

Abstract
Somatostatin and the stable octapeptide analogues, octreotide and angiopeptin, were examined for their ability to stimulate the release of tritium from [3H]-arachidonic acid pre-loaded CHO-K1 cells expressing human recombinant sst2, sst3 or sst5 receptors.
Somatostatin stimulated tritium release (pEC50) through the sst2 (7.8±0.1) and sst5 (7.3±0.2), but not the sst3 receptor. Octreotide behaved as a full (sst2 receptor) or partial agonist (sst5 receptor), whereas angiopeptin behaved as a weak partial agonist at both receptor types.
Maximum responses to somatostatin through both receptor types were significantly reduced by pertussis toxin, whereas pEC50 estimates were unaffected.
Inhibition of MEK1 or Src, but not PKA, PI 3-kinases or tyrosine kinases, by reportedly selective inhibitors reduced sst2-mediated responses by somatostatin, but not angiopeptin. A selective inhibitor of PKC (Ro-31-8220) reduced both somatostatin and angiopeptin responses.
These data provide further evidence for partial agonist activity of synthetic peptides of somatostatin. Furthermore, the somatostatin receptor signalling mechanisms which mediate arachidonic acid mobilization appear to be multiple and complex.
Keywords: Somatostatin; angiopeptin, octreotide, arachidonic acid; sst2 receptor; sst5 receptor
Introduction
Somatostatin (SRIF; somatotrophin release inhibiting factor) receptors (sst1 – sst5) couple to multiple transduction pathways, including the inhibition of adenylate cyclase (Patel et al., 1994; Carruthers et al., 1999), the stimulation of Ca2+ mobilization and inositol phosphate (IP) production (Akbar et al., 1994; Wilkinson et al., 1997a,1997b), and the activation of protein tyrosine phosphatases (Buscail et al., 1994; Florio et al., 1994; Reardon et al., 1997). Additionally, there are reports that somatostatin receptors couple to the mobilization of arachidonic acid. Thus studies have demonstrated that activation of recombinant somatostatin receptors, such as the rat sst4 (Sakanaka et al., 1994) and the human sst5 receptor (Carruthers et al., 1999), can lead to arachidonic acid release. Somatostatin-stimulated increases in neuronal M-currents have been suggested to be mediated through the generation of an arachidonic acid metabolite(s), such as LTC4, in CA1 pyramidal cells (Schweitzer et al., 1990) and in the hippocampus (Lammers et al., 1996). Similarly, the stimulation by somatostatin of BKCa channel activity in rat pituitary tumour cells, possibly through sst2 receptor activation, is reportedly via the production of a leukotriene, possibly also LTC4 (Duerson et al., 1996). However, the mobilization of arachidonic acid through the sst5 receptor by somatostatin occurs at agonist concentrations a thousand fold higher than required for inhibition of adenylate cyclase, and similar to those for the stimulation of cyclic AMP (adenosine 3′, 5′ cyclic monophosphate) accumulation (Carruthers et al., 1999), Ca2+ mobilization (Wilkinson et al., 1996), inositol phosphate production (Wilkinson et al., 1997a) and GTPγS binding (Williams et al., 1997).
On the basis of structural and pharmacological similarities, somatostatin receptors (sst1 – sst5) can be sub-divided into two groups, the SRIF1 group, comprising the sst2, sst3 and sst5 receptors, and the SRIF2 group, comprising the sst1 and sst4 receptor (see Hoyer et al., 1995; Humphrey et al., 1998). Angiopeptin and octreotide, both stable octapeptide somatostatin receptor analogues, are selective for the SRIF1 group of receptors (Patel & Srikant, 1994). Both of these analogues have generated recent interest concerning their potential use therapeutically for the prevention of angiogenesis (Woltering et al., 1997; Albini et al., 1999) and restenosis following balloon angioplasty (Emanuelsson et al., 1995; von Essen et al., 1997). Although angiopeptin and octreotide both behave as potent and full agonists to inhibit adenylate cyclase or stimulate extracellular acidification through the sst2 or sst5 receptor (Patel et al., 1994; Taylor et al., 1996), the ability of these peptides to mobilize arachidonic acid has not been reported. The aims of this study were to characterize the effects of somatostatin, angiopeptin and octreotide on arachidonic acid mobilization through human recombinant receptors, comprising the SRIF1 receptor group, and to examine some of the potential transduction mechanisms involved.
