

Abstract
Recent studies demonstrated that the cardiac calpain system is activated during ischaemic events and is involved in cardiomyocyte injury. The aim of this study was to investigate the contribution of AT1 and AT2 receptors in the regulation of calpain-mediated myocardial damage following myocardial infarction (MI).
Infarcted animals were treated either with placebo, the ACE inhibitor ramipril (1 mg kg−1 d−1), the AT1 receptor antagonist valsartan (10 mg kg−1 d−1) or the AT2 receptor antagonist PD 123319 (30 mg kg−1 d−1). Treatment was started 7 days prior to surgery. On day 1, 3, 7 and 14 after MI, gene expression and protein levels of calpain I, II and calpastatin were determined in left ventricular free wall (LVFW) and interventricular septum (IS). At day 3 and 14 post MI, morphological investigations were performed.
Calpain I mRNA expression and protein levels were increased in IS 14 days post MI, whereas mRNA expression and protein levels of calpain II were maximally increased in LVFW 3 days post MI. Ramipril and valsartan decreased mRNA and protein up-regulation of calpain I and II, and reduced infarct size and interstitial fibrosis. PD 123319 did not affect calpain I or II up-regulation in the infarcted myocardium, but decreased interstitial fibrosis. Calpastatin expression and translation were not affected by AT receptor antagonists or ACE inhibitor.
Our data demonstrate a distinct, temporary-spatial up-regulation of calpain I and II following MI confer with the hypothesis of calpain I being involved in cardiac remodelling in the late and calpain II contributing to cardiac tissue damage in the early phase of MI. The up-regulation of calpain I and II is partly mediated via the AT1 receptor and can be reduced by ACE inhibitors and AT1 receptor antagonists.
Keywords: Myocardial infarction, renin-angiotensin-system, calpain-system
Introduction
The calpain system is an intracellular, strongly Ca2+-dependent, neutral cysteine protease system existing in all mammalian and in some non-mammalian cells. The calpain family consists of ubiquitous and tissue specific isoforms of calpains and of the naturally occurring endogenous inhibitor, calpastatin. The best characterized calpains are the widely distributed isoenzymes, calpain I or μ-calpain, which requires micromolar intracellular calcium ([Ca2+]i)-concentrations for half-maximum activity, and calpain II or m-calpain, which requires millimolar [Ca2+]i-concentrations for activation, respectively. These calpains are heterodimeric molecules consisting of a large 80 kDa catalytic and a small 30 kDa regulatory subunit. The tissue specific calpain isoenzymes are the skeletal muscle calpain III (Sorimachi et al., 1989), the stomach calpain nCl-2 and smooth muscle calpain nCl-2 (Sorimachi et al., 1994), the testis, liver, trachea, colon and kidney calpain htra-3 (Dear et al., 1997), the placenta calpain VI (Mugita et al., 1997) and the lens calpain Lp82 (Ma et al., 1998). The endogenous inhibitor, calpastatin, has been shown to fully inhibit proteolytic activity of all calpain isoforms at a 1 : 1 ratio of calpain to calpastatin (Suzuki et al., 1987).
Although the precise intracellular functions of the calpains have not been fully defined, a number of studies indicate their potential importance in regulated proteolysis of key enzymes and structural proteins as well as in apoptotic processes (Rechsteiner & Rogers, 1996). The calpains have also been demonstrated to play a harmful role in a variety of pathological states which are associated with an Ca2+-overload, such as Duchenne's muscular dystrophy, Alzheimer's disease, multiple sclerosis and development of cataract (see review; Wang & Yuen, 1994). The injury sustained by cardiomyocytes during anoxia or ischaemia displays similarities to that observed in neurons following cerebral ischaemia. The rise in [Ca2+]i in myocytes during myocardial ischaemia (Nayler, 1981; Steenbergen et al., 1990) has been considered to be a pivotal event in the activation of calpains leading to cardiac cell death and structural damage of myocardium (Toyo-oka & Ross, 1981; Steenbergen et al., 1987). The involvement of calpain in the breakdown of myofibrillar proteins such as myosin heavy chain, troponin T and I, tropomyosin A and α-actinin (Ishiura et al., 1991) indicates that this enzyme contributes to the anoxic or ischaemic cell death in the heart.
A major pathogenic factor contributing to functional and structural alterations in hypertensive as well as in ischaemic heart disease is angiotensin II, the effector peptide of the renin-angiotensin-system (RAS). At present, it is not clear whether the calpain system is affected by the RAS in ischaemic heart disease, such as after myocardial infarction (MI). Evidence has been presented that the RAS is activated after MI associated with increased mRNA and protein levels for angiotensinogen (Lindpainter et al., 1993), angiotensin-converting-enzyme (ACE) (Hirsch et al., 1991) and angiotensin receptors (Meggs et al., 1993; Nio et al., 1995) in rat hearts, and angiotensin II is considered to have a profound effect on ventricular remodeling following MI (Sladek et al., 1996). The angiotensin receptors include at least two different subtypes: the AT1 receptor is the predominant receptor in the adult rat heart and is considered to be the major mediator of the angiotensin II-induced effects in the cardiovascular system (Timmermans et al., 1993). In contrast, AT2 receptor activation has been reported to inhibit cell proliferation (Stoll et al., 1995), to induce differentiation (Meffert et al., 1996), apoptosis (Yamada et al., 1996; Gallinat et al., 1999), and regeneration (Lucius et al., 1998), and to be involved in the control of voltage-sensitive ion currents (Kang et al., 1993).
The present study was undertaken to investigate possible interactions of the RAS with the cardiac calpain system following MI and the contribution of the calpains on cardiac structural remodelling process in the infarcted myocardium. In particular, the differential role of angiotensin AT1 and AT2 receptor subtypes in the regulation of the members of the calpain system as well as on the calpain-mediated myocardial damage following MI was addressed. For this purpose, we examined the influence of pretreatment with the ACE inhibitor, ramipril, the angiotensin AT1 receptor antagonist, valsartan, and the angiotensin AT2 receptor antagonist, PD 123319, on calpain I, calpain II and calpastatin mRNA expression and respective protein levels in several regions of the rat myocardium at different time points after induction of MI. The effects of the three drugs on calpain-mediated structural remodelling process of the infarcted heart was determined by measurement of infarct size and interstitial collagen content of the non-infarcted myocardium.
Methods
Animals and treatment
Male normotensive Wistar rats (Charles River Viga, Sulzfeld, Germany) initially weighing 220 – 230 g were used in all experiments. The animals were housed individually at controlled temperature and humidity under a 12 h light/dark cycle and had free access to a standard diet (Altromin®, Altromin International GmbH, Lage-Lippe, Germany) and to drinking water. The study was performed in accordance with the German law on animal protection as released in its new version in 1993.
The animals were randomly divided into seventeen groups: Group 1: sham operation without treatment; Groups 2 – 5: myocardial infarction (MI) subjected to placebo treatment (0.9% saline); Groups 6 – 9: MI subjected to ramipril treatment (1 mg kg−1 d−1); Groups 10 – 13: MI subjected to valsartan treatment (10 mg kg−1 d−1); Groups 14 – 17: MI subjected to PD 123319 treatment (30 mg kg−1 d−1). Treatment was begun 1 week prior to induction of MI (7 days pre) in four separate groups per each treatment and continued until sacrifice on day 1, 3, 7 and 14 after induction of MI. The number of animals in each of the 17 groups was 6 – 8.
For surgical procedures, rats were anaesthetized by injection of methohexital-Na+ (10 mg kg−1 i.v.) and artificially ventilated (70 ventilations min−1, 200 mmH2O, 2.5 ml ventilation−1) to perform a left thoracotomy. MI was induced by permanent ligation of the left coronary artery as previously described (Sandmann et al., 1998). On the day of sacrifice, infarcted and sham-operated animals were decapitated and the hearts were rapidly excised and placed on a preparation chamber at 4°C. The left ventricle (LV) was separated from the atria and the right ventricle (RV) and then divided into the interventricular septum (IS) and the left ventricular free wall (LVFW) including scar tissue and area at risk. The tissue samples were divided into two parts of equal size, rapidly frozen in liquid nitrogen and separately stored at −80°C. One aliquot of each tissue sample was used to investigate the mRNA expression of calpain I, calpain II and calpastatin, whereas the other part was used to determine the protein levels.
Drugs
The ACE inhibitor, ramipril, was given daily via gastric gavage and the AT1 and AT2 receptor antagonists, valsartan and PD 123319, via osmotic minipumps. The drug dosages for ramipril (Gohlke et al., 1994), valsartan (Hayashi et al., 1997) and PD 123319 (Gohlke et al., 1998) were adjusted to the individual body weight of each rat to ensure effective receptor- or enzyme-blockade, respectively. Ramipril was dissolved in water. Valsartan and PD 123319 were dissolved in 1 N NaOH and buffered with HCl (pH 7.4) before filling into minipumps.
RNA extraction and reverse transcription-polymerase chain reaction (RT – PCR)
Total RNA from RV, IS and LVFW of sham-operated and infarcted animals with placebo- ramipril-, valsartan- or PD 123319-treatment was extracted from an aliquot of the fresh frozen tissue using a single step isolation method described by Chomczynski & Sacchi (1987). Integrity of the RNA was confirmed by agarose gel electrophoresis, and the concentration was determined by densitometric measurement of UV absorption at 260 nm. Poly(A)+-RNA was isolated from total RNA by using Oligotex mRNA Mini Kit (QIAGEN, Hilden, Germany). Trizol solution, molecular size standards, Super Script Preamplification Systems and Taq Polymerase were purchased from Gibco BRL (Eggenstein, Germany).
Primers were designed using software supplied by the German Cancer Research Center (DKFZ, Heidelberg, Germany) and were obtained from Pharmacia, (Pharmacia Biotech, Cambridge, U.K.). Details concerning primers used are given in Table 1. The reverse transcription of total RNA (5 μg) was performed with oligo (dT) primers using Prime-It II Random Primer Kit reagents (Stratege, Heidelberg, Germany). In order to exclude genomic DNA contaminations, aliquots of RNA preparations were subjected to RT – PCR without the use of reverse transcriptase. The absence of amplification products served as negative control and confirmed the purity of the total RNA used. For experiments with amplification of GAPDH mRNA as house keeping gene, the reaction mixture was equally split into two tubes before specific primers and PCR reagents were added. The GAPDH signals used as an internal control have been shown not to be changed in rat heart after MI (Ju et al., 1998). To optimize the RT – PCR assay, we determined the relationship of signal strength to the number of PCR cycles and the amount of input cDNA (1 to 5 μl) for all PCR products. Exact PCR protocols for the various amplification reactions are as following: (1) Calpain I: 5 μl PCR buffer, 1.5 μl of 1.5 mM MgCl2, 2 μl of 0.2 mM dNTP's, 1 μl of 20 μM primers, 0.25 μl of 2.5 units Taq polymerase, 62°C annealing temperature, 26 PCR cycles; (2) Calpain II: 5 μl PCR buffer, 1.5 μl of 1.5 mM MgCl2, 2 μl of 0.2 mM dNTP's, 1 μl of 20 μM primers, 0.25 μl of 2.5 units Taq polymerase, 59°C annealing temperature, 26 PCR cycles; (3) Calpastatin: 5 μl PCR buffer, 2 μl of 1.5 mM MgCl2, 2 μl of 0.2 mM dNTP's, 1 μl of 20 μM primers, 0.25 μl of 2.5 units Taq polymerase, 57°C annealing temperature, 25 PCR cycles; (4) GAPDH: 5 μl PCR buffer, 3 μl of 1.5 mM MgCl2, 1 μl of 0.2 mM dNTP's, 1 μl of 20 μM primers, 0.25 μl of 2.5 units Taq polymerase, 60°C annealing temperature, 23 PCR cycles. Each mixture was denatured at 95°C for 1 min, and samples were amplified in a programmable thermal controller (PTC-100, MJ Research). At the end of amplification reactions, PCR products were incubated at 72°C for 2 min for a final extension. Aliquots of each sample (10 μl) together with the respective negative controls were subjected to electrophoresis and were run next to each other in the same 1.2% agarose gel and stained with ethidium bromide. The identity of PCR products was verified by sequence analysis (data not shown). To ensure a high reproducibility, each sample was subjected to four independent PCR reactions for calpain I, calpain II, calpastatin and GAPDH.
Table 1.
Primer sequences for amplification of calpain I (Calp I), calpain II (Calp II), calpastatin (Calpast) and GAPDH

Northern blot analysis and mRNA quantification
Eight μg of Poly(A)+-RNA from each tissue sample was denatured with formaldehyde, fractionated by electrophoresis through 1.2% agarose gel, and transferred to a nylon membrane (Hybond-N Membrane, Amersham, Braunschweig, Germany) using a capillary blotting technique. RNA was fixed on the membrane for 2 h at 80°C. RNA size markers (0.24 – 9.5 kb ladder, Ambion) were visualized with UV light after staining with ethidium bromide. Hybridizations were carried out in a Rapid-Hyb solution (Amersham, Braunschweig, Germany) using 32P-labelled probes. Membranes were washed using high stringency conditions: one time for 30 min at 22°C in 1×SSC-0.1% SDS and one time for 60 min at 60°C in 0.1 SSC – 0.1% SDS. The blots were then examined by autoradiography. Exposure times were 24 h for Northern blots probed with calpain I or II, 20 h for calpastatin probes and 4 h for GAPDH probes. The fragment used for labelling of rat calpain 1 mRNA was 233 – 634 bp of the rat calpain I cDNA sequence corresponding to the coding region. The fragment for probing the calpain II message was 633 – 822 bp, the probe for calpastatin was 462 – 723 bp and the probe for GAPDH was 568 – 989 bp. The cDNA fragments were radiolabelled with [α-32P]-dCTP (1×109 cpm μg−1 of DNA, Amersham, Braunschweig, Germany) using the random primer procedure (Strategene). An optical scanner (EPSON GT-8000, Seiko, Tokyo, Japan) was used for digitization of autoradiograms and pictures in order to measure mRNA levels. The densities of bands in digitized images were measured using the public domain NIH image program on a computer. For all RNA samples, the density of an individual mRNA band was divided by that of the GAPDH mRNA band to correct for differences in RNA loading and/or transfer.
Protein extraction and Western blot analysis
The second aliquots of the three tissue samples (RV, IS, LVFW) were separately suspended in 5 ml of ice-cooled lysis buffer containing (mM)-Tris-HCl 20 (pH 7.4), EDTA 1, NaCl 150, DTT 1, 2-mercaptoethanol 10, freshly added proteinase inhibitor (0.1 mg ml−1 E64, 100 μM pepstatin, 10 μM antipain, 1 mM PMSF, 100 μg ml−1 leupeptin) and disrupted by using a WHEATON-tissue homogenisator (neoLab, Hamburg, Germany). The particulate material was discarded by centrifugation at 100,000×g at 4°C for 1 h in a Beckmann-L8-Ultracentrifuge. The clear supernatant of each tissue sample was collected and aliquots were frozen at −80°C until use. Protein concentration was determined by the method of Bradford (1976) using bovine serum albumin as a standard. All preparations were carried out at 4°C. For Western blotting, 40 μg of total protein solubilized for 5 min at 95°C in one volume loading buffer (1% SDS, 30% glycerol, 0.8 M DTT, 1 mM Tris-HCl pH 6.8, 2% bromophenol-blue) was loaded per lane onto a 10%/5% SDS – PAGE gel. Electrophoresis was performed for 1 h at 150 mA according to the procedure of Laemmli (1970). Proteins were transferred onto Immobilon-P transfer membrane (Millipore, Bedford, MA, U.S.A.) for 1.5 h at 0.8 mA cm−2 in a 20% methanol containing cathodes buffer. To ensure protein loading, the lane containing the control peptide was cut off from each blotted membrane and stained with Ponceau-solution (Sigma, Deisenhofen, Germany) and scanned. The membrane was washed three times for 20 min in TTBS (0.1% Tween 20, 100 mM Tris-HCL, 150 mM NaCl, pH 7.5), blocked for 1 h in 5% nonfat milk-TTBS and incubated with the primary antibody (Chemicon, Hofheim, Germany) for calpain I (mouse monoclonal), calpain II (rabbit polyclonal) or calpastatin (mouse monoclonal). The primary antibody were used in a 1 : 1000 dilution in TTBS. After washing three times in TTBS for 15 min, the membrane was incubated with a 1 : 10,000 dilution of the horseradish-peroxidase coupled secondary antibody (anti-mouse for calpain I and calpastatin, anti-rabbit for calpain II; Amersham, Braunschweig, Germany) for 30 min at room temperature. Following extensive washes (one time 15 min and three times 5 min) in TTBS, the calpains were detected using ECL-reagents (Amersham, Braunschweig, Germany) and exposed to ECL-film according to the manufacturer's instructions. Each membrane was counter blotted with 1 : 5000 dilution of monoclonal anti-β-actin antibodies (Sigma, Deisenhofen, Germany) to ensure same amounts of protein loading on the membranes.
As β-actin has been shown not to be changed in ischaemic rat hearts the signals can be used as an internal control (Gallinat et al., 1998). To measure protein levels, the Western blots were scanned and digitized on an optical scanner (EPSON GT-8000, Seiko, Tokyo, Japan). Quantification of Western blots was done on a computer using the NIH image analysis system (Scion Corporation, Frederick, Maryland, U.S.A.). For all Western blots, the density of the target protein band was divided by that of the β-actin band to correct differences in protein loading and/or transfer.
Haemodynamics and cardiac morphology
In a second set of the study, sham-operated animals received an intravenous infusion of angiotensin II to investigate its effects on transcriptional and translational regulation of calpain I, calpain II and calpastatin. In these experiments, the mRNA expression and protein levels of the three calpains have been determined 3 days after sham surgery in angiotensin II-infused and non-infused rats. To exclude angiotensin II-induced blood pressure-dependent effects on regulation of the cardiac calpains, we found an angiotensin II infusion rate of 1 μg kg−1 h−1 that not significantly increased mean arterial blood pressure (MAP) in normotensive animals. The infusion was started 7 days before surgery via chronically implanted femoral venous catheters connected to subcutaneously implanted osmotic minipumps. At day 3 post sham-operation, MAP was measured in conscious animals 24 h after chronically implanted catheters into the femoral artery using a procedure as described previously (Sandmann et al., 1998). When haemodynamic parameters were recorded the hearts were quickly excised and dissected into the three tissue samples RV, IS and LVFW. Each tissue sample was divided into two parts of equal size to investigate mRNA expression and protein levels of calpain I, calpain II and calpastatin. The number of animals per group was 6 – 8.
To investigate the effects of the ACE-inhibitor and the AT1- or AT2-receptor antagonists on haemodynamic parameters and cardiac morphology in the late phase after induction of MI, MAP of sham-operated and infarcted animals with placebo-, ramipril-, valsartan- or PD 123319-pretreatment was measured 14 days post MI. At this time point, the animals were sacrificed and the hearts were fixed by infusion of 4% phosphate buffered formaldehyde in 0.15 M NaCl, rapidly excised and stored in the same formaldehyde solution for measurement of infarct size and interstitial collagen content of the non-infarcted myocardium. According to the method described by Sandmann et al. (2000), the hearts were cut in a standardized fashion into four transversal slices, embedded in paraffin and stained with the collagen specific stain picrosirius red (Sirius Red, C.I. 3570, Polysciences, Warrington, PA, U.S.A., in aqueous picric acid). The interstitial collagen content of the non-infarcted myocardium was measured using a computerized surface determination method (Quantimet 570 morphometer, Leica, Cambridge Instruments, Ltd., U.K.) and was calculated as the sum of all connective tissue areas divided by the sum of all connective tissue and muscle areas of the non-infarcted myocardium (Junqueira et al., 1979). For measurement of infarct size, the borderline between the infarcted area and the remaining myocardial muscle was marked exactly with a pointer. Infarct size was calculated by the computer program as the percentage of the LV circumference.
Statistics
Computer assisted programs based upon square fitting methods served to analyse RT – PCR standard curves. Statistical evaluation was determined using one-way analysis of variance with repeated measurements (ANOVA). Means shown to be different between individual groups were compared using the post hoc unpaired Student's t-test or the Bonferroni test when appropriate. A probability of P<0.05 or less was considered as significant. Results are expressed as means±standard error of the mean (s.e.mean). Further details of statistical analysis are given in the legends to the figures.
Results
Transcriptional and translational regulation of calpain I, calpain II and calpastatin in the infarcted myocardium
To investigate temporal and regional changes in the transcription and translation of cardiac calpain I, calpain II and calpastatin we performed RT – PCR, Northern blot analysis and Western blot analysis for the three calpains in the tissue samples RV, IS and LVFW 1 day, 3 days, 7 days and 14 days after induction of MI.
Reverse transcription followed by PCR amplification of total RNA resulted in single bands of the predicted size for the three calpains as well as for the house-keeping gene, GAPDH. As shown in Figure 1a, calpain I mRNA abundance steadily increased in the IS of infarcted rat hearts throughout the time course following MI compared to sham-operated rats (upper lane). The message of calpain I was significantly increased on day 3 post MI (about 2 fold) and reached its maximum at 14 days post MI (about 3 fold) compared to sham-operated rats. No significant differences in calpain I mRNA expression were observed in the RV and LVFW (data not shown). In contrast, calpain II mRNA expression in placebo-treated MI animals was increased on day 1 post MI, reached maximum expression (2 fold) on day 3 post MI and remained elevated for 7 days post MI in the LVFW compared to sham-operated rats (Figure 1a, middle lane). Calpain II mRNA expression was unchanged in RV and IS at any time point measured following MI (data not shown). GAPDH mRNA expression of infarcted animals was not affected in the three tissue samples at the four time points studied compared to non-infarcted animals (Figure 1a, lower lane).
Figure 1.
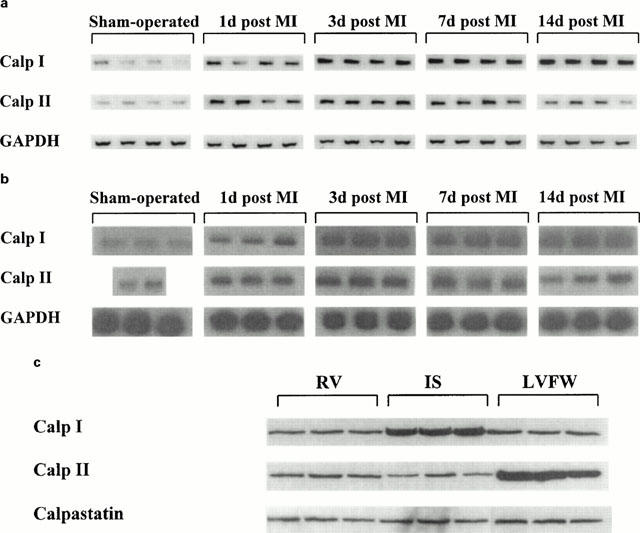
(a) Representative experiments using RT – PCR amplification showing mRNA expression of calpain I (Calp I, upper lane) in the interventricular septum (IS), of calpain II (Calp II, middle lane) in left ventricular free wall (LVFW) and of GAPDH (lower lane) of placebo-treated infarcted animals on day 1, 3, 7 and 14 post MI compared to sham-operated animals (n=6 – 8). (b) Representative experiments using Northern blot analysis showing mRNA expression of calpain I (Calp I, upper lane) in the interventricular septum (IS), of calpain II (Calp II, middle lane) in left ventricular free wall (LVFW) and of GAPDH (lower lane) of placebo-treated infarcted animals on day 1, 3, 7 and 14 post MI compared to sham-operated animals (n=6 – 8). (c) Representative experiments using Western blot analysis showing protein of calpain I 14 days post MI (Calp I, upper lane), of calpain II 3 days post MI (Calp II, middle lane) and of calpastatin (lower lane) in right ventricle (RV), interventricular septum (IS) and left ventricular free wall (LVFW) of placebo-treated infarcted animals (n=6 – 8).
The expression patterns for the three calpains observed using RT – PCR were checked by Northern blot analysis and yielded identical data. The mRNA levels for calpain I showed maximum values 14 days post MI in the IS (Figure 1b, upper lane), but were unchanged in the RV and LVFW. Calpain II mRNA abundance was at most detectable in the LVFW 3 days post MI (Figure 1b, middle lane), whereas no significant differences were observed in RV and IS. GAPDH mRNA was constantly expressed in the three tissue samples at all four time points measured (Figure 1b, lower lane). Calpastatin mRNA expression of infarcted rats studied by RT – PCR and Northern blot analysis was unchanged in the three tissue samples throughout the 14 days post MI period when compared to sham-operated rats (data not shown).
To determine whether alterations in calpain I, calpain II and calpastatin expression after MI result in changes of protein levels, Western blot analysis was performed. In preliminary experiments, the amount of 40 μg of total protein loading was determined to detect relative differences in translational expression of the three proteins. The density of the digitized signals was quantified with respect to the density of the β-actin signals. Immunostaining of the nylon-fixed proteins probed with specific primary antibodies for the large subunit of calpain I and II resulted in a single band of 80 kDa for each protein. As demonstrated for mRNA expression, calpain I protein levels were maximally increased in the IS of the infarcted rat heart 14 days after MI (Figure 1c, upper lane), whereas calpain II protein levels were highest detectable in the LVFW on day 3 post MI (Figure 1c, middle lane). Western blotting of calpastatin showed a single band of 70 kDa. No differences in protein levels for calpastatin were found in RV, IS and LVFW of sham-operated and infarcted animals at any time point measured (Figure 1c, lower lane). The protein amount of β-actin was unchanged in RV, IS and LVFW of infarcted rat hearts at the four time points measured.
Effects of angiotensin II on transcriptional and translational regulation of cardiac calpain I, calpain II and calpastatin
To investigate whether the mRNA and protein up-regulation of calpain I and calpain II depends on MI-induced stimulation of the renin-angiotensin-system (RAS), sham-operated rats were infused with angiotensin II. As shown in Figure 2, calpain I mRNA expression and protein levels (upper lanes) were significantly increased in the IS 3 days after surgery in chronic angiotensin II-infused sham-operated animals (right panel) compared to sham-operated animals without angiotensin II infusion (left panel). Additionally, Calpain II mRNA expression and protein levels (Figure 2, lower lanes) were increase in LVFW 3 days post operation in angiotensin II-infused sham-operated animals (right panel) compared to non-angiotensin II-infused sham-operated animals (left panel). In these experiments, a non blood pressure increasing infusion rate of angiotensin II (1 μg kg−1 h−1) was used to prevent angiotensin II-induced blood pressure-dependent effects on transcriptional and translational regulation of calpain I and II (Figure 2). These results demonstrate that angiotensin II stimulates the up-regulation of calpain I and II in the rat myocardium.
Figure 2.

Representative experiments showing mRNA expression (upper lane, PCR) and protein levels (lower lane, WB) of Calpain I (Calp I) in interventricular septum (IS) and of calpain II (Calp II) in left ventricular free wall (LVFW) 3 days post MI of sham-operated animals without (left panel) or with (right panel) infusion of angiotensin II. Using an infusion rate of 1 μg kg−1 h−1 angiotensin II, mean arterial blood pressure (MAP) was not significantly increased in sham-operated animals (n=6 – 8).
Effects of ACE inhibitor, AT1 and AT2 receptor antagonists on cardiac calpain I, calpain II and calpastatin mRNA expression
To investigate which angiotensin receptor subtype was linked to the up-regulation of calpain I and II mRNA expression in the infarcted myocardium, animals were pretreated 1 week before and up to sacrifice post MI either with the ACE inhibitor, ramipril, or with the selective AT1 and AT2 receptor antagonists, valsartan and PD 123319, respectively. The results illustrated in Figure 3a indicate that, in the IS, the increase of mRNA expression of calpain I was significantly reduced by ramipril and valsartan on day 14 post MI but not by PD 123319 compared to placebo-treated MI animals. As shown in Figure 3b, pretreatment for 1 week followed by treatment post MI up to sacrifice with the ACE inhibitor, ramipril, and the AT1 receptor antagonist, valsartan, totally abolished calpain II mRNA up-regulation in LVFW on day 3 post MI, whereas the AT2 receptor antagonist, PD 123319, did not affect the upregulation of calpain II mRNA expression in LVFW of infarcted animals 3 days post MI. These results illustrate that calpain I and II up-regulation in the infarcted and non-infarcted myocardium is mediated via the AT1 receptor, whereas the AT2 receptor seems not to be involved in the up-regulation of the cardiac calpains.
Figure 3.
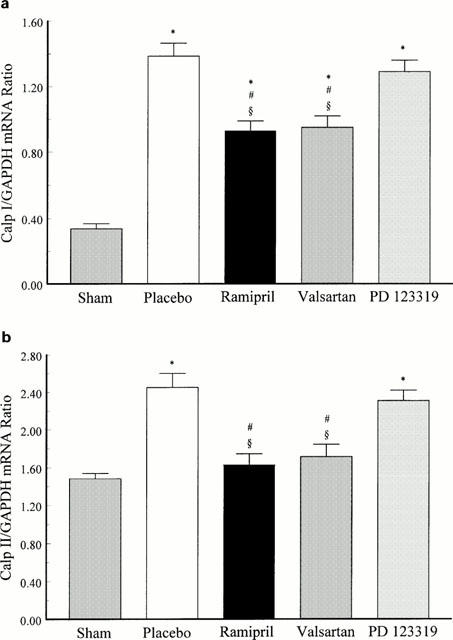
(a) Relative changes of calpain I (Calp I) mRNA expression in the interventricular septum (IS) determined by densitometric analysis and normalized to GAPDH signals of sham-operated and infarcted animals treated either with the ACE inhibitor ramipril (1 mg kg−1 d−1), the AT1 receptor antagonist valsartan (10 mg kg−1 d−1) and the AT2 receptor antagonist PD 123319 (30 mg kg−1 d−1) 14 days post MI. *P<0.05 compared to sham; □num;P<0.05 compared to placebo; §P<0.05 compared to PD 123319. Data represent mean±s.e.mean.; n=6 – 8. (b) Relative changes of calpain II (Calp II) mRNA expression in the left ventricular free wall (LVFW) determined by densitometric analysis and normalized to GAPDH signals of sham-operated and infarcted animals treated either with the ACE inhibitor ramipril (1 mg kg−1 d−1), the AT1 receptor antagonist valsartan (10 mg kg−1 d−1) and the AT2 receptor antagonist PD 123319 (30 mg kg−1 d−1) 3 days post MI. *P<0.05 compared to sham; □num;P<0.05 compared to placebo; §P<0.05 compared to PD 123319. Data represent mean±s.e.mean.; n=6 – 8.
The message of calpastatin was unaffected by treatment with ramipril, valsartan and PD 123319 compared to placebo treatment (data not shown).
Effects of ACE inhibitor, AT1 and AT2 receptor antagonists on protein levels of cardiac calpain I, calpain II and calpastatin
As demonstrated for calpain I mRNA expression, pretreatment of infarcted animals with the ACE inhibitor, ramipril, and the AT1 receptor antagonist, valsartan, but not with the AT2 receptor antagonist, PD 123319, reduced the post MI up-regulation of the calpain I protein in IS 14 days post MI compared to placebo-treated animals (Figure 4a). Similarly to mRNA expression, the increase in calpain II protein levels in the LVFW of infarcted rat hearts 3 days post MI was attenuated by ramipril and valsartan, but not by PD 123319 compared to placebo (Figure 4b). These results show that ACE inhibitor and AT1 receptor antagonist reduced translational up-regulation of cardiac calpain I and II following MI.
Figure 4.
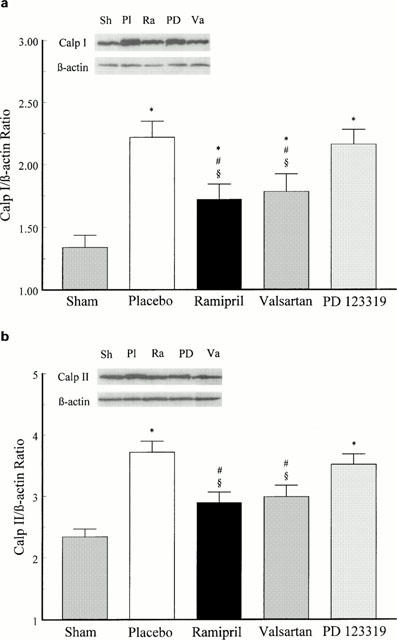
(a) Effects of chronic treatment with the ACE inhibitor ramipril (1 mg kg−1 d−1), the AT1 receptor antagonist valsartan (10 mg kg−1 d−1) and the AT2 receptor antagonist PD 123319 (30 mg kg−1 d−1) on calpain I (Calp I) protein levels in the interventricular septum (IS) 14 days post MI compared to sham-operated and placebo-treated infarcted animals. *P<0.05 compared to sham; #P<0.05 compared to placebo; §P<0.05 compared to PD 123319. Data represent mean±s.e.mean; n=6 – 8. (b) Effects of chronic treatment with the ACE inhibitor ramipril (1 mg kg−1 d−1), the AT1 receptor antagonist valsartan (10 mg kg−1 d−1) and the AT2 receptor antagonist PD 123319 (30 mg kg−1 d−1) on calpain II (Calp II) protein levels in the left ventricular free wall (LVFW) 3 days post MI compared to sham-operated and placebo-treated infarcted animals. *P<0.05 compared to sham; #P<0.05 compared to placebo; §P<0.05 compared to PD 123319. Data represent mean±s.e.mean; n=6 – 8.
Calpastatin protein concentration in the myocardium was unaffected by chronic treatment with ramipril, valsartan or PD 123319 (data not shown).
Effects of ACE inhibitor, AT1 and AT2 receptor antagonists on haemodynamics and cardiac morphology
Fourteen days post MI, a decrease in mean arterial blood pressure (MAP) was observed in all infarcted animals (placebo-, ramipril-, valsartan- and PD 123319-treated) when compared to sham-operated animals indicating that LV dysfunction was present in these animals following MI (Figure 5). The fact, that MAP of infarcted animals chronically treated with ramipril, valsartan and PD 123319 was significantly elevated compared to placebo-treated animals suggests that pretreatment with these drugs preserved cardiac function post MI (Figure 5).
Figure 5.
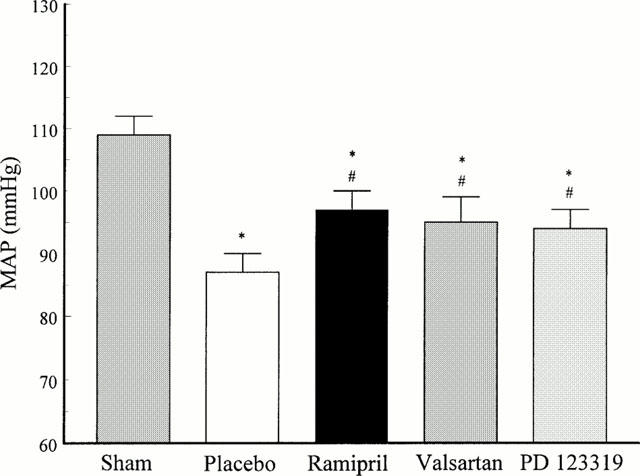
Mean arterial blood pressure (MAP) 14 days post MI of sham-operated and infarcted animals with placebo-, ramipril-, valsartan- and PD 123319-treatment. *P<0.05 compared to sham; #P<0.05 compared to placebo; Data represent mean±s.e.mean; n=6 – 8.
When haemodynamics were recorded, infarct size and interstitial collagen content of the non-infarcted myocardium were investigated to determine the degree of structural cardiac remodeling 14 days post MI. The histology of picrosirius red stained sections of slice 3 showed a transmural MI in all animals. Placebo-treated MI animals had an infarct size of 50%. Fourteen days post MI, infarct size of ramipril- and valsartan-treated infarcted animals was significantly reduced compared to placebo-treated animals. Treatment of MI animals with PD 123319 had no effect on infarct size (Figure 6a). The interstitial collagen content of the non-infarcted myocardium was increased in placebo-treated MI animals compared to sham-operated animals. Chronic treatment of infarcted animals with ramipril, valsartan and PD 123319 decreased the collagen content in the interstitium of the non-infarcted myocardium (Figure 6b). These results demonstrate that ACE inhibition and AT1 receptor blockade reduced infarct size and interstitial fibrosis whereas AT2 receptor blockade only decreased interstitial fibrosis post MI.
Figure 6.
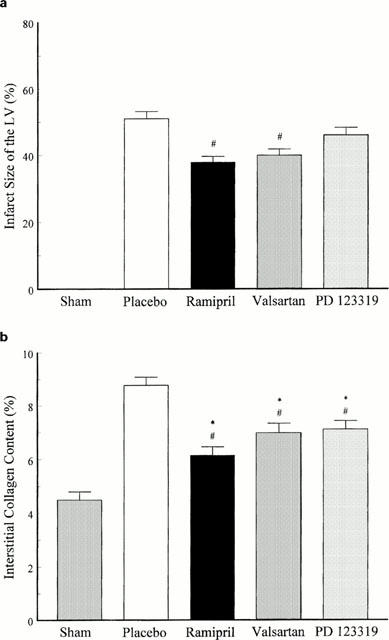
(a) Infarct size determined as percentage of the left ventricle (LV) 14 days post MI of placebo-, ramipril-, valsartan- and PD 123319-treated MI animals. #P<0.05 compared to placebo; Data represent mean±s.e.mean; n=6 – 8. (b) Interstitial collagen content in the non-infarcted myocardium 14 days post MI of placebo-, ramipril-, valsartan- and PD 123319-treated MI animals. #P<0.05 compared to placebo; Data represent mean±s.e.mean; n=6 – 8.
Discussion
In the present study we demonstrate for the first time a differential temporal and regional up-regulation of calpain I and II in the rat heart after myocardial infarction (MI). Our data show that MI induced an increase in calpain I gene expression and protein levels in the non-ischaemic septal region during the chronic state of MI (14 days post MI). In contrast, transcriptional and translational up-regulation of calpain II occurred at an earlier stage post MI in the ischaemic region of the infarcted myocardium with highest values 3 days post MI. These results indicate that cardiac calpain I and II appear to be differentially regulated at the gene and protein level after the ischaemic insult. This finding correlates with previous studies demonstrating that calpain I is up-regulated by sustained stimuli and contributes to the regulated protein turn-over during differentiation or hypertrophy (Pussard et al., 1993; Arthur & Belcastro, 1997). In view of earlier experiments demonstrating that MI induces cardiac hypertrophy of the non-infarcted myocardium (Sandmann et al., 1998) our data are consisting with the idea that calpain I may be involved in the structural remodelling process of the heart in the late phase after MI. It has been speculated that the elevated left ventricular diastolic and/or systolic pressure during heart failure which determines the inner ventricular wall stress of the infarcted heart might be responsible for transcriptional and translational up-regulation of calpain I in the non-infarcted area of the myocardium (Contard et al., 1991; Pfeffer & Braunwald, 1990).
In contrast to calpain I, calpain II mRNA and protein levels were increased in the ischaemic myocardium of the infarcted heart during the early phase after MI. Calpain II has been shown to be up-regulated by processes which are associated with elevated intracellular free Ca2+-concentrations ([Ca2+]i) (Pussard et al., 1993). The ligation of the left coronary artery, as performed in the present experiments, is thought to be associated with an intracellular accumulation of Ca2+ within the ischaemic region of the myocardium (Katz & Reuter, 1979). Thus, the MI-induced rise in myocardial [Ca2+]i as a result of increased Ca2+-influx through calcium channels and/or elevated Ca2+-release by the sarcoplasmatic reticulum (Wier, 1990) might be responsible for the increased expression of calpain II within the ischaemic myocardium. Additionally, intracellular acidosis stimulates pH-regulatory ion transporters such as Na+-H+-exchanger, which induce the Na+-dependent activation of Na+-Ca2+-exchanger leading to an intracellular Ca2+-overload (Siffert & Akkerman, 1989). Ca2+-overload has been shown to act as a specific mechanism to activate calpain II (Toyo-oka & Ross, 1981; Steenbergen et al., 1987). It is possible that the MI-induced increase in calpain II protein production as well as the rise of [Ca2+]i leads to an abnormal calpain II activity in the ischaemic myocardium which, in turn, accelerates tissue injury following MI. Indeed, calcium channel blockers have been demonstrated to reduce calpain activity during ischaemia via inhibition of Ca2+-influx through L-type calcium channels (Yoshida et al., 1993). Additionally, calpain inhibitors have been found to effectively protect the myocardium from damage after MI via reduction of calpain activity (Urthaler et al., 1997; Iwamoto et al., 1999).
A central aim of our study was to investigate the role of the RAS in the regulation of calpain I, calpain II and calpastatin by using an in vivo animal model of MI. The background of the present study was that the angiotensin receptors subtypes, referred to as AT1 and AT2, have been shown to be up-regulated in the rat myocardium during the early phase after MI (Nio et al., 1995). ACE inhibition has been shown to have an influence on cardiac remodelling and to prolong survival in experimental MI (Sladek et al., 1996; Fleetwood et al., 1991) as well as in patients with LV dysfunction (Pfeffer et al., 1992; The AIRE Study Investigators, 1993). Further, many reports suggest that the AT1 receptor subtype plays a major role in cardiac pathophysiology, and the administration of AT1 receptor antagonists have been shown to reduce infarct size (Jalowy et al., 1998), left ventricular loading (Ford et al., 1998) and to improve haemodynamics and coronary angiogenesis (Sladek et al., 1996; Kuizinga et al., 1998) after MI. In isolated ischaemic rat hearts, the cardioprotective effect of AT1 receptor blockade may be mediated in part by endogenously released angiotensin II via AT2 receptor stimulation (Wiemer et al., 1993). However, the role of cardiac AT2 receptors in the control of heart function and cardiac structure remains to be clarified.
Our data point to a participation of the angiotensin AT1 receptor, but not of the angiotensin AT2 receptor, in the regulation of the calpains in the ischaemic myocardium. The exact mechanism underlying these events has not been established at present. However, our observation that an angiotensin II infusion induced mRNA and protein up-regulation of cardiac calpain I and II and that the ACE inhibitor, ramipril, inhibited the up-regulation of the calpains supports the hypothesis that angiotensin II is involved in transcriptional and translational control of the calpain system in the ischaemic heart. Our present data show that the angiotensin II-induced stimulation of calpain I and II message and protein is mediated in part via activation of the AT1 receptor as evidenced by the fact that AT1 receptor blockade by valsartan reduced calpain I and II up-regulation. These results indicate that angiotensin II, through the AT1 receptor, is crucial for the enhanced expression of both calpain isoforms post MI. The inhibited up-regulation of calpain I in the non-infarcted myocardium and of calpain II in the LVFW by ramipril and valsartan correlated with the limiting effect of these two drugs on interstitial fibrosis and infarct size. This effect was accompanied by an increase in MAP in these animals. Thus, our results suggest that chronic ACE inhibition and AT1 receptor blockade reduced cardiac remodelling and improved cardiac function following MI. On the other hand, the up-regulation of the calpains was not affected by the AT2 receptor antagonist, PD 123319, suggesting that this angiotensin receptor subtype does not contribute to the regulation of calpain expression. These findings implicate a tissue protective effect of ACE inhibitors and AT1 receptor antagonists via attenuation of MI-induced up-regulation and/or activation of cardiac calpains.
Calpastatin expression, on the other hand, was not up-regulated whether in the ischaemic nor in the non-ischaemic myocardium, at mRNA or protein levels suggesting that transcriptional and translational expression of the naturally occurring calpain inhibitor was not affected in MI-induced heart failure. These results are in keeping with findings of Sorimachi et al. (1997) showing that cardiac calpastatin was not up-regulated in the ischaemic-reperfused rat heart model. On the other hand, calpastatin has been shown to be a substrate for calpain (Doumit & Koohmaraie, 1999) implying that the ischaemia-induced increase in calpain expression and activity seems to be responsible for abnormal calpastatin degradation in the myocardium. Thus, it can be speculated that the calpain – calpastatin-ratio might be shifted to more calpain leading to abnormal protein degradation and increased myocardial damage during cardiac ischaemia. Therefore, the reduction of calpain gene and protein expression by ACE inhibition and AT1 receptor antagonism might be cardioprotective via decreasing the amount of activable calpain levels within the myocardial tissue and preserve cardiac function in MI-induced heart failure via prevention of myocardial protein degradation and possible limitation of cardiac remodelling and infarct size.
In conclusion, the results of the present study demonstrate first, that calpain I mRNA and protein levels are increased in the late phase of MI in the cardiac interventricular septum but not in the left ventricular free wall and right ventricle. In contrast, calpain II is up-regulated during the acute phase after MI in the ischaemic myocardium. Second, treatment with an ACE inhibitor or AT1 receptor antagonist effectively reduces the increased expression and translation of calpain I and II whereas treatment with AT2 receptor antagonist has no effect on the up-regulation of the calpains in the infarcted heart. The results suggest that the calpain I-induced structural remodelling of the failing heart and the calpain II-dependent myocardial cell injury in the ischemic region of the infarcted heart are under the control of angiotensin II via its AT1 receptors and can be prevented either by ACE inhibition or AT1 receptor blockade. Additionally, our findngs suggest that the cardioprotective action of ACE inhibition and AT1 receptor blockade in the MI-induced heart failure can be partially explained by reduction of calpain-mediated cardiac damage and remodelling.
Acknowledgments
The authors thank Aventis (Frankfurt/Main, Germany), Novartis (Basel, Switzerland) and Park Davis (Ann Arbor, MI, U.S.A.) for their kind gifts of ramipril, valsartan and PD 123319, respectively.
Abbreviations
- ACE
angiotensin converting enzyme
- AT1
angiotensin type 1 receptor
- AT2
angiotensin type 2 receptor
- [Ca2+]i
intracellular calcium concentration
- Calp I
calpain isoform I or μ-calpain
- Calp II
calpain isoform II or m-calpain
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- IS
interventricular septum
- LV
left ventricle
- LVFW
left ventricular free wall
- MAP
mean arterial blood pressure
- MI
myocardial infarction
- RAS
renin-angiotensin system
- RV
right ventricle
References
- ARTHUR G.D., BELCASTRO A.N. A calcium stimulated cysteine protease involved in isoproterenol induced cardiac hypertrophy. Mol. Cell. Biochem. 1997;176:241–248. [PubMed] [Google Scholar]
- BRADFORD M.M. A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- CHOMCZYNSKI P., SACCHI N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- CONTARD F., KOTELIANSKY V., MAROTTE F., DUBUD I., RAPPAPORT L., SAMUEL J.L. Specific alterations in the distribution of extracellular matrix components within rat myocardium during the development of pressure overload. Lab. Invest. 1991;64:65–75. [PubMed] [Google Scholar]
- DEAR N., MATENA K., VINGRON M., BOEHM T. A new subfamily of vertebrate calpains lacking a calmodulin-like domain: implications for calpain regulation and evolution. Genomics. 1997;45:175–184. doi: 10.1006/geno.1997.4870. [DOI] [PubMed] [Google Scholar]
- DOUMIT M.E., KOOHMARAIE M. Immunoblot analysis of calpastatin degradation: evidence for cleavage by calpain in post mortem muscle. J. Anim. Sci. 1999;77:1467–1473. doi: 10.2527/1999.7761467x. [DOI] [PubMed] [Google Scholar]
- FLEETWOOD G., BOUTINET S., MEIER M., WOOD J.M. Involvement of the renin-angiotensin system in ischemic damage and reperfusion arrhythmias in the isolated perfused rat heart. J. Cardiovasc. Pharmacol. 1991;17:351–356. doi: 10.1097/00005344-199103000-00001. [DOI] [PubMed] [Google Scholar]
- FORD W.R., KHAN M.I., JUGDUTT B.I. Effect of the novel angiotensin II type-1 receptor antagonist L-158,809 on acute infarct expansion and acute anterior myocardial infarction in the dog. Can. J. Cardiol. 1998;14:73–80. [PubMed] [Google Scholar]
- GALLINAT S., MINGHUAN Y., DORST A., UNGER Th., HERDEGEN Th. Sciatic nerve transection evokes lasting up-regulation of angiotensin AT2 and AT1 receptor mRNA in adult rat dorsal root ganglia and sciatic nerves. Mol. Brain. Res. 1998;57:111–122. doi: 10.1016/s0169-328x(98)00079-5. [DOI] [PubMed] [Google Scholar]
- GALLINAT S., BUSCHE S., SCHÜTZE S., KRÖNKE M., UNGER Th. AT2 receptor stimulation induces generation of ceramides in PC 12W cells. FEBS Lett. 1999;443:75–79. doi: 10.1016/s0014-5793(98)01675-5. [DOI] [PubMed] [Google Scholar]
- GOHLKE P., LINZ W., SCHÖLKENS B.A., WIEMER G., MARTORANA P., VAN EVEN P., UNGER Th. Effect of chronic high- and low-dose ACE inhibitor treatment on cardiac and vascular hypertrophy and vascular function in spontaneously hypertensive rats. Exp. Nephrol. 1994;2:93. [PubMed] [Google Scholar]
- GOHLKE P., PEES C., UNGER Th. AT2 receptor stimulation increases aortic cyclic GMP in SHRSP by a kinin-dependent mechanism. Hypertension. 1998;31:349–355. doi: 10.1161/01.hyp.31.1.349. [DOI] [PubMed] [Google Scholar]
- HAYASHI N., YAMAMOTO S., KOMETANI M., NAKAO K. Pharmacological profile of valsartan, a non-peptide angiotensin II type 1 receptor antagonist. Third communication: hemodynamic effects of valsartan in rats and dogs. Arzneimittelforschung. 1997;47:620–625. [PubMed] [Google Scholar]
- HIRSCH A.T., TALSNESS C.E., SCHUNKERT H., PAUL M., DZAU V.J. Tissue-specific activation of cardiac angiotensin converting enzyme in experimental heart failure. Circ. Res. 1991;69:475–482. doi: 10.1161/01.res.69.2.475. [DOI] [PubMed] [Google Scholar]
- ISHIURA S., SUGITA H.Duchenne muscular dystrophy No To Shinkei. 199143405–409.(5) [PubMed] [Google Scholar]
- IWAMOTO H., MIURA T., OKAMURA T., SHIRAKAWA K., IWATATE M., KAWAMURA S., TATSUNO H., IKEDA Y., MATSUZAKI M. Calpain inhibitor-1 reduces infarct size and DNA fragmentation of myocardium in ischemia/reperfusion rat heart. J. Cardiovasc. Pharmacol. 1999;33:580–586. doi: 10.1097/00005344-199904000-00010. [DOI] [PubMed] [Google Scholar]
- JALOWY A., SCHULZ R., DORGE H., BEHRENDS M., HEUSCH G. Infarct size reduction by AT1-receptor blockade through a signal cascade of AT2-receptor activation, bradykinin and prostaglandins in pigs. J. Am. Coll. Cardiol. 1998;32:1787–1796. doi: 10.1016/s0735-1097(98)00441-0. [DOI] [PubMed] [Google Scholar]
- JU H., ZHAO S., TAPPIA P.S., PANAGIA V., DIXON I.M. Expression of Gq alpha and PLC-beta in scar and border tissue in heart failure due to myocardial infarction. Circulation. 1998;97:892–899. doi: 10.1161/01.cir.97.9.892. [DOI] [PubMed] [Google Scholar]
- JUNQUEIRA L.C., BIGNOLAS G., BRENTANI R.R. Red sirius staining plus polarizing microscopy: a specific method for collagen detection in tissue sections. Histochem. J. 1979;79:445–447. doi: 10.1007/BF01002772. [DOI] [PubMed] [Google Scholar]
- KANG J., SUMNERS C., POSNER P. Angiotensin II type 2 receptor-modulated changes in potassium currents in cultured neurons. Am. J. Physiol. 1993;265:C607–C616. doi: 10.1152/ajpcell.1993.265.3.C607. [DOI] [PubMed] [Google Scholar]
- KATZ A.M., REUTER H. Cellular calcium and cardiac cell death. Am. J. Cardiol. 1979;44 Suppl. 1:188–190. doi: 10.1016/0002-9149(79)90270-4. [DOI] [PubMed] [Google Scholar]
- KUIZINGA M.C., SMITS J.F.M., ARENDS J.W., DAEMEN M.J.A.P. AT2 receptor blockade reduces cardiac interstitial cell DNA synthesis and cardiac function after rat myocardial infarction. J. Mol. Cell. Cardiol. 1998;30:425–434. doi: 10.1006/jmcc.1997.0607. [DOI] [PubMed] [Google Scholar]
- LAEMMLI U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- LINDPAINTER K., LU W., NEIDERMAIER N., SCHIEFFER B., JUST H., GANTEN D., DREXLER H. Selective activation of cardiac angiotensinogen gene expression in post-infarction ventricular remodeling in the rat. J. Mol. Cell. Cardiol. 1993;25:133–143. doi: 10.1006/jmcc.1993.1017. [DOI] [PubMed] [Google Scholar]
- LUCIUS R., GALLINAT S., ROSENSTIEL P., HERDEGEN Th., SIEVERS J., UNGER Th. The angiotensin II type 2 (AT2) receptor promotes axonal regeneration in the optic nerve of adult rats. J. Exp. Med. 1998;188:661–670. doi: 10.1084/jem.188.4.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MA H., FUKIAGE C., AZUMA M., SHEARER T.R. Cloning and expression of mRNA for calpain Lp82 from rat lens: splice variant of p94. Invest. Ophthalmol. Visual. Sci. 1998;39:454–461. [PubMed] [Google Scholar]
- MEFFERT S., STOLL M., STECKELINGS U.M., BOTTARI S.P., UNGER Th. The angiotensin II AT2 receptors inhibits proliferation and promotes differentiation in PC 12W cells. Mol. Cell. Endocr. 1996;112:59–67. doi: 10.1016/0303-7207(96)03873-7. [DOI] [PubMed] [Google Scholar]
- MEGGS L.G., COUPET J., HUANG H., CHENG W., LI P., CAPASSO J.M., HOMCY C.J., ANVERSA P. Regulation of angiotensin II receptors on ventricular myocytes after myocardial infarction in rats. Circ. Res. 1993;72:1149–1162. doi: 10.1161/01.res.72.6.1149. [DOI] [PubMed] [Google Scholar]
- MUGITA N., KIMURA Y., OGAWA M., SAYA H., NAKAO M. Identification of a novel, tissue-specific calpain htra-3; a human homologue of the Caenorhabditis elegans sex determination gene. Biochem. Biophys. Res. Commun. 1997;239:845–850. doi: 10.1006/bbrc.1997.7571. [DOI] [PubMed] [Google Scholar]
- NAYLER W.G. The role of calcium in the ischemic myocardium. Am. J. Pathol. 1981;102:262–270. [PMC free article] [PubMed] [Google Scholar]
- NIO Y., MATSUBARA H., MURASAWA S., KANASAKI M., INADA M. Regulation of gene transcription of angiotensin II receptor subtypes in myocardial infarction. J. Clin. Invest. 1995;95:46–54. doi: 10.1172/JCI117675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PFEFFER M.A., BRAUNWALD E. Ventricular remodeling after myocardial infarction. Circulation. 1990;81:1161–1172. doi: 10.1161/01.cir.81.4.1161. [DOI] [PubMed] [Google Scholar]
- PFEFFER M.A., BRAUNWALD E., MOYE L.A., BASTA L., BROWN E.J., Jr, CUDDY T.E., DAVIS B.R., GELTMAN E.M., GOLDMAN S., FLAKER G.C., KLEIN M., LAMAS G.A., PACKER M., ROULEAU J., ROULEAU J.L., RUTHERFORD J., WERTHEIMER J.H., HAWKINS C.M. Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. Results of the survival and ventricular enlargement trial. The SAVE Investigators. N. Engl. J. Med. 1992;327:669–677. doi: 10.1056/NEJM199209033271001. [DOI] [PubMed] [Google Scholar]
- PUSSARD S., COTTIN P., BRUSTIS J.J., TALMAT S., ELAMRANI N., DUCASTAING A. Quantitative measurement of calpain I and II mRNAs in differentiating rat muscle cells using a competitive polymerase chain reaction method. Biochemistry. 1993;75:885–890. doi: 10.1016/0300-9084(93)90044-s. [DOI] [PubMed] [Google Scholar]
- RECHSTEINER M., ROGERS S.W. PEST sequences and regulation by proteolysis. Trends Biochem. Sci. 1996;21:267–271. [PubMed] [Google Scholar]
- SANDMANN S., SPITZNAGEL H., CHUNG O., XIA Q.-G., ILLNER S., JÄNICHEN G., ROSSIUS B., DAEMEN M.J.A.P., UNGER Th. Effects of the calcium channel blocker mibefradil on haemodynamic and morphological parameters in myocardial infarction-induced cardiac failure in rats. Cardiovasc. Res. 1998;39:339–350. doi: 10.1016/s0008-6363(98)00087-x. [DOI] [PubMed] [Google Scholar]
- SANDMANN S., BOHLE R.M., DREYER Th., UNGER Th. The T-type calcium channel blocker mibefradil reduced interstitial and perivascular fibrosis and improved haemodynamic parameters in myocardial infarction-induced cardiac failure in rats. Virchows Arch. 2000;436:147–157. doi: 10.1007/pl00008215. [DOI] [PubMed] [Google Scholar]
- SIFFERT W., AKKERMAN J.W.N. Na+/H+ exchange and Ca2+ influx. FEBS Let. 1989;259:1–4. doi: 10.1016/0014-5793(89)81480-2. [DOI] [PubMed] [Google Scholar]
- SLADEK T., SLADKOVA J., KOLAR F., PAPOUSEK F., CICUTTI N., KORECKY B., RAKUSAN K. The effect of AT1 receptor antagonist on chronic cardiac response to coronary artery ligation in rats. Cardiovasc. Res. 1996;31:568–576. [PubMed] [Google Scholar]
- SORIMACHI H., IMAJOH-OHMI S., EMORI Y., KAWASAKI H., OHNO S., MINAMI Y., SUZUKI K. Molecular cloning of a novel mammalian calcium-dependent protease distinct from both m- and mu-type: specific expression of the mRNA in skeletal muscle. J. Biol. Chem. 1989;264:20106–20111. [PubMed] [Google Scholar]
- SORIMACHI H., SAIDO T.C., SUZUKI K. New era of calpain research. FEBS Lett. 1994;343:1–5. doi: 10.1016/0014-5793(94)80595-4. [DOI] [PubMed] [Google Scholar]
- SORIMACHI Y., HARADA K., SAIDO T.C., ONO T., KAWASHIMA S., YOSHIDA K. Downregulation of calpastatin in rat heart after brief ischemia and reperfusion. J. Biochem. (Tokyo) 1997;122:743–748. doi: 10.1093/oxfordjournals.jbchem.a021818. [DOI] [PubMed] [Google Scholar]
- STEENBERGEN C., MURPHY E., LEVY L., LONDON R.E. Elevation of cytosolic free calcium concentration early in myocardial ischemia in perfused rat heart. Circ. Res. 1987;60:700–707. doi: 10.1161/01.res.60.5.700. [DOI] [PubMed] [Google Scholar]
- STEENBERGEN C., MURPHY E., WATTS J.A., LONDON R.E. Correlation between cytosolic free calcium, contracture, ATP, and irreversible ischemic injury in perfused rat hearts. Circ. Res. 1990;66:135–146. doi: 10.1161/01.res.66.1.135. [DOI] [PubMed] [Google Scholar]
- STOLL M., STECKELINGS U.M., PAUL M., BOTTARI S.P., METZGER R., UNGER Th. The angiotensin AT2-receptor mediates inhibition of cell proliferation in coronary endothelial cells. J. Clin. Invest. 1995;95:651–657. doi: 10.1172/JCI117710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUZUKI K., IMAJOH S., EMORI Y., KAWASAKI H., MINAMI Y., OHNO S. Calcium activated neutral protease and its endogenous inhibitor: activation at the cell membrane and biological function. FEBS Lett. 1987;220:271–277. doi: 10.1016/0014-5793(87)80828-1. [DOI] [PubMed] [Google Scholar]
- The AIRE Study Investigators Effect of ramipril on mortality and morbidity of survivors of acute myocardial infarction with clinical evidence of heart failure. Lancet. 1993;342:821–828. [PubMed] [Google Scholar]
- TIMMERMANS P.B.M.W.M., WONG P.C., CHIU A.T., HERBLIN W.F., BENFIELD P., CARINI D.J., LEE R.J., WEXLER R.R., SAYE J.A.M., SMITH R.D. Angiotensin II receptors and angiotensin II receptor antagonists. Pharmacol. Rev. 1993;45:205–251. [PubMed] [Google Scholar]
- TOYO-OKA T., ROSS J.R. Ca2+ sensitivity change and troponin loss in cardiac natural actomyosin after coronary occlusion. Am. J. Physiol. 1981;240:H704–H708. doi: 10.1152/ajpheart.1981.240.5.H704. [DOI] [PubMed] [Google Scholar]
- URTHALER F., WOLKOWICZ P.E., DIGERNESS S.B., HARRIS K.D., WALKER A.A. MDL-28170, a membrane-permeant calpain inhibitor, attenuates stunning and PKCε proteolysis in reperfused ferret hearts. Cardiovasc. Res. 1997;35:60–67. doi: 10.1016/s0008-6363(97)00099-0. [DOI] [PubMed] [Google Scholar]
- WANG K.K.W., YUEN P. Calpain inhibitors: an overview of its therapeutic potential. TiPS. 1994;15:412–419. doi: 10.1016/0165-6147(94)90090-6. [DOI] [PubMed] [Google Scholar]
- WIEMER G., SCHÖLKENS B.A., WAGNER A., HEITSCH H., LINZ W. The possible role of Ang II subtype AT2 receptors in endothelial cells and isolated ischemic rat hearts. J. Hypertens. 1993;11 Suppl. 5:S234–S235. [PubMed] [Google Scholar]
- WIER W.G. Cytosolic [Ca2+] in mammalian ventricle: dynamic control by cellular process. Ann. Rev. Physiol. 1990;52:467–485. doi: 10.1146/annurev.ph.52.030190.002343. [DOI] [PubMed] [Google Scholar]
- YAMADA T., HORIUCHI M., DZAU V.J. Angiotensin II type 2 receptor mediates programmed cell death. Proc. Natl. Acad. Sci. U.S.A. 1996;93:156–160. doi: 10.1073/pnas.93.1.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YOSHIDA K., YAMASAKI Y., KAWASHIMA S. Calpain activity alters in rat myocardial subfractions after ischemia or reperfusion. Biochim. Biophys. Acta. 1993;1182:215–220. doi: 10.1016/0925-4439(93)90143-o. [DOI] [PubMed] [Google Scholar]