

Abstract
Bradykinin (BK) effect on the [Ca2+]i response to 1 nM angiotensin II was examined in muscular juxtamedullary efferent arterioles (EA) of rat kidney.
BK (10 nM) applied during the angiotensin II-stimulated [Ca2+]i increase, induced a [Ca2+]i drop (73±2%). This drop was prevented by de-endothelialization and suppressed by HOE 140, a B2 receptor antagonist. It was neither affected by L-NAME or indomethacin, nor mimicked by sodium nitroprusside, 8-bromo-cyclic GMP or PGI2. The BK effect did not occur when the [Ca2+]i increase was caused by 100 mM KCl-induced membrane depolarization and was abolished by 0.1 μM charybdotoxin, a K+ channel blocker.
Although proadifen prevented the BK-caused [Ca2+]i fall, more selective cytochrome P450 inhibitors, 17-octadecynoic acid (50 μM) and 7-ethoxyresorufin (10 μM) were without effect.
Increasing extracellular potassium from 5 to 15 mM during angiotensin II stimulation caused a [Ca2+]i decrease (26±4%) smaller than BK which was charybdotoxin-insensitive. Inhibition of inward rectifying K+ channels by 30 μM BaCl2 and/or of Na+/K+ ATPase by 1 mM ouabain abolished the [Ca2+]i decrease elicited by potassium but not by BK.
A voltage-operated calcium channel blocker, nifedipine (1 μM) did not prevent the BK effect but reduced the [Ca2+]i drop.
These results indicate that the BK-induced [Ca2+]i decrease in angiotensin II-stimulated muscular EA is mediated by an EDHF which activates charybdotoxin-sensitive K+ channels. In these vessels, EDHF seems to be neither a cytochrome P450-derived arachidonic acid metabolite nor K+ itself. The closure of voltage-operated calcium channels is not the only cellular mechanism involved in this EDHF-mediated [Ca2+]i decrease.
Keywords: Glomerular efferent arterioles, [Ca2+]i, bradykinin, EDHF, K+ channels, potassium, cytochrome P450, voltage-operated calcium channels
Introduction
Several lines of evidence indicate that renal microcirculation is controlled by endocrine, autocrine and paracrine mechanisms which interact to regulate vascular tone. For examples, angiotensin II is able to increase intrarenal bradykinin (BK) level which, in turn, counteracts the vasoconstrictor effect of angiotensin II (Siragy et al., 1996). Similarly, as demonstrated by Nobes et al., 1991, the effect of angiotensin II to enhance papillary blood flow appear to be due to angiotensin II-stimulated production of kinins in the renal medulla. Moreover, the observation that a B2-kinin receptor antagonist reduces basal papillary blood flow without altering the cortical flow (Roman et al., 1988) indicates that kinins affect medullary microcirculation more than cortical microcirculation. It is generally agreed that blood flow to renal medulla is almost exclusively supplied through juxtamedullary efferent arterioles (EA) which divide into several branches to form vasa recta (Pallone et al., 1990). These arterioles called muscular EA (Helou & Marchetti, 1997) may, therefore, be a target for hormonal and neural factors involved in the control of medullary microcirculation and the interplay between angiotensin II and BK effects might be an essential process in this control.
The vasoconstrictor responses to angiotensin II are generally thought to be mediated by an increase in intracellular calcium ([Ca2+]i). Vascular smooth muscle relaxation is supposed to result mainly from a [Ca2+]i decrease although a reduction in the calcium sensitivity of the contractile apparatus may also be involved (Bolz et al., 1999). We have previously reported first that angiotensin II elevates [Ca2+]i in the muscular cells of isolated muscular EA (Helou & Marchetti, 1997) and second, that BK also increases [Ca2+]i in these arterioles (Praddaude et al., 1999). However, the BK-induced increase occurs in endothelium cells, the main target for BK in blood vessels. BK can indirectly cause a relaxation of smooth muscle cells in various blood vessels by releasing several mediators from endothelium (Busse & Fleming, 1996; Mombouli & Vanhoutte, 1995; 1997; Quilley et al., 1997). These mediators include nitric oxide, prostacyclin and endothelium-derived hyperpolarizing factor(s) (EDHF). The chemical nature of the latter is not yet established. BK acts very likely, on the muscular EA by releasing one or several of these agents.
The present study aimed firstly at investigating the ability of BK to attenuate the [Ca2+]i response to angiotensin II in muscular EA by releasing an endothelium-derived factor, and then to specify the nature of mediator(s) and the cellular mechanisms involved. The results of our experiments indicate that: (a) BK decreases [Ca2+]i by releasing an EDHF which is able to activate charybdotoxin-sensitive K+ channels; (b) this EDHF would be neither a cytochrome P450-derived arachidonic metabolite nor the K+ ion; and (c) the closure of voltage-operated calcium channels (VOCC) is not the only cellular mechanism involved in the [Ca2+]i decrease induced by BK via EDHF.
Methods
Microdissection of muscular juxtamedullary EA
Experiments were conducted on glomerular arterioles isolated from rat kidney by using the previously described protocol (Helou & Marchetti, 1997). Male Sprague-Dawley rats (180 – 240 g) obtained from Iffa-Credo (L'Arbresle, France) were anaesthetized with a single dose of pentobarbital sodium (50 mg kg−1 i.p.). The left kidney was infused through cannulation of the abdominal aorta, with 3 – 5 ml of a standard solution (4°C) containing (in mM): NaCl 127, KCl 5, MgSO4 0.8, Na2HPO4 0.33, KH2PO4 0.44, MgCl2 1, NaHCO3 4, CaCl2 2, D-Glucose 5, CH3CO2Na 10, HEPES (pH 7.4) 20 and 0.1% BSA. The kidney was then infused with 5 ml of the same solution containing 8 mg Collagenase A and immediately removed, decapsulated and longitudinally sliced. Small pyramids were cut out and incubated in the presence of collagenase (1 mg ml−1) for 8 min at 30°C before they were washed with the standard solution to eliminate collagenase. The muscular juxtamedullary EA were microdissected from inner cortex at 4°C under a stereomicroscope (SZ3, Olympus, Tokyo, Japan) and isolated with the glomerulus. They were easily recognized by their thick, muscular and regular wall and by the presence of side branches that made it possible to distinguish them from AA. Before [Ca2+]i measurements, each sample was transferred on a thin glass slide in 1 μl of a standard solution containing 1% agarose (type IX). Agarose was set on by cooling the slide 1 min on ice. Arterioles were then loaded by addition of 1 μl of 10 μM fura 2-AM (acetoxymethyl ester) and incubated for 1 h at room temperature in darkness.
Measurements of [Ca2+]i
In accordance with previous experiments (Marchetti et al., 1995), [Ca2+]i was evaluated by using a Photoscan II microfluorimeter (Photon Technology International, Kontron, France). The glass slide with the sample was fixed at the bottom of a superfusion chamber which was then put on the stage of an inverted fluorescent microscope (Nikon). The sample was continuously superfused with the standard solution (0.8 ml min−1, 37°C) carrying or not the test substances. The microscope was fitted out with a quartz illumination system and a 40 fold magnification fluorescence objective. The sample was alternatively excited at 340 and 380 nm (4 s cycle−1), and the fluorescence emitted at 510 nm from an area defined by an adjustable window (about 25×30 μm) was measured.
At the end of several experiments, autofluorescence was measured at 340 and 380 nm from the selected arteriolar area after quenching the fura 2-AM fluorescence with 1 mM of a solution of MnCl2 in the presence of 10 μM ionomycin. The obtained values were very similar to the background emission from the same measurement window. Background was routinely recorded for both wavelengths at the end of each experiment and subtracted from all the measurements. [Ca2+]i was calculated from the following equation (Grynkiewicz et al., 1985): [[Ca2+]i=KD×∂ (R−Rmin)/(Rmax−R)], where KD (dissociation constant for the fura 2-Ca2+ complex) is 224 nM, R is the ratio of fluorescence emitted for each wavelength (340/380 nm), Rmax is the maximal ratio emitted in the presence of saturating calcium (2 mM), Rmin is the minimal ratio measured in absence of calcium (0 mM), and ∂ is the ratio of fluorescence obtained at 384 nm in absence and in presence of 2 mM calcium. The values of Rmin, Rmax and ∂ were periodically determined by external calibration using a buffer that mimicked intracellular medium (Marchetti et al., 1995).
Arteriolar de-endothelialization
The technique reported by Beierwaltes (1990) was used to de-endothelialize renal microvessels without altering the arteriolar smooth muscle layer. Kidneys were infused in situ with standard buffer to clear them from blood, and then infused with a solution of 10 mM H2O2 for 2 min. After eliminating residual H2O2 by infusing 20 ml standard buffer, kidneys were infused with the collagenase solution and prepared for microdissection. In each experiment, the integrity of smooth muscular layer of de-endothelialized arterioles was checked by comparing their arteriolar [Ca2+]i response to angiotensin II to that of control arterioles. De-endothelialization was confirmed by the absence of [Ca2+]i response to 1 μM acetylcholine, a well established endothelial stimulator (Furchgott & Vanhoutte, 1989).
Experimental protocols
The effect of a maximal dose (10 nM) of BK (Praddaude et al, 1999) was investigated on the maximal [Ca2+]i increase induced by 1 nM angiotensin II (Helou & Marchetti, 1997). BK was added during the plateau phase of the response to angiotensin II, 5 – 8 min after the beginning of angiotensin II application. The drugs used to characterize BK effect were superfused for 5 or 10 min before and during BK application. When 17-ODYA and 7-ethoxyresorufin were tested, additional experiments were performed by preincubating EA with either drug for 40 min. We always checked whether these drugs changed basal [Ca2+]i level when superfused alone.
To induce cell membrane depolarization, the potassium concentration of superfusion medium was increased from 5 to 100 mM by replacing 95 mM of sodium with 95 mM of potassium. The solutions containing 15 mM of potassium were prepared by replacing 10 mM of sodium by 10 mM of potassium.
Drugs
Collagenase A (Clostridium Histolyticum, 1.1 PZ u mg−1) was purchased from Serva (France), Fura 2-AM from Molecular Probe (Leiden The Netherlands), HOE 140 (D Arg0[Hyp3,Thi5,DTic7,Oic8]-BK) from Hoechst (Germany), dimethyl sulphoxide (DMSO), agarose, H2O2, angiotensin II, bradykinin, acetylcholine, Nω-nitro-L-arginine methyl ester (L-NAME), Sodium nitroprusside (SNP), 8-bromo-cyclic GMP, indomethacin, BaCl2, apamin, iberiotoxin, 4-aminopyridine (4-AP), glybenclamide, charybdotoxin, proadifen, 17-octadecynoic acid (17-ODYA), 7-ethoxyresorufin and nifedipine from Sigma (France).
Stock solutions of indomethacin (10 mM) and nifedipine (10 mM) were prepared in absolute ethanol (Merck) while those of Fura 2-AM (10 mM), glybenclamide (10 mM) were made up in DMSO. Stock solutions of all other drugs were prepared in distilled water. In the superfusion solutions, the concentrations of absolute ethanol and DMSO were equal or inferior to 0.1%. The superfusion solutions containing 0.1% ethanol or DMSO modified neither basal [Ca2+]i level nor the [Ca2+]i response to angiotensin II and did not alter BK effect.
Statistics
Results are reported as mean±s.e.mean. When each arteriole was its own control, significance was obtained by paired Student's t-test. Differences between two groups were analysed using unpaired Student's t-test. Values were considered significantly different at P<0.05.
Results
Effect of BK on the angiotensin II-induced [Ca2+]i increase
The magnitude of the plateau phase of the [Ca2+]i response to 1 nM angiotensin II, was of 117±6 nM (n=66). Addition of 10 nM BK caused a rapid drop in [Ca2+]i by 90±7 nM. This drop was reached 1.4±0.1 min after BK application and represented 73±2% of [Ca2+]i increase induced by angiotensin II (Figure 1). In 42 out of 66 muscular EA, the fall in [Ca2+]i was transient and the BK effect lasted 2.1±0.1 min. In these arterioles, [Ca2+]i either returned directly back to a level very similar to that before BK (Figure 1a) or exceeded it briefly before decreasing (Figure 1b). In the remaining 24 arterioles, the BK effect was sustained and the [Ca2+]i response to angiotensin II remained partially inhibited by 60±4% (Figure 1c).
Figure 1.
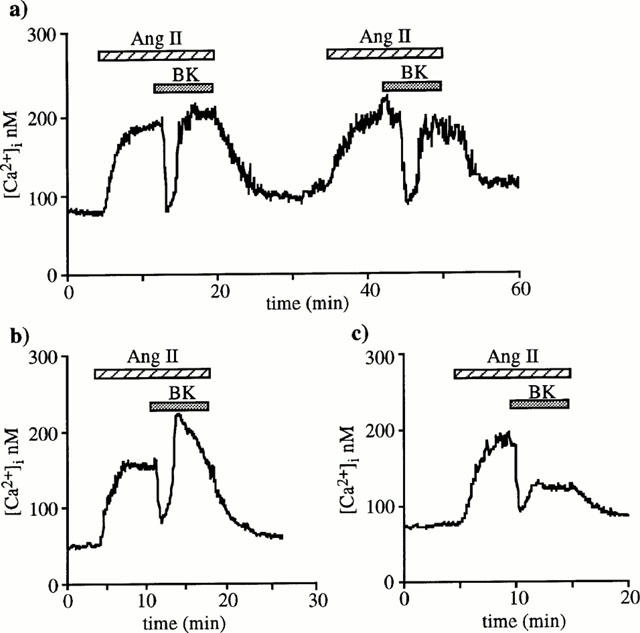
BK effect on [Ca2+]i responses to angiotensin II in muscular EA. Original [Ca2+]i tracings illustrating the different time-courses observed when 10 nM BK was added during the plateau phase of 1 nM angiotensin II-induced [Ca2+]i elevations in 66 different arterioles. After dropping, [Ca2+]i returned directly to plateau in 31 arterioles (a), exceeded plateau level before stabilizing close to prestimulation values in 17 (b) or did not completely reverse in 24 arterioles (c).
Figure 1a also shows that it was possible to reproduce on the same arteriole at least twice the inhibitory effect of BK on angiotensin II-induced increase in [Ca2+]i indicating the absence of desensitization phenomenon.
Characterization of the receptors involved in theBK-induced [Ca2+]i effect
To localize the BK receptors responsible for the [Ca2+]i decrease, BK effect was investigated on de-endothelialized EA unresponsive to acetylcholine. The [Ca2+]i response to angiotensin II was similar but it was no longer altered by BK (Figure 2a). These results indicate that the BK receptors responsible for the [Ca2+]i decrease in intact arterioles are located on the endothelial layer.
Figure 2.
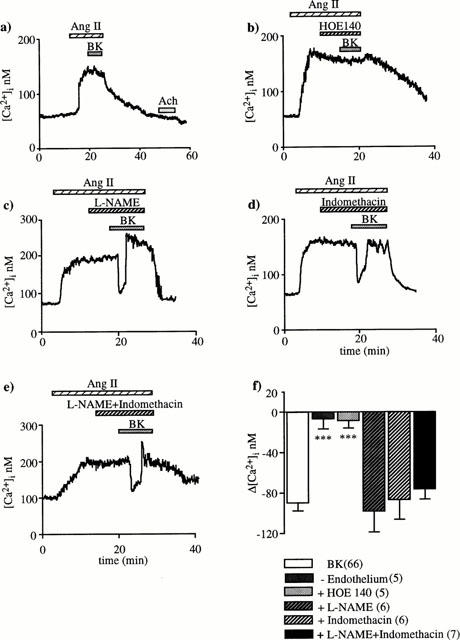
Effects of arteriolar de-endothelialization (a), B2 specific receptor antagonist HOE 140 (b), L-NAME (c), indomethacin (d) and mixture L-NAME plus indomethacin (e) on the BK-induced [Ca2+]i drop. De-endothelialization was confirmed by the absence of [Ca2+]i response to 1 μM acetylcholine (Ach). Either 0.1 μM HOE 140, 10 μM L-NAME or 10 μM indomethacin was applied during the plateau phase of the [Ca2+]i response to 1 nM angiotensin II, 10 min before and during 10 nM BK superfusion. In (f), the columns represent the means±s.e.mean of [Ca2+]i decreases induced by BK under control and experimental conditions; numbers of tested arterioles is in brackets. ***P<0.001: significant difference from BK response (unpaired Student's t-test).
The type of BK receptors involved was examined using a specific B2 receptor antagonist (HOE 140). Superfusion of 0.1 μM HOE 140 prevented the BK-induced fall in [Ca2+]i indicating that the BK receptors are of the B2 type (Figure 2b).
BK-induced [Ca2+]i decrease is independent of NO and PGI2
Neither L-NAME (10 μM) nor indomethacin (10 μM) nor mixture of both altered the BK effect on the [Ca2+]i response to angiotensin II (Figure 2c – e). SNP, 8-bromo-cyclic GMP and PGI2 elicited a decrease in [Ca2+]i when they were applied during the plateau phase of the response to angiotensin II. However, their effects differed from those of BK in that they were always sustained and of much smaller magnitude. The [Ca2+]i decrease caused by 1 μM SNP, 100 μM 8-bromo-cyclic GMP and 10 μM PGI2 was of only 15±3 nM (n=6), 31±6 nM (n=7) and 23±9 nM (n=6), respectively (Figure 3a – c). When these drugs were superfused alone at the beginning of experiments, they did not elicit changes in the [Ca2+]i basal level. Taken together, these results indicate that neither NO nor PGI2 mediate the BK-induced [Ca2+]i fall.
Figure 3.
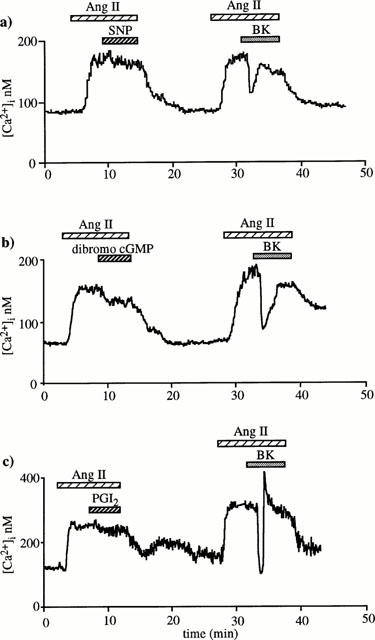
Effects of sodium nitroprusside (SNP), 8-bromo-cyclic GMP and PGI2 on the [Ca2+]i response to angiotensin II. Two successive applications of 1 nM angiotensin II were performed at interval of 15 min on a same muscular EA. Either 1 μM SNP (a) (n=6) or 100 μM 8-bromo-cyclic GMP (b) (n=7) or 10 μM PGI2 (c) (n=6) was applied during the plateau phase of the first response to angiotensin II, whereas 10 nM BK was superfused during the second response.
Effect of BK on 100 mM KCl-induced [Ca2+]i increase
To test the hypothesis that BK response might involve an EDHF, we examined the effect of BK on the [Ca2+]i increase caused by high external K+ (100 mM) susceptible to prevent membrane hyperpolarization. In these experiments, BK did not cause a [Ca2+]i decrease, but a significant enhancement of 45±7 nM (n=6) supporting the hypothesis that BK effect might be due to an hyperpolarizing factor (Figure 4).
Figure 4.
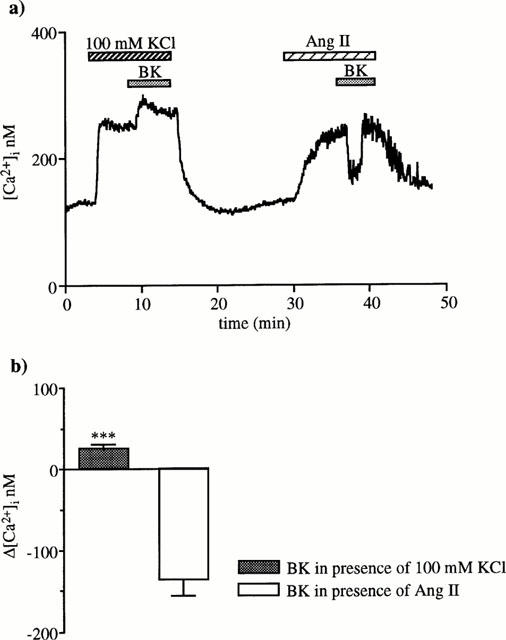
BK effect on the [Ca2+]i increase induced by membrane depolarization provoked by elevating K+ concentration in superfusate to 100 mM. (a) BK (10 nM) was added during the plateau phase of the K+-caused [Ca2+]i increase and, after 15 min washing, on a [Ca2+]i response to 1 nM angiotensin II. The trace is one of six similar experiments. (b) The columns represent [Ca2+]i changes induced by BK (means±s.e.mean) during KCl or angiotensin II (***P<0.001, paired Student's t-test).
Effect of K+ channel blockade on the BK-induced [Ca2+]i drop
To investigate the type of K+ channels that are activated by the hyperpolarizing factor, different blockers were tested. None of used blockers changed the basal [Ca2+]i level. As indicated in Table 1, 1 μM apamin, 0.1 μM iberiotoxin, 10 μM glybenclamide, and 1 mM 4-aminopyridine did not affect the BK response. Only 0.1 μM charybdotoxin prevented the [Ca2+]i fall caused by BK. Under this condition, BK produced a 37±9 nM significant increase (n=6, P<0.01, Figure 5).
Table 1.
Effects of K+ channel inhibitors on BK-induced [Ca2+]i decrease

Figure 5.
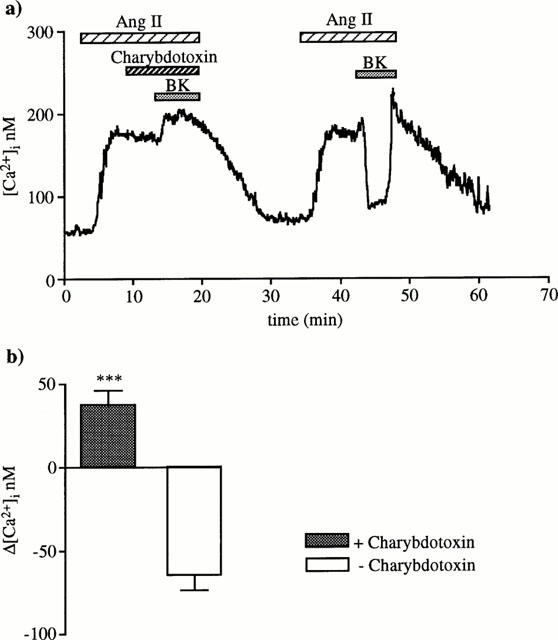
Charybdotoxin effect on BK-induced [Ca2+]i drop. First, 0.1 μM charybdotoxin was added during the plateau phase of the [Ca2+]i response to angiotensin II, 5 min before and during 10 nM BK superfusion. Second, BK effect was checked in the absence of charbdotoxin. This trace is representative of six similar experiments. (b) The columns represent [Ca2+]i changes induced by BK (means±s.e.mean) in the presence or in the absence of charybdotoxin (***P<0.001, paired Student's t-test).
Effects of cytochrome P450 inhibitors on BK-induced [Ca2+]i decrease
The studies indicating that cytochrome P450-derived arachidonic acid metabolites could be EDHF, prompted us to examine the effects of structurally different cytochrome P450 mono-oxygenase inhibitors (proadifen, 17-ODYA and 7-ethoxyresorufin). In the presence of 10 μM proadifen (lower concentrations were inefficient), BK failed to reduce the [Ca2+]i response to angiotensin II but rather caused a 49±10 nM [Ca2+]i increase (n=7, Figure 6a). In contrast, 17-ODYA (50 μM) and 7-ethoxyresorufin (10 μM) did not prevent BK-induced [Ca2+]i decrease (79±17, n=6 and 66±19 nM, n=6; Figure 6b,c). Note that the three compounds did not modify the basal [Ca2+]i, but proadifen produced a small significant decrease (25±3 nM, P<0.01) of the [Ca2+]i response to angiotensin II (Figure 6a).
Figure 6.
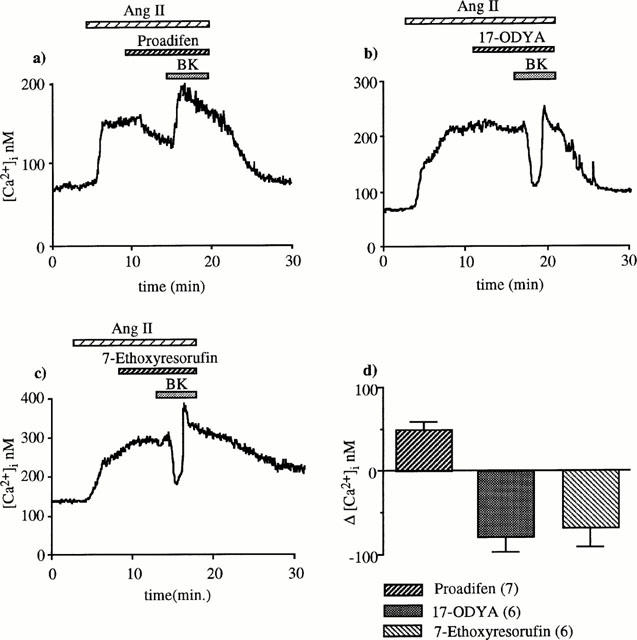
Effect of three structurally different cytochrome P450 inhibitors on BK-induced [Ca2+]i decrease. (a) In the presence of 10 μM proadifen, BK did not reduce [Ca2+]i but caused a significant increase (49±10 nM). (b,c) BK-induced [Ca2+]i drop was not prevented by 17-ODYA (50 μM) or ethoxyresorufin. (d) The columns represent BK-induced [Ca2+]i changes.
Comparison between the effects of low potassium and BK
It has been recently proposed that EDHF could be K+ susceptible to cause hyperpolarization of smooth muscle cells by activating Ba2+-sensitive inward rectifier K+ channels and/or Na+/K+ ATPase, (Edwards et al., 1998). To test this hypothesis, we compared the effects of non-depolarizing low KCl concentrations with those of BK. Trials performed with 10, 15 and 20 mM KCl showed that basal [Ca2+]i level was not altered by 10 and 15 mM but 20 mM KCl caused a slight 14±3 nM increase (n=6, P<0.05). When 10 or 15 mM KCl were applied during the plateau phase of the response to angiotensin II, only 15 mM KCl induced a small, slow and transitory decrease (Figure 7a). It was about one third of the decrease induced by 10−8 M BK (23±3 vs 66±15 nM, n=6, P<0.02) and was not sensitive to 0.1 μM charybdotoxin (Figure 7b).
Figure 7.
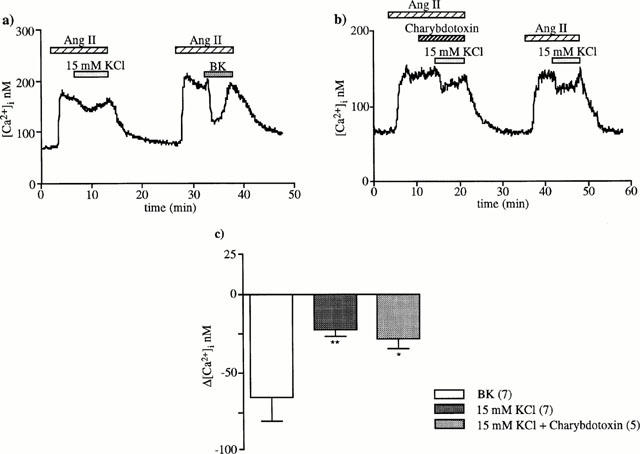
[Ca2+]i decrease in response to 15 mM external K+ and effect of charybdotoxin. Fifteen mM K+ was added during the plateau phase of the [Ca2+]i response to 1 nM angiotensin II (a) without (n=7) and (b) with 0.1 μM charybdotoxin (n=5). (c) The columns represent [Ca2+]i decreases (means±s.e.mean) induced by BK and by 15 mM KCl in the absence or in the presence of charybdotoxin. **P<0.02, *P<0.05 compared to BK-induced [Ca2+]i decrease (unpaired Student's t-test).
When 30 μM BaCl2 (an inward rectifier K+ channel blocker) was applied during the plateau phase of the response to angiotensin II, addition of 15 mM K+ elicited no significant decrease in [Ca2+]i whereas BK induced always a [Ca2+]i drop (Figure 8a,c).
Figure 8.
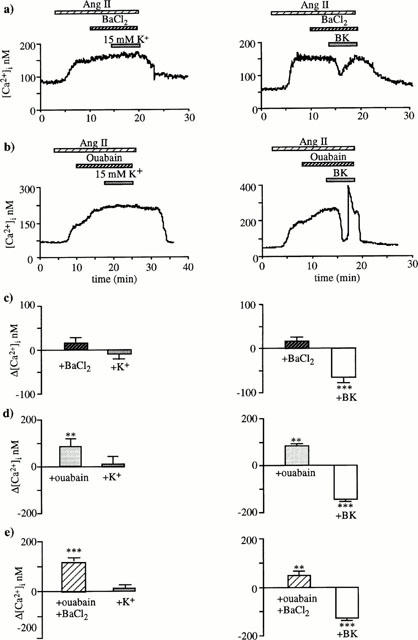
Effects of BaCl2 or/and ouabain on the [Ca2+]i decreases induced by 15 mM KCl or 10 nM BK. (a,b) Illustrate the typical [Ca2+]i tracings obtained when K+ (left) or BK (right) was superfusate either in the presence of 30 μM BaCl2 (a) or 1 mM ouabain (b). These agents were added during the plateau phase of the response to 1 nM angiotensin II, 5 min before KCl or BK. BaCl2 and ouabain prevented [Ca2+]i decrease induced by 15 mM KCl but not that induced by BK (a,b). The magnitudes of [Ca2+]i variations (means±s.e.mean) are shown graphically in (c) and (d), respectively. (e) Shows that combination of ouabain and BaCl2 prevented the [Ca2+]i decrease induced by 15 mM KCl but not by 10 nM BK. For each protocol, 5 – 7 arterioles were tested. Note that addition of ouabain at the plateau of the response to 1 nM angiotensin II increased significantly [Ca2+]i (b,d,e), **P<0.01, ***P<0.001 (paired Student's t-test).
As shown in Figure 8b,d, application of 1 mM ouabain resulted in an abrupt additional rise of [Ca2+]i from 175±8 to 250±7 nM. This increase of 75±10 nM (n=22, P<0.001) was very likely due to an additional membrane depolarization consecutive to Na+/K+ ATPase inhibition. When 15 mM K+ was superfused after ouabain or after successive applications of ouabain and BaCl2, it did not alter [Ca2+]i. By contrast, BK induced always a rapid drop in [Ca2+]i (Figure 8b,d,e). After ouabain and ouabain plus BaCl2, the magnitude of the BK effect was significantly higher than the initial [Ca2+]i increase caused by angiotensin II alone (136±7 vs 110±10 nM, n=10, P<0.05). It was also significantly greater than the decrease observed in the absence of ouabain (P<0.02). These observations indicate that BK also counteracts the ouabain-induced increase. Note that BaCl2 (30 μM) and ouabain (1 mM), added alone or together had no effect on the basal [Ca2+]i level.
Effect of BK after closure of VOCC by nifedipine
It is generally agreed that endothelium-dependent hyperpolarization results in closure of VOCC (Garland et al., 1995). To investigate whether this mechanism is involved, BK was tested when L-type VOCC were blocked by nifedipine. Nifedipine (1 μM) alone provoked a sustained decrease of 30±4 nM (n=7) in the [Ca2+]i response to angiotensin II. A subsequent addition of BK still produced a transitory 27±5 nM drop in [Ca2+]i (n=7, P<0.01, Figure 9). This decrease was much smaller than that observed in the absence of nifedipine (90±7 nM, n=66, P<0.001). This remaining effect of BK after nifedipine was inhibited by neither L-NAME (Figure 9b) nor indomethacin (results not shown).
Figure 9.
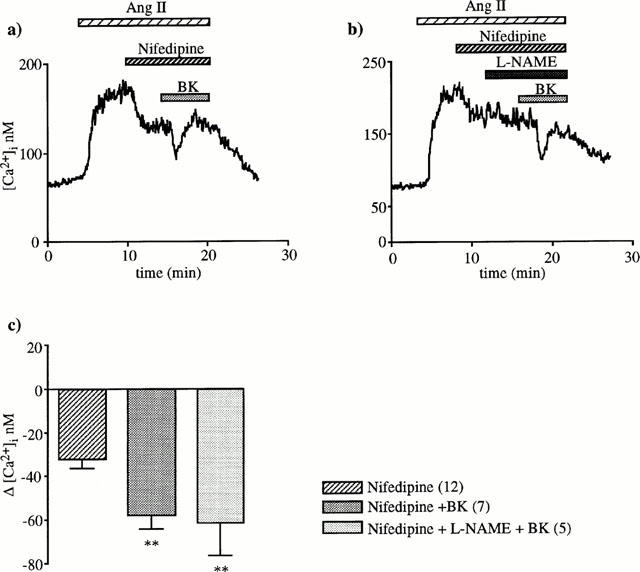
BK effect in the presence of nifedipine. Application of 1 μM nifedipine during the plateau phase of the [Ca2+]i response to 1 nM angiotensin II caused a [Ca2+]i decrease consecutive to closure VOCC previously opened by angiotensin II. Subsequent addition of 10 nM BK still produced a significant drop (a). This fall in [Ca2+]i was not inhibited by L-NAME (b). The columns represent [Ca2+]i decreases (means±s.e.mean) caused by nifedipine alone or nifedipine plus BK or nifedipine plus L-NAME plus BK, **P<0.01 compared to the [Ca2+]i decrease induced by nifedipine alone (unpaired Student's t-test).
Discussion
This work demonstrates that BK induces a [Ca2+]i drop secondary to release of an EDHF, in juxtamedullary EA stimulated by angiotensin II. This effect is not altered by selective inhibitors of cytochrome P450 and could not be mimicked by elevating external K+. BK still induces a small [Ca2+]i decrease after VOCC closure by nifedipine.
It is generally agreed that BK relaxes vascular muscle cells by releasing vasodilator mediators such as NO, PGI2 and EDHF from the endothelium (Mombouli & Vanhoute, 1995; Hornig & Drexler, 1997) that in turn decrease [Ca2+]i to produce relaxation. However, only a few studies have directly examined [Ca2+]i variation (Blatter & Wier, 1994; Hirano & Kanaide, 1993; Bolz et al., 1999). To our knowledge, this is the first work describing the [Ca2+]i time-course during the application of a relaxant substance to a glomerular arteriole. In our experiments, BK caused a decrease in [Ca2+]i only when [Ca2+]i was previously elevated by angiotensin II. BK-induced [Ca2+]i drop required both an intact endothelium and B2 BK receptor activation, indicating that BK acts through endothelial B2 receptors to release a diffusible substance from endothelium which reduces [Ca2+]i in the smooth muscle cells. This substance was neither NO nor PGI2 since L-NAME and indomethacin failed to inhibit BK-induced [Ca2+]i decrease, and since SNP, cyclic GMP or PGI2 did not mimic the BK effect. The reduction in the [Ca2+]i response to angiotensin II could therefore be attributed to some hyperpolarizing factor (Mombouli & Vanhoute, 1997). This possibility is supported by lack of BK response when [Ca2+]i was increased with high external K+ (100 mM) which would also prevent EDHF-induced K+ efflux and membrane hyperpolarization. Although no measurement of membrane potential was done in this study, these observations strongly suggest that the mediator of BK effect is an EDHF in the muscular EA. In the case of afferent arterioles (Yu et al., 1998) and interlobar renal arteries (Ihara et al., 2000), other mediators, such as metabolites of cyclo-oxygenase pathway and NO, could also be involved in BK effect.
In the present study when BK response could not develop (presence of high external K+, charybdotoxin or proadifen), BK rather elicited a small significant [Ca2+]i increase whose magnitude and time-course were similar to those observed on muscular EA without angiotensin II pretreatment. This BK-induced [Ca2+]i increase which takes place in the endothelium (Praddaude et al., 1999) and may be essential for the release of EDHF (Chen & Suzuki, 1990; Nagao & Vanhoute, 1993; Higuchi et al., 1996) was very likely concealed in intact angiotensin II-prestimulated arterioles by the [Ca2+]i drop occuring in muscular cells.
The chemical nature of EDHF is still controversial. It was firstly reported that EDHF could be a cytochrome P450-derived arachidonic acid metabolite (Hecker et al., 1994; Campbell et al., 1996). More recently, Edwards et al. (1998) proposed that K+ ion would itself be the EDHF. In our study, we found that EDHF was not a cytochrome P450-derived arachidonic acid metabolite because 17-ODYA, an irreversible suicide-substrate inhibitor which selectively affects cytochrome P450 mono-oxygenases (Zou et al., 1994), failed to alter BK action. We also found that 7-ethoxyresofurin, another substrate of cytochrome P450, had no detectable effect on the BK-induced [Ca2+]i decrease, as observed by Eckman et al., 1998, during acetylcholine-induced relaxation and hyperpolarization in guinea-pig coronary artery. Only proadifen, a reversible inhibitor of cytochrome P450 prevented the BK effect. However, this inhibitor has additional effects, unrelated to inhibition of cytochrome P450 (Edwards et al., 1996; Fukao et al., 1997; Eckman et al., 1998). Thus, it can alter ATP-sensitive K+ channel (Zygmunt et al., 1996), Ca2+-activated K+ channels (Alvarez et al., 1992) and charybdotoxin binding sites (Brugnara et al., 1993). Consequently, the inhibitory effect of proadifen, found in our experiments, could be explained rather by a blockade of charybdotoxin-sensitive K+ channels (see below) than by a reduced endothelial EDHF synthesis.
In support of the proposition that K+ ion could itself be the EDHF (Edwards et al., 1998) is the fact that slight and non-depolarizing increases in extracellular K+ are able to cause membrane hyperpolarization and relaxation in coronary and cerebral arteries (Knot et al., 1996) which can be prevented by an inward rectifier K+ channel blocker, barium. So, in rat hepatic artery, Edwards et al. (1998) demonstrated that acetylcholine provoked an efflux of K+ ions through endothelial K+ channels and K+ effect could account for the entire hyperpolarizing and relaxing effects of acetylcholine (Edwards et al., 1999b). This effect results from activation, in the muscular cells, of the barium-sensitive inwardly rectifying K+ conductance and of Na+/K+ pump. In our experiments, 15 mM K+ decreased the [Ca2+]i response to angiotensin II. However the K+ effect differed from that of BK in that it was of a much smaller magnitude, not sensitive to charybdotoxin and sensitive to barium, ouabain or combination of both whereas barium and ouabain had no effect on the BK response. These differences indicate that K+ itself is not the EDHF in muscular EA. However, a limited participation of K+ ion to the BK effect cannot be completely ruled out since K+ induced a small [Ca2+]i decrease. Our results in muscular EA are in accordance with those of Quignard et al. (1999), in guinea-pig carotid and porcine coronary arteries. Like Quignard et al. (1999), we therefore think that the chemical nature of EDHF is different according to the type of vessels.
It is believed that EDHF induces membrane hyperpolarization of vascular smooth muscle cell by opening K+ channels whose nature differs according to blood vessel types (Petersson et al., 1997). In the present study, charybdotoxin, an inhibitor of BKCa (large conductance Ca-sensitive K+ channel) and of some Kv (voltage-sensitive K+ channels), prevented BK-induced [Ca2+]i drop. However, iberiotoxin, a more selective inhibitor of BKCa (Edwards & Weston, 1994) and 4-aminopyridine, an inhibitor of Kv, had no effect. Thus, as in rat hepatic artery (Zygmunt et al., 1997), the targets in muscular EA for the BK-produced EDHF might be other K+ channel types structurally related to both BKCa and Kv or intermediate-conductance calcium-activated K+ channels (IKCa) which are also sensitive to charybdotoxin (Andersson et al., 2000). It is also possible that gap junctions may contribute in the electronic propagation of hyperpolarization from the endothelial to the smooth muscle cells (Edwards et al., 1999a).
With regard to the link between vascular smooth muscle cell hyperpolarization and relaxation, it is generally agreed that hyperpolarization provokes a closure of VOCC to decrease [Ca2+]i and relax vessels, but to our knowledge, no [Ca2+]i measurement had been yet performed to support this hypothesis. In this study, BK was still able to produce a small decrease in [Ca2+]i after the closure of all angiotensin II-activated VOCCs by nifedipine (Helou & Marchetti, 1997). Thus, BK effect involves not only VOCC closure but also requires other cellular mechanisms which could be a reduction of Ca2+ release from internal stores, an increase in Ca2+ uptake by the sarcoplasmic reticulum, an increase in Ca2+ extrusion by plasma membrane Ca2+-ATPase or a decrease of Ca2+ influx through voltage-independent Ca2+ channel. In support of such a hypothesis, it has been reported that BK-induced relaxation is not sensitive to nifedipine in bovine coronary artery (Drummond & Cocks, 1996) and a relaxation can be produced in arteries preconstricted with the thromboxane-mimetic U46619 which does not activate VOCC (Kilpatrick & Cocks, 1994).
To date, we can only speculate on the functional significance of the [Ca2+]i drop because data concerning the contractile response are lacking for the muscular EA. If this response consists of a sustained relaxation, BK has to produce other effect(s) such as reduction in the Ca2+ sensitivity of muscular contractile elements (Bolz et al., 1999; Tomioka et al., 1999) in addition of reducing [Ca2+]i, because the [Ca2+]i decrease was only transient in most of the tested muscular EA. Note that different relaxant agonists induce a sustained relaxation in a variety of blood vessels though the associated hyperpolarization generally occurs in a transient fashion (Garland et al., 1995). The role of hyperpolarization and of resulting [Ca2+]i drop would therefore be to rapidly initiate the relaxation response. Consequently, our results suggest that BK could in this way rapidly reverse the vasoconstriction induced by angiotensin II and document the cellular basis for the interplay between the kallikrein-kinin and renin-angiotensin systems in the control of renal medullary microcirculation (Navar et al., 1996; Siragy et al., 1996).
In conclusion, this study clearly shows that the BK-induced [Ca2+]i drop in muscular EA stimulated by angiotensin II is mediated neither by NO nor by PGI2 but involves an EDHF able to activate charybdotoxin-sensitive K+ channels. It also demonstrates that BK-released EDHF is neither a cytochrome P450-derived arachidonic acid metabolite nor the K+ ion itself. The EDHF-induced [Ca2+]i drop involves VOCC closure amongst other cellular mechanisms that remain to be investigated.
Acknowledgments
We acknowledge the skilful technical assistance of Catherine Chollet. This work was supported by INSERM and by a grant from the Bristol Myer-Squibb Institute for Medical Research (Princeton, NJ, U.S.A.).
Abbreviations
- BK
bradykinin
- [Ca2+]i
intracellular calcium concentration
- EA
efferent arteriole
- EDHF
endothelium-derived hyperpolarizing factor
- HEPES
N-2-hydroxyethylpiperazine-N′-2-ethanesulphonic acid
- L-NAME
Nω-nitro-L-arginine methyl ester
- 17-ODYA
17-octadecynoic acid
- SNP
sodium nitroprusside
- VOCC
voltage-operated calcium channel
References
- ALVAREZ J., MONTERO M., GARCIA-SANCHO J. High affinity inhibition of Ca2+-dependent K+ channels by cytochrome P450 inhibitors. J. Biol. Chem. 1992;267:11789–11793. [PubMed] [Google Scholar]
- ANDERSSON D.A., ZYGMUNT P.M., MOVAHED P., ANDERSSON T.L.G., HOGESTATT E.D. Effects of inhibitors of small- and intermediate-conductance calcium-activated potassium channels, inwardly-rectifying potassium channels and Na+/K+ ATPase on EDHF relaxations in the rat hepatic artery. Br. J. Pharmacol. 2000;129:1490–1496. doi: 10.1038/sj.bjp.0703226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BEIERWALTES W.H. Possible endothelial modulation of prostaglandin-stimulated renin release. Am. J. Physiol. 1990;258:F1363–F1371. doi: 10.1152/ajprenal.1990.258.5.F1363. [DOI] [PubMed] [Google Scholar]
- BLATTER L.A., WIER W.G. Nitric oxide decreases Ca2+i in vascular smooth muscle by inhibition of the calcium current. Cell Calcium. 1994;15:122–131. doi: 10.1016/0143-4160(94)90051-5. [DOI] [PubMed] [Google Scholar]
- BOLZ S.S., DE WIT C., POHL U. Endothelium-derived hyperpolarizing factor but not NO reduces smooth muscle Ca2+ during acetylcholine-induced dilation of microvessels. Br. J. Pharmacol. 1999;128:124–134. doi: 10.1038/sj.bjp.0702775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRUGNARA C., DE FRANCESCHI L., ALPER S.L. Inhibition of Ca2+-dependent K+ transport and cell dehydration in sickle erythrocytes by clotrimazole and other imidazole derivatives. J. Clin. Invest. 1993;92:520–526. doi: 10.1172/JCI116597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUSSE R., FLEMING I. Molecular responses of endothelial tissue to kinins. Diabetes. 1996;45:S8–S13. doi: 10.2337/diab.45.1.s8. [DOI] [PubMed] [Google Scholar]
- CAMPBELL W.B., GEBREMEDHIN D., PRATT P.F., HARDER D.R. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ. Res. 1996;78:415–423. doi: 10.1161/01.res.78.3.415. [DOI] [PubMed] [Google Scholar]
- CHEN G., SUZUKI H. Calcium dependency of the endothelium-dependent hyperpolarization in smooth muscle cells of the rabbit carotid artery. J. Physiol. 1990;421:521–534. doi: 10.1113/jphysiol.1990.sp017959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DRUMMOND G.R., COCKS T.M. Evidence for mediation by endothelium-derived hyperpolarizing factor of relaxation to bradykinin in the bovine isolated coronary artery independently of voltage-operated Ca2+ channels. Br. J. Pharmacol. 1996;117:1035–1040. doi: 10.1111/j.1476-5381.1996.tb16693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ECKMAN D.M., HOPKINS N., MCBRIDE C., KEEF K.D. Endothelium-dependent relaxation and hyperpolarization in guinea-pig coronary artery: role of epoxyeicosatrienoic acid. Br. J. Pharmacol. 1998;124:181–189. doi: 10.1038/sj.bjp.0701778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDWARDS G., DORA K.A., GARDENER M.J., GARLAND C.J., WESTON A.H. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- EDWARDS G., FELETOU M., GARDENER M.J., THOLLON C., VANHOUTTE P.M., WESTON A.H. Role of gap junctions in the responses to EDHF in rat and guinea-pig small arteries. Br. J. Pharmacol. 1999a;128:1788–1794. doi: 10.1038/sj.bjp.0703009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDWARDS G., GARDENER M.J., FELETOU M., BRADY G., VANHOUTTE P.M., WESTON A.H. Further investigation of endothelium-derived hyperpolarizing factor (EDHF) in rat hepatic artery: studies using 1-EBIO and ouabain. Br. J. Pharmacol. 1999b;128:1064–1070. doi: 10.1038/sj.bjp.0702916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDWARDS G., WESTON A.H. K ATP-fact or artefact? New thoughts on the mode of action of the potassium channel openers. Cardiovasc. Res. 1994;28:735–737. doi: 10.1093/cvr/28.6.735. [DOI] [PubMed] [Google Scholar]
- EDWARDS G., ZYGMUNT P.M., HOGESTATT E.D., WESTON A.H. Effects of cytochrome P450 inhibitors on potassium currents and mechanical activity in rat portal vein. Br. J. Pharmacol. 1996;119:691–701. doi: 10.1111/j.1476-5381.1996.tb15728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FUKAO M., HATTORI Y., KANNO M., SAKUMA I., KITABATAKE A. Evidence against a role of cytochrome P450-derived arachidonic acid metabolites in endothelium-dependent hyperpolarization by acetylcholine in rat isolated mesenteric artery. Br. J. Pharmacol. 1997;120:439–446. doi: 10.1038/sj.bjp.0700932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FURCHGOTT R.F., VANHOUTTE P.M. Endothelium-derived relaxing and contracting factors. FASEB J. 1989;3:2007–2018. [PubMed] [Google Scholar]
- GARLAND C.J., PLANE F., KEMP B.K., COCKS T.M. Endothelium-dependent hyperpolarization: a role in the control of vascular tone. Trends Pharmacol. Sci. 1995;16:23–30. doi: 10.1016/s0165-6147(00)88969-5. [DOI] [PubMed] [Google Scholar]
- GRYNKIEWICZ G., POENIE M., TSIEN R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- HECKER M., BARA A.T., BAUERSACHS J., BUSSE R. Characterization of endothelium-derived hyperpolarizing factor as a cytochrome P450-derived arachidonic acid metabolite in mammals. J. Physiol. 1994;481:407–414. doi: 10.1113/jphysiol.1994.sp020449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HELOU C.M., MARCHETTI J. Morphological heterogeneity of renal glomerular arterioles and distinct [Ca2+]i responses to ANG II. Am. J. Physiol. 1997;273:F84–F96. doi: 10.1152/ajprenal.1997.273.1.F84. [DOI] [PubMed] [Google Scholar]
- HIGUCHI T., NISHIMURA J., KOBAYASHI S., KANAIDE H. CPA induces a sustained release in [Ca2+]i of endothelial cells in situ and relaxes porcine coronary artery. Am. J. Physiol. 1996;270:H2038–H2049. doi: 10.1152/ajpheart.1996.270.6.H2038. [DOI] [PubMed] [Google Scholar]
- HIRANO K., KANAIDE H. Cytosolic Ca2+ transients in endothelium-dependent relaxation of pig coronary artery, and effects of captopril. Eur. J. Pharmacol. 1993;250:439–446. doi: 10.1016/0014-2999(93)90031-c. [DOI] [PubMed] [Google Scholar]
- HORNIG B., DREXLER H. Endothelial function and bradykinin in humans. Drugs. 1997;54 Suppl. 5:42–47. doi: 10.2165/00003495-199700545-00007. [DOI] [PubMed] [Google Scholar]
- IHARA E., HIRANO H., DERKACH D.M., NISHIMURA J., NAWATA H., KANAIDE H. The mechanism of bradykinin-induced endothelium-dependent contraction and relaxation in the porcine interlobar renal artery. Br. J. Pharmacol. 2000;129:943–952. doi: 10.1038/sj.bjp.0703141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KILPATRICK E.V., COCKS T.M. Evidence for differential roles of nitric oxide (NO) and hyperpolarization in endothelium-dependent relaxation of pig isolated coronary artery. Br. J. Pharmacol. 1994;112:557–565. doi: 10.1111/j.1476-5381.1994.tb13110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KNOT H.J., ZIMMERMANN P.A., NELSON M.T. Extracellular K+-induced hyperpolarizations and dilatations of rat coronary and cerebral arteries involve inward rectifier K+ channels. J. Physiol. 1996;492:419–430. doi: 10.1113/jphysiol.1996.sp021318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARCHETTI J., MENETON P., LEBRUN F., BLOCH-FAURE M., RAJERISON R.M. The parietal sheet of Bowman' capsule of rat renal glomerulus: a target of endothelin and PAF. Am. J. Physiol. 1995;268:F1053–F1061. doi: 10.1152/ajprenal.1995.268.6.F1053. [DOI] [PubMed] [Google Scholar]
- MOMBOULI J.V., VANHOUTTE P.M. Kinins and endothelial control of vascular smooth muscle. Annu. Rev. Pharmacol. Toxicol. 1995;35:679–705. doi: 10.1146/annurev.pa.35.040195.003335. [DOI] [PubMed] [Google Scholar]
- MOMBOULI J.V., VANHOUTTE P.M. Endothelium-derived hyperpolarizing factor(s): updating the unknown. Trends Pharmacol. Sci. 1997;18:252–256. [PubMed] [Google Scholar]
- NAGAO T., VANHOUTTE P.M. Endothelium-derived hyperpolarizing factor and endothelium-dependent relaxations. Am. J. Respir. Cell. Mol. Biol. 1993;8:1–6. doi: 10.1165/ajrcmb/8.1.1. [DOI] [PubMed] [Google Scholar]
- NAVAR L.G., INSCHO E.W., MAJID D.S.A., IMIG J.D., HARRISON-BERNARD L.M., MITCHELL K.D. Paracrine regulation of the renal microcirculation. Physiol. Rev. 1996;76:425–536. doi: 10.1152/physrev.1996.76.2.425. [DOI] [PubMed] [Google Scholar]
- NOBES M.S., HARRIS P.J., YAMADA H., MENDELSOHN F.A.O. Effects of angiotensin on renal cortical and papillary blood flows measured by laser-Doppler flowmetry. Am. J. Physiol. 1991;261:F998–F1006. doi: 10.1152/ajprenal.1991.261.6.F998. [DOI] [PubMed] [Google Scholar]
- PALLONE T.L., ROBERTSON C.R., JAMISON R.L. Renal medullary microcirculation. Physiol. Rev. 1990;70:885–920. doi: 10.1152/physrev.1990.70.3.885. [DOI] [PubMed] [Google Scholar]
- PETERSSON J., ZYGMUNT P.M., HÖGESTÄTT E.D. Characterization of the potassium channels involved in EDHF-mediated relaxation in cerebral arteries. Br. J. Pharmacol. 1997;120:1344–1350. doi: 10.1038/sj.bjp.0701032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRADDAUDE F., MARCHETTI J., ALHENC-GELAS F., ADER J-L. Dissimilar mechanisms of Ca2+ response to bradykinin in different types of juxtamedullary glomerular arterioles. Am. J. Physiol. 1999;277:F697–F705. doi: 10.1152/ajprenal.1999.277.5.F697. [DOI] [PubMed] [Google Scholar]
- QUIGNARD J.-F., FELETOU M., THOLLON C., VILAINE J.-P., DUHAULT J., VANHOUTTE P.M. Potassium ions and endothelium-derived hyperpolarizing factor in guinea-pig carotid and porcine coronary arteries. Br. J. Pharmacol. 1999;127:27–34. doi: 10.1038/sj.bjp.0702493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QUILLEY J., FULTON D., McGIFF J.C. Hyperpolarizing factors. Biochem. Pharmacol. 1997;54:1059–1070. doi: 10.1016/s0006-2952(97)00039-7. [DOI] [PubMed] [Google Scholar]
- ROMAN R.J., KALDUNSKY M.L., SCICLI A.G., CARRETERO O.A. Influence of kinins and angiotensin II on the regulation of papillary blood flow. Am. J. Physiol. 1988;255:F690–F698. doi: 10.1152/ajprenal.1988.255.4.F690. [DOI] [PubMed] [Google Scholar]
- SIRAGY H.M., JAFFA A.A., MARGOLIUS H.S., CAREY R.M. Renin-angiotensin system modulates renal bradykinin production. Am. J. Physiol. 1996;271:R1090–R1095. doi: 10.1152/ajpregu.1996.271.4.R1090. [DOI] [PubMed] [Google Scholar]
- TOMIOKA H., HATTORI Y., FUKAO M., SATO A., LIU M.Y., SAKUMA I., KITABATAKE A., KANNO M. Relaxation in different-sized rat blood vessels mediated by endothelium-derived hyperpolarizing factor: importance of processes mediating precontractions. J. Vasc. Res. 1999;36:311–320. doi: 10.1159/000025659. [DOI] [PubMed] [Google Scholar]
- YU H., CARRETERO O.A., JUNCOS L.A., GARVIN J.L. Biphasic effect of bradykinin on rabbit afferent arterioles. Hypertension. 1998;32:287–292. doi: 10.1161/01.hyp.32.2.287. [DOI] [PubMed] [Google Scholar]
- ZOU A.P., MA Y.H., SUI Z.H., ORTIZ DE MONTELLANO P.R., CLARK J.E., MASTERS B.S., ROMAN R.J. Effects of 17-octadecynoic acid, a suicide-substrate inhibitor of cytochrome P450 fatty acid ω-hydrolase, on renal function in rats. J. Pharmacol. Exp. Ther. 1994;268:474–481. [PubMed] [Google Scholar]
- ZYGMUNT P.M., EDWARDS G., WESTON A.H., DAVIS C., HÖGESTÄTT E.D. Effects of cytochrome P450 inhibitors on EDHF-mediated relaxation in the rat hepatic artery. Br. J. Pharmacol. 1996;118:1147–1152. doi: 10.1111/j.1476-5381.1996.tb15517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZYGMUNT P.M., EDWARDS G., WESTON A.H., LARSSON B., HÖGESTÄTT E.D. Involvement of voltage-dependent potassium channels in the EDHF-mediated relaxation of rat hepatic artery. Br. J. Pharmacol. 1997;121:141–149. doi: 10.1038/sj.bjp.0701108. [DOI] [PMC free article] [PubMed] [Google Scholar]