that all calcium uptake studies to date that have been performed with cell lines stably transfected with VR1 receptors, including the present investigation, suggest that anandamide and indeed AM404 are full agonists at this receptor (Zygmunt et al., 1999; Smart et al., 2000; Jerman et al., 2000). However, the possibility remains that in native systems with lower expression levels of VR1 receptors these compounds may behave as partial agonists. Indeed, in electrophysiological studies of dorsal root ganglion neurones, which express native vanilloid receptors, anandamide has already been shown to produce an inward current with a peak amplitude significantly smaller (<50%) than that of capsaicin (Zygmunt et al., 1999; Smart et al., 2000). Moreover, the maximal contractile effect of anandamide on the guinea-pig bronchus has been reported to be markedly less than that of capsaicin (Spina et al., 2000). As this effect of anandamide was absent in capsaicin-desensitised tissues, these data lend further support to the hypothesis that anandamide has lower efficacy than capsaicin at vanilloid receptors. However, it is notable that none of the above studies included PMSF raising the possibility that the apparent low efficacy of anandamide may be due to its rapid intracellular hydrolysis.
Table 4.
Comparison of the inhibition constant (Ki) from radioligand binding and calcium uptake potency (EC50) of vanilloids and cannabinoids
Our experiments with the mouse vas deferens were performed in order to investigate the possibility that the inhibitory effect of anandamide on evoked contractions of this preparation is mediated by vanilloid receptors as well as by CB1 receptors. As a first step, we looked for evidence that, as already seems likely for the rat vas deferens (Maggi et al., 1993), the mouse vas deferens contains vanilloid receptors that can mediate inhibition of electrically-evoked contractions. The results we obtained support the presence of such receptors. First, it was found that capsaicin and RTX are both potent inhibitors of evoked contractions of the mouse vas deferens. Second, RTX was 218 times more potent than capsaicin, a relative potency value similar to that obtained by us in VR1 transfected cells (Table 1) and to previously published relative potency data for these two agonists (Szallasi & Blumberg, 1999). Third, the inhibitory effects of capsaicin and RTX on evoked contractions of the vas deferens were markedly attenuated by capsazepine. Moreover, the KB values of capsazepine for antagonism of these agonists (66 and 119 nM respectively; Table 2) are not significantly different from each other. They also approximate to KB values of capsazepine against capsaicin and RTX (107 and 148 nM respectively) that have been determined in experiments with cultured dorsal root ganglion neurones in which the measured response was 86Rb+ efflux (Bevan et al., 1992).
Capsaicin and RTX do not seem to act on CB1 receptors in the mouse vas deferens as neither compound was antagonized by SR141716A, even at a concentration of 1 μM that did significantly attenuate the inhibitory effects of established cannabinoid receptor agonists in this tissue preparation (Table 3). In contrast, the inhibitory effect of the cannabinoid receptor agonist, (+)-WIN55212, on evoked contractions seems to be mediated by CB1 receptors but not by vanilloid receptors. Thus (+)-WIN55212 was readily antagonized by SR141716A but was not antagonized by capsazepine at a concentration (10 μM) that was effective against capsaicin and RTX (Table 2).
The ability of anandamide to inhibit evoked contractions of the mouse vas deferens was attenuated by SR141716A, confirming the results of Rinaldi-Carmona et al., (1994). However, anandamide was also antagonized by capsazepine, supporting the hypothesis that both CB1 and VR1 receptors contribute to the inhibitory effect of anandamide on the mouse vas deferens. SR141716A was significantly more potent against the CB1-selective agonist, methanandamide, than against anandamide (Table 3), whilst capsazepine was significantly more potent against capsaicin than against anandamide (Table 2). This finding differs from that of previous investigations (Zygmunt et al., 1999; Smart et al., 2000), in which both capsaicin and anandamide have been shown to be equally susceptible to antagonism by capsazepine. These differences presumably reflect the fact that, in contrast to the blood vessels and dorsal root ganglion neurones used in other investigations (Zygmunt et al., 1999; Smart et al., 2000), the vas deferens may contain both CB1 and VR1 receptors which mediate inhibition of the same measured response (electrically-evoked contractions). It is also notable that, in the vas deferens, anandamide appears to be eliciting VR1 receptor mediated effects at concentrations lower than those required for interaction of anandamide with the VR1 receptor in the calcium uptake and radioligand binding assay. We are investigating the hypothesis that in this tissue, CB1 and VR1 receptors may synergise when activated by anandamide. An alternative explanation for this discrepancy would be a species difference between the rat and mouse. CB1 and VR1 receptors may also both contribute to the inhibitory effect on evoked contractions of arvanil as its susceptibility to antagonism by SR141716A and capsazepine in the vas deferens is similar to that of anandamide. Further experiments are required to establish whether anandamide and arvanil-induced inhibition of evoked contractions of the mouse vas deferens is mediated solely by CB1 and VR1 receptors.
As to methanandamide, the KB value of SR141716A against this fatty acid amide (0.27 nM; Table 3) is significantly lower than that obtained against anandamide. This suggests that the inhibitory effect of methanandamide on the vas deferens is mediated mainly by CB1 receptors. Even so, a minor component of its inhibitory effect does seem to depend on an interaction with vanilloid receptors in this tissue as methanandamide was marginally but significantly antagonized by capsazepine (Table 2). The apparent KB value of capsazepine against methanandamide (4.05 μM) was 4.4 times greater than its apparent KB value against anandamide (0.92 μM) (Table 2). However, this difference was not statistically significant.
It is known that capsaicin causes a rapid desensitization of vanilloid receptors (Szallasi & Blumberg, 1999). This may well also be true of anandamide and arvanil in the mouse vas deferens. Consequently, the possibility that these compounds induce vanilloid receptor desensitization is the subject of ongoing investigations.
At present there is much debate about the relative concentrations at which anandamide activates CB1 and VR1 receptors, and hence about the physiological relevance of its interaction with both these receptor types (see Szolcsanyi, 2000; Smart & Jerman, 2000). The data from transfected cells presented here and by others (Zygmunt et al., 1999; Smart et al., 2000) suggest that the endogenous cannabinoid has about 20 – 50 fold lower affinity for and potency at VR1 receptors than that previously demonstrated for this compound at the CB1 receptor (Pertwee, 1997). However, it is notable that in the mouse vas deferens preparation, in which CB1 and VR1 receptors seem to be capable of mediating inhibition of electrically-evoked contractions, anandamide appears to interact with both these receptors types in the same concentration range. Thus responses to the same nanomolar concentration of anandamide can be attenuated by the CB1 receptor antagonist, SR141716A, and by the VR1 receptor antagonist, capsazepine. As discussed earlier this may reflect a synergism or ‘cross-talk' between these two receptor systems and this is now the subject of ongoing investigations.
Acknowledgments
This work was supported by grant 047980 from the Wellcome Trust (to R.A. Ross and R.G. Pertwee), by grant DA09789 (to R.G. Pertwee) and by Pfizer (to R.G. Pertwee and R.A. Ross). We thank Dr Iain James of Novartis for the gift of the rVR1 transfected cells and Sanofi Recherche and the National Institute on Drug Abuse for SR141716A.
Abbreviations
- AEA (22 : 4)
docosatetraenyl ethanolamide, n-6
- AEA (18 : 2)
linoleyl ethanolamide, n-6
- AM404
4-hydroxyphenyl arachidonyl ethanolamide
- Anandamide
arachidonyl ethanolamide
- Arvanil
3-methoxy-4-hydroxy benzyl arachidonyl amide
- Capsaicin
3-methoxy-4-hydroxy benzyl-8-methyl-6-nonenamide
- FAAH
fatty acid amide hydrolase
- MAFP
methyl arachidonyl fluorophosphonate
- Mead acid ethanolamide
eicosa-5Z,8Z,11Z-trienylethanolamide
- PEA
palmitylethanolamide
- PMSF
phenylmethylsulphonyl fluoride
- RTX
resiniferatoxin
- VR1
vanilloid receptor
References
- BEVAN S., HOTHI S., HUGHES G., JAMES I.F., RANG H.P., SHAH K., WALPOLE C.S.J., YEATS J.C. Capsazepine–a competitive antagonist of the sensory neuron excitant capsaicin. Br. J. Pharmacol. 1992;107:544–552. doi: 10.1111/j.1476-5381.1992.tb12781.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHENG Y.-C., PRUSOFF W.H. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (IC50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- COLQUHOUN D. Lectures on Biostatistics. Oxford: Oxford University Press; 1971. [Google Scholar]
- FERNANDO S.R., PERTWEE R.G. Evidence that methyl arachidonyl fluorophosphonate is an irreversible cannabinoid receptor antagonist. Br. J. Pharmacol. 1997;121:1716–1720. doi: 10.1038/sj.bjp.0701303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HWANG S.W., HAWOON C., KWAK J., LEE S.-Y., KANG C.-J., JUNG J., CHO S., MIN K.H., SUH Y.-G., KIM G., OH U. Direct activation of capsaicin receptors by products of lipoxygenase: Endogenous capsaicin-like substances. Proc. Natl. Acad. Sci. U.S.A. 2000;97:6155–6160. doi: 10.1073/pnas.97.11.6155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JERMAN J.C., BROUGH S.J., DAVIS J.B., MIDDLEMISS D.N., SMART D. The anandamide transport inhibitor AM404 is an agonist at the rat vanilloid receptor (VR1) Br. J. Pharmacol. 2000;129 Suppl.:73P. [Google Scholar]
- JUNG J., HWANG S.W., KWAK J., LEE S.Y., KANG C.J., KIM W.B., KIM D., OH U. Capsaicin binds to the intracellular domain of the capsaicin-activated ion channel. J. Neurosci. 1999;19:529–538. doi: 10.1523/JNEUROSCI.19-02-00529.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KENAKIN T. Pharmacological Analysis of Drug Receptor Interaction 1997Raven Press: New York, USA; p.2712nd edn [Google Scholar]
- LANG W.S., QIN C., LIN S.Y., KHANOLKAR A.D., GOUTOPOULOS A., FAN P.S., ABOUZID K., MENG Z.X., BIEGEL D., MAKRIYANNIS A. Substrate specificity and stereoselectivity of rat brain microsomal anandamide amidohydrolase. J. Med. Chem. 1999;42:896–902. doi: 10.1021/jm980461j. [DOI] [PubMed] [Google Scholar]
- MAGGI C.A., BEVAN S., WALPOLE C.S.J., RANG H.P., GIULIANI S. A comparison of capsazepine and ruthenium red as capsaicin antagonists in the rat isolated urinary bladder and vas deferens. Br. J. Pharmacol. 1993;108:801–805. doi: 10.1111/j.1476-5381.1993.tb12881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARTIN B.R., MECHOULAM R., RAZDAN R.K. Discovery and characterization of endogenous cannabinoids. Life Sci. 1999;65:573–595. doi: 10.1016/s0024-3205(99)00281-7. [DOI] [PubMed] [Google Scholar]
- MELCK D., BISOGNO T., DE PETROCELLIS L., CHUANG H.H., JULIUS D., BIFULCO M., DI MARZO V. Unsaturated long-chain N-acyl-vanillyl-amides (N-AVAMs): vanilloid receptor ligands that inhibit anandamide-facilitated transport and bind to CB1 cannabinoid receptors. Biochem. Biophys. Res. Comm. 1999;262:275–284. doi: 10.1006/bbrc.1999.1105. [DOI] [PubMed] [Google Scholar]
- PERTWEE R.G. Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol. Ther. 1997;74:129–180. doi: 10.1016/s0163-7258(97)82001-3. [DOI] [PubMed] [Google Scholar]
- PERTWEE R.G. Pharmacology of cannabinoid receptor ligands. Curr. Med. Chem. 1999;6:635–664. [PubMed] [Google Scholar]
- PERTWEE R.G. Cannabinoid receptor ligands: clinical and neuropharmacological considerations, relevant to future drug discovery and development. Exp. Opin. Invest. Drugs. 2000;9:1553–1571. doi: 10.1517/13543784.9.7.1553. [DOI] [PubMed] [Google Scholar]
- PERTWEE R.G.Cannabinoid receptors and pain Prog. Neurobiol 2001. in press [DOI] [PubMed]
- PERTWEE R.G., FERNANDO S.R., GRIFFIN G., ABADJI V., MAKRIYANNIS A. Effect of phenylmethylsulphonyl fluoride on the potency of anandamide as an inhibitor of electrically evoked contractions in two isolated tissue preparations. Eur. J. Pharmacol. 1995;272:73–78. doi: 10.1016/0014-2999(94)00618-h. [DOI] [PubMed] [Google Scholar]
- PRILLER J., BRILEY E.M., MANSOURI J., DEVANE W.A., MACKIE K., FELDER C.C. Mead ethanolamide, a novel eicosanoid, is an agonist for the central (CB1) and peripheral (CB2) cannabinoid receptors. Mol. Pharmacol. 1995;48:288–292. [PubMed] [Google Scholar]
- RINALDI-CARMONA M., BARTH F., HÉAULME M., SHIRE D., CALANDRA B., CONGY C., MARTINEZ S., MARUANI J., NÉLIAT G., CAPUT D., FERRARA P., SOUBRIÉ P., BRELIÉRE J.C., LE FUR G. SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Letts. 1994;350:240–244. doi: 10.1016/0014-5793(94)00773-x. [DOI] [PubMed] [Google Scholar]
- SMART D., GUNTHORPE M.J., JERMAN J.C., NASIR S., GRAY J., MUIR A.I., CHAMBERS J.K., RANDALL A.D., DAVIS J.B. The endogenous lipid anandamide is a full agonist at the human vanilloid receptor (hVR1) Br. J. Pharmacol. 2000;129:227–230. doi: 10.1038/sj.bjp.0703050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SMART D., JERMAN J.C. Anandamide: an endogenous activator of the vanilloid receptor. Trends Pharmacol. Sci. 2000;21:134. doi: 10.1016/s0165-6147(00)01459-0. [DOI] [PubMed] [Google Scholar]
- SPINA D., TUCKER R.C., PAGE C.P. Effect of the putative vanilloid receptor agonist, anandamide on the baseline tone in the guinea-pig bronchus. Proc. Aust. Soc. Clin. Exp. Pharmacol. Toxicol. 2000;7:60. [Google Scholar]
- SZALLASI A., BLUMBERG P.M. Vanilloid (capsaicin) receptors and mechanisms. Pharmacol. Rev. 1999;51:159–211. [PubMed] [Google Scholar]
- SZOLCSANYI J. Anandamide and the question of its functional role for activation of capsaicin receptors. Trends Pharmacol. Sci. 2000;21:203–204. doi: 10.1016/s0165-6147(00)01484-x. [DOI] [PubMed] [Google Scholar]
- TALLARIDA R.J., COWAN A., ADLER M.W. pA2 and receptor differentiation: a statistical analysis of competitive antagonism. Life Sci. 1979;25:637–654. doi: 10.1016/0024-3205(79)90505-8. [DOI] [PubMed] [Google Scholar]
- ZYGMUNT P.M., CHUANG H.-H., MOVAHED P., JULIUS D., HÖGESTÄTT E.D. The anandamide transport inhibitor AM404 activates vanilloid receptors. Eur. J. Pharmacol. 2000;396:39–42. doi: 10.1016/s0014-2999(00)00207-7. [DOI] [PubMed] [Google Scholar]
- ZYGMUNT P.M., PETERSSON J., ANDERSSON D.A., CHUANG H., SØRGÅRD M., DI MARZO V., JULIUS D., HÖGESTÄTT E.D. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature. 1999;400:452–457. doi: 10.1038/22761. [DOI] [PubMed] [Google Scholar]