Methods
Cell culture
Chinese hamster ovary cells (CHO-K1 cells) stably expressing either recombinant human sst2 (CHO h sst2), sst3 (CHO h sst3) or sst5 (CHO h sst5) receptors were used as previously described (see Wilkinson et al., 1996). Following single cell dilution cloning, levels of receptor expression in all the CHO-K1 cell lines used were of the same order (mean Bmax estimates (n=3) in pmol mg−1 protein, h sst2 6.2±0.5; h sst3 12.7±2.2; h sst5 5.6±0.9). All recombinant CHO-K1 cell lines were maintained in Dulbecco's modified Eagles Medium/Hams F-12 nutrient (1 : 1) mix supplemented with Glutamax I, 10% foetal calf serum (FCS) and 0.5 mg ml−1 G418 sulphate (Geneticin) as the selection agent (Life Technologies, Paisley, Scotland). Cells were maintained in the appropriate medium at 37°C in humidified air containing 5% carbon dioxide.
[3H]-arachidonic acid assay
Cells were seeded into 24-well plates at a density of 5×104 and were incubated with 0.5 μCi ml−1 of [5,6,8,9,11,12,14,15-3H]-arachidonic acid (215 Ci mmol−1; Amersham International Ltd) in normal cell culture medium 18 h prior to experimental use. Arachidonic acid is rapidly taken up and almost completely stored in phospholipids (see Washizaki et al., 1994). Cells were then washed four times with 20 mM HEPES-buffered Krebs (mM : NaCl 125; KCl 5.4; NaHCO3 16.2; D-glucose 5.5; HEPES 20; NaH2PO4 1 and CaCl2 1.3, pH 7.4), supplemented with 0.1% protease-free BSA and left to equilibrate at 37°C for 10 min. The wash buffer was removed and replaced with fresh buffer (1 ml) containing varying agonist concentrations. An aliquot (600 μl) of the buffer was then removed and counted on a Canberra Packard 2500TR Liquid Scintillation Analyser following the addition of 2 ml of Ultima Gold XR scintillant (Packard). In experiments where the effects of pertussis toxin were to be determined, the toxin was added at the same time as the [3H]-arachidonic acid. Experiments to investigate the susceptibility of tritium release to a variety of enzyme inhibitors were modified to include a 120 min pre-incubation of the cells in the presence or absence (control) of the relevant enzyme inhibitor at 37°C. After pre-incubation, the buffer was removed and replaced with fresh buffer containing enzyme inhibitor±1 μM somatostatin (basal and agonist-stimulated tritium release for each enzyme concentration).
Chemicals
Somatostatin was obtained from Peninsula Laboratories Europe Ltd (St. Helens, Merseyside, U.K.). Pertussis toxin, genistein, forskolin, PD 98059, Ro-31-8220, PP1 and LY-294002 were purchased from Calbiochem-Novabiochem Ltd (Beeston, Nottingham, U.K.). Angiopeptin, indomethacin, eicosatriynoic acid (ETI), arachidonic acid, PGE2, quinacrine and the PKA inhibitor amide 14 – 22 were obtained from Sigma-Aldrich Company Ltd (Poole, Dorset, U.K.). Octreotide was obtained from Novartis Pharma AG (Basel, Switzerland).
Data analysis
All drug responses were initially measured as disintegrations per minute (d.p.m.) well−1 and have been expressed either as a per cent over the basal d.p.m. well−1 or as a percentage of the response to 1 μM somatostatin. For all concentration-effect curves, pEC50 values (negative log of the concentration of each individual agonist to produce half its own maximum stimulation) were calculated for the curves by non-linear regression analysis of the data and fitting to a four parameter logistic function, using the curve-fitting programme GraphPad Prism. Statistical significance was determined (where appropriate) using Student's unpaired t-test with P<0.05 as the level of significance.
Results
Effects of somatostatin peptide analogues on tritium release from [3H]-arachidonic acidpreloaded CHO-K1 cells
In CHO h sst2 cells, somatostatin stimulated the release of tritium in a concentration-dependent manner with a pEC50 of 7.8±0.13 and maximum stimulation of 985±91% over basal (Figure 1A). Octreotide did not reach a maximum response at 1 μM, although the response at this concentration (827±25%; estimated pEC50 value, 6.33±0.23) did not differ significantly (P>0.05) from that of somatostatin. In contrast, angiopeptin (pEC50, 6.57±0.15) acted as a partial agonist, with a maximum response of 423±111% at 3 μM (Figure 1A). Neither somatostatin (31±13%), angiopeptin (13±22%) or octreotide (22±41%) were able to elicit the release of tritium significantly above that of basal (0%) from CHO h sst3 cells at concentrations up to 10 μM (Figure 1B). In CHO h sst5 cells, somatostatin stimulated tritium release (pEC50, 7.29±0.17; maximum response 221±8%). Octreotide also stimulated the release of tritium (pEC50, 7.44±0.29) but produced a lower (P<0.05) maximum stimulation than that of somatostatin (123±44%). Angiopeptin did not significantly stimulate the release of tritium above that of basal (42±27% at 10 μM; Figure 1C). The estimated pEC50 values, maximum stimulation and Hill slopes obtained with CHO h sst2 or CHO h sst5 cells are summarized in Table 1.
Figure 1.
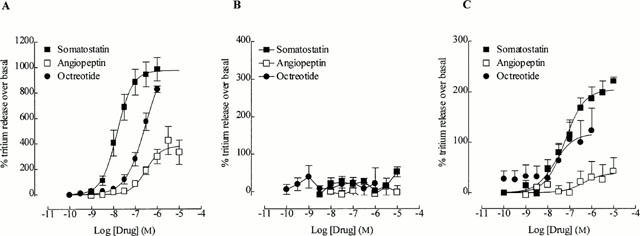
The effect of somatostatin, angiopeptin and octreotide on the release of tritium on (A) CHO h sst2 cells, (B) CHO h sst3 cells and (C) CHO h sst5 cells. All cells were pre-loaded with [3H]-arachidonic acid for 18 h before experimentation and were incubated with drug or vehicle for 1 h. All values are normalized as a percentage over the basal response (0%) and are the mean from 5 – 7 experiments. Vertical lines represent the s.e.mean. Where no error bar is shown, the s.e.mean lies within the symbol.
Table 1.
A summary of pEC50 values, the Hill slopes (nH) and the maximum responses to somatostatin, angiopeptin or octreotide in CHO h sst2 or CHO h sst5 cells pre-loaded with [3H]-arachidonic acid

Effects of pertussis toxin
Compared to the responses without pertussis toxin pre-treatment, the maximum response to somatostatin (percentage over basal) after pre-incubation of cells with pertussis toxin (100 ng ml−1 for 18 h) was reduced in both CHO h sst2 (1127±96% and 613±120%, respectively; Figure 2A) and CHO h sst5 cells (199±23 and 108±8%, respectively; Figure 2C) by approximately 50%. However, there was no significant change between the pEC50 values for somatostatin before and after pertussis toxin pre-treatment of CHO h sst2 cells (7.65±0.08 and 7.53±0.06, respectively), or CHO h sst5 cells (7.15±0.17 and 7.02±0.28, respectively). Pre-treatment of CHO h sst3 cells with pertussis toxin did not alter the inability of somatostatin to stimulate tritium release (maximum response 33±20% over basal with 10 μM somatostatin after pertussis toxin; Figure 2B).
Figure 2.
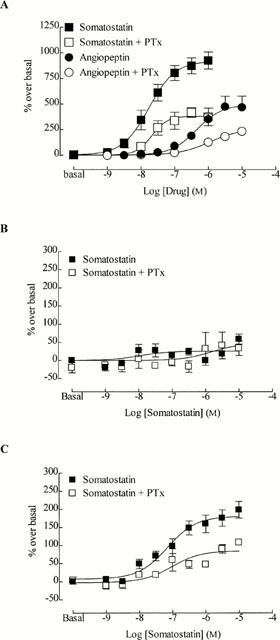
The effect of pertussis toxin (PTx; 100 ng.ml−1, 18 h) upon somatostatin-stimulated release of tritium. (A) CHO h sst2, (B) CHO h sst3, and (C) CHO h sst5 cells pre-loaded with [3H]-arachidonic acid. Cells were pre-incubated with pertussis toxin for 18 h. Vertical lines represent the s.e.mean (n=4 – 6). Where no error bar is shown, s.e.mean lies within the symbol.
In order to determine whether the partial agonism of angiopeptin in CHO h sst2 cells was due to its inability to stimulate coupling to specific G proteins, the effect of pertussis toxin on the ability of angiopeptin to release tritium was examined. The potency (pEC50 values) of angiopeptin was similar before and after pertussis toxin treatment (6.40±0.18 and 6.01±0.36, respectively), whereas the maximum response was significantly reduced (474±104% and 230±26%, respectively; P<0.05; Figure 2A).
Mechanism of tritium release through the sst2 receptor
The mechanism of somatostatin-stimulated tritium release via the sst2 receptor was further investigated. Quinacrine (1 or 10 μM), a non-selective inhibitor of PLA2, or PGE2 (1 nM to 10 μM) had no effect on the basal or somatostatin (1 μM)-stimulated release of tritium (see Table 2 for values). The selective MEK1 inhibitor, PD 98059 (40 μM), had no effect on the basal tritium release (8.5±1.1% and 9.3±1.2%, respectively; values are expressed as a per cent of the 1 μM somatostatin response), but reduced the somatostatin (1 μM)-stimulated release of tritium to 61.9±3.0% (Figure 3A). A higher concentration of PD 98059 (60 μM) had no further effect (data not shown). Surprisingly, the response to the partial agonist angiopeptin in CHO h sst2 cells was unaffected by PD 98059 (42.5±10.4% and 50.1±3.4%, respectively). After pre-treatment of CHO h sst2 cells with pertussis toxin, the ability of somatostatin (34.1±2.5%) or angiopeptin (20.4±1.9%) to stimulate tritium release was unaffected by the PD 98059 compound (33.1±1.0% and 25.2±0.8%, respectively; Figure 3A).
Table 2.
Effects of inhibitors on the release of tritium from CHO h sst2 cells
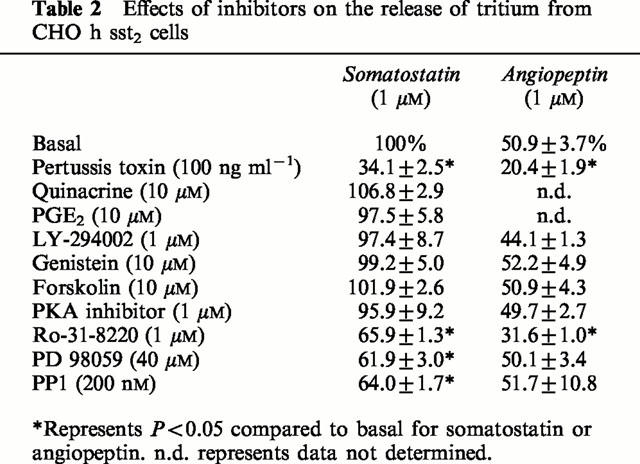
Figure 3.
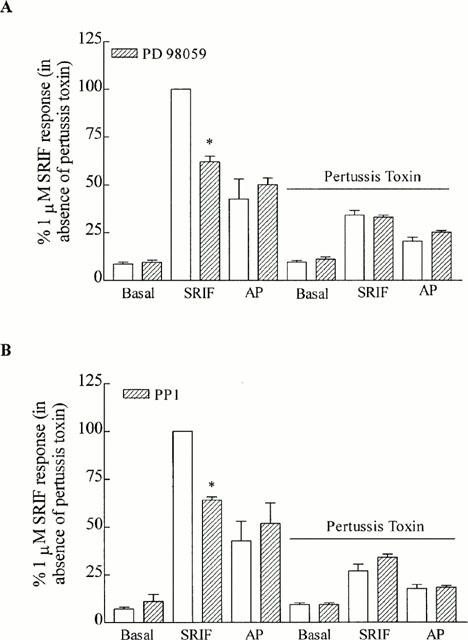
The inhibition of MEK and Src. The effect of (A) the selective MEK1 inhibitor, PD 98059 (40 μM), and (B) the Src inhibitor, PP1 (200 nM), on the release of tritium from CHO h sst2 cells stimulated by somatostatin (SRIF) and angiopeptin (AP; both 1 μM) in the presence and absence of pertussis toxin (100 ng ml−1; 18 h). Results are expressed as a percentage of the somatostatin response in the absence of pertussis toxin. *Significantly different from somatostatin alone (P<0.05). Vertical lines represent the s.e.mean (n=5).
Similar to PD 98059, a selective inhibitor of Src, PP1 (200 nM), had no effect on the basal release of tritium (7.1±0.9% and 11.0±3.7% before and after PP1, respectively), but inhibited somatostatin (1 μM)-stimulated release (100% and 64.0±1.7%, respectively; Figure 3B). In contrast, the responses to angiopeptin (1 μM) were unaffected by PP1 (42.5±10.4% and 51.7±10.8%, respectively). In the presence of pertussis toxin, somatostatin- and angiopeptin-stimulated tritium release (26.8±3.7% and 17.7±2.2%, respectively) was unaffected by PP1 (34.0±1.7% and 18.4±0.8%, respectively; Figure 3B).
In the presence of PD 98059 and PP1 combined, somatostatin-stimulated tritium release was reduced to 65.6±3.4%, no different to the effect produced by either inhibitor alone on the somatostatin response (see above). As expected, the inhibitory effect of both inhibitors combined upon somatostatin was abolished after pertussis toxin pre-treatment (data not shown).
Genistein (10 μM), a non-selective inhibitor of protein tyrosine kinases, LY 294004 (1 μM), a selective PI 3-kinase inhibitor, the adenylate cyclase activator, forskolin (10 μM), and a PKA inhibitor amide 14 – 22 (1 μM), all had no effect on responses to either somatostatin or angiopeptin (values shown in Table 2).
Basal tritium release before (5.6±0.4%) or after (6.8±0.2%) pertussis toxin pre-treatment was unaffected by the protein kinase C (PKC) inhibitor, Ro-31-8220 (1 μM; 5.1±0.1% and 4.3±0.2%, respectively). In both the absence and presence of pertussis toxin pre-treatment, the response to somatostatin (100% and 28.3±0.6, respectively) or angiopeptin (43.2±1.0% and 17.1±1.3, respectively) were reduced by Ro-31-8220 (65.9±1.3% and 13.8±0.5% for somatostatin; 31.6±1.0% and 9.5±1.1% for angiopeptin, respectively; Figure 4).
Figure 4.
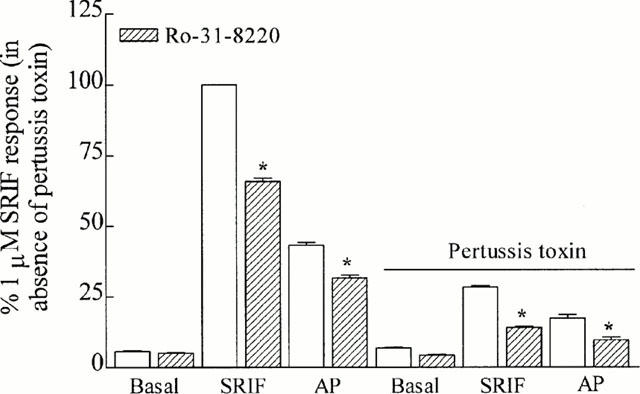
The inhibition of PKC. The effect of a PKC inhibitor, Ro-31-8220 (1 μM), on basal, somatostatin- or angiopeptin (both 1 μM)-stimulated tritium release from CHO h sst2 cells pre-loaded with [3H]-arachidonic acid. The effects of Ro-31-8220 to block tritium release were examined in the absence and presence of pertussis toxin (100 ng ml−1; 18 h). Results are expressed as a percentage of the somatostatin response in the absence of pertussis toxin. *Significantly different from peptide in absence of Ro-31-8220 (P<0.05). Vertical lines represent the s.e.mean (n=5).
Discussion
We have characterized agonist-stimulated tritium release from various CHO-K1 cell lines expressing recombinant receptor types of the SRIF1 group and pre-loaded with [3H]-arachidonic acid. Although few studies to date have examined recombinant somatostatin receptor activation and effects on arachidonic acid mobilization, several groups have used a similar assay to study other recombinant G protein-coupled receptors, such as substance P (Garcia et al., 1994), dopamine (Nilsson et al., 1998), 5-hydroxytryptamine (Berg et al., 1998) and adrenoceptors (Xing & Insel, 1996).
Using recombinant somatostatin receptor systems, the rat sst4 and the human sst5 receptor have been identified as coupling to arachidonic acid mobilization (Bito et al., 1994; Carruthers et al., 1999). These observations have been extended in this study by demonstrating that, under similar experimental conditions, agonist-stimulated tritium release occurs through the recombinant sst2 and sst5 receptor, but not the sst3 receptor. Somatostatin receptor density (Bmax) was similar for CHO h sst2 (6.2) and CHO h sst5 cells (5.6) and only slightly higher in CHO h sst3 cells (12.7). Although variation in receptor density can dramatically alter agonist activity (see Kenakin, 1996; Hermans et al., 1999), the distinct differences seen in the present study within the SRIF1 receptor group to mediate arachidonic acid release do not appear to be attributable to differences in receptor expression levels.
The tritiated product released by somatostatin receptor activation in the current study was not identified. Prostaglandin E2 represents only a relatively small proportion of the released product through recombinant sst5 receptor activation (Carruthers et al., 1999). Nilsson et al. (1998) report that 90 – 99% of the tritiated product measured from CHO cells expressing D2 receptors is attributable to free arachidonic acid release. It would be of great interest to confirm whether or not arachidonic acid itself represents the major releasable product upon somatostatin receptor activation since the particular eicosanoid(s) released may have dramatic consequences for the physiological relevance of activation of the arachidonic acid cascade (see Shimizu & Wolfe, 1990; Piomelli, 1993). Although experiments using the same CHO h sst5 cell line have demonstrated the existence of functional prostanoid receptors positively linked to adenylate cyclase (Carruthers et al., 1999), exogenous PGE2 had no effect on the ability of somatostatin to stimulate arachidonic acid release through sst2 or sst5 receptors (this study). This contrasts with the findings of Di Marzo & Piomelli (1992), who report that in CHO-K1 cells expressing dopamine D2 receptors, PGE2 increased D2 receptor-mediated arachidonic acid release.
In this study, both the sst2 and sst5 receptor coupled to pertussis toxin-sensitive and -insensitive G proteins to stimulate tritium release. In contrast, the rat sst4 receptor reportedly mediates arachidonic acid release exclusively through Gi/o proteins (Bito et al., 1994). Although the maximum response to somatostatin through the sst2 and sst5 receptors was markedly reduced by pertussis toxin, there was no change in the estimated pEC50 values, implying that both receptor-types couple equally efficiently to pertussis toxin-sensitive and -insensitive G proteins to stimulate the release of tritium. The particular G proteins involved in the mobilization of arachidonic acid by somatostatin have yet to be determined. Previous work in our laboratory suggests that the pertussis toxin-insensitive release of arachidonic acid through the sst5 receptor is not mediated through a Gαs protein (Carruthers et al., 1999), indicating the possibility of the involvement of Gαq or Gα12 proteins.
Several studies have employed inhibition of forskolin-stimulated cyclic AMP as a functional assay to determine agonist activity of octreotide and angiopeptin, where both peptides act as full and potent agonists (for example, Patel et al., 1994). This is in agreement with unpublished data from our own laboratory using these cell lines. However, because of the efficient coupling of somatostatin receptors to the inhibition of adenylate cyclase, even a very weak partial agonist may display full agonist activity (Carruthers et al., 1999). We have now shown that octreotide and angiopeptin are actually considerably weaker than somatostatin at mobilizing arachidonic acid even although both agonists bind with high affinity at the sst2 receptor (IC50 values of 0.36 nM and 0.26 nM, respectively) and sst5 receptor (pIC50 values of 20.5 nM and 6.92 nM, respectively). In particular, angiopeptin had low intrinsic activity at both the sst2 and the sst5 receptor when employing arachidonic acid mobilization as an index of activity. The hypothesis that the partial agonism of angiopeptin at the sst2 receptor was due to differential stimulation of pertussis toxin-sensitive and -insensitive G proteins was discounted since angiopeptin behaved as a partial agonist with similar pEC50 values both before and after pertussis toxin treatment, although the maximum response was reduced with pertussis toxin treatment.
In light of the ability of a receptor to couple to multiple G proteins with differing affinities for the receptor, it has been postulated that the intrinsic efficacy of a drug/receptor is neither constant nor independent of downstream G proteins, but rather may have multiple estimated values (see Kenakin, 1996; Berg et al., 1998). Since it is apparent that agonist-activity is dependent in part upon the downstream response measured (see Kenakin, 1996 for review) it becomes necessary to know the particular intracellular mechanisms which couple to the receptor and are involved in mediating the response measured. Thus, theoretically, the development of a clinically useful agonist ought not to be targeted against the receptor type alone, but the receptor-G protein complex implicated in mediating the response considered a target for therapeutic intervention. Although it is yet uncertain whether somatostatin receptors differentially couple to multiple G proteins in vivo, there are important implications clinically if this is found to be so. For example, the inhibition of proliferation in rat vascular smooth muscle cells is reported to be through the sst5 receptor (Lauder et al., 1997). Supposing this effect were mediated in vivo through the inhibition of adenylate cyclase (efficiently-coupled), octreotide or angiopeptin (both full agonists), may be prove to be effective antiproliferative agents. However, were the antiproliferative effect mediated through an sst5-stimulated release of arachidonic acid (poorly-coupled), it would be predicted that both peptides would be ineffective therapeutic agents.
The selective MEK1 inhibitor, PD98059, and the Src inhibitor, PP1, reduced somatostain sst2 receptor-mediated responses, although these effects were observed only in the absence of pertussis toxin. This suggests that both p42/44 MAP kinase and Src are involved exclusively in the Gi/o protein-mediated release of tritium. Intriguingly, the MEK and Src inhibitors reduced the responses to somatostatin but not those to angiopeptin. A possible explanation for this may be that somatostatin (a full agonist) is able to recruit a comparatively greater number of different downstream signalling molecules to mobilize arachidonic acid release than angiopeptin (a partial agonist). Assuming this to be the case, the effects of specific inhibitory compounds, such as PD 98059 or PP1, would be more readily observed against a somatostatin rather than an angiopeptin response, which was indeed found to be so.
Somatostatin-induced tritium release via the sst2 receptor was insensitive to the non-selective PLA2 inhibitor, quinacrine, even at high concentrations (10 μM), or the selective PI 3-kinase inhibitor, LY-294002. In contrast, sst4 receptor-mediated mobilization of AA is reportedly dependent upon both PLA2 and PI 3-kinase (Bito et al., 1994; Sakanaka et al., 1994). This suggests that there are discrete differences between somatostatin receptor types to mobilize AA release. The lack of effect of quinacrine in our study may reflect a situation where arachidonic acid is not synthesized de novo, but rather exogenous [3H]-arachidonic acid is loosely stored in intracellular/plasma membrane ‘pools' and is easily releasable upon receptor activation (see Chilton, 1989). Somatostatin sst2 receptor-mediated AA release was independent of cyclic AMP since neither forskolin nor a selective PKA inhibitor affected responses. Conversely, the ability of the PKC inhibitor, Ro-31-8220, to reduce responses to somatostain or angiopeptin in the presence or absence of pertussis toxin, indicates a possible role for PKC in the moblization of arachidonic acid via the sst2 receptor.
In conclusion, this study has characterized the ability of the somatostatin receptor types comprising the SRIF1 group to mobilize tritium from CHO-K1 cells pre-loaded with [3H]-arachidonic acid. The signalling pathways utilized by the sst2 receptor to release arachidonic acid and/or its metabolites remain to be further characterized, but appear to involve PKC and p42/44 MAP kinase. Perhaps most notably, the somatostatin receptor peptide analogues, octreotide and angiopeptin, have low intrinsic activity at the sst2 and sst5 receptors which may have important implications for their potential as therapeutic agents, and highlights the need for rigorous analyses of agonist activity.
Abbreviations
- AA
arachidonic acid
- CHO-K1
Chinese hamster ovary cell
- cyclic AMP
adenosine 3′, 5′ cyclic monophosphate
- IP
inositol phosphate
- MAP
mitogen activated protein
- MEK1
mitogen activated kinase 1
- PGE2
prostaglandin E2
- PKA
protein kinase A
- PKC
protein kinase C
- PLA2
phospholipase A2
- SRIF
somatotrophin release inhibiting factor
- sst
somatostatin receptor
References
- AKBAR M., OKAJIMA F., TOMURA H., MAFID M.A., YAMADA Y., SEINO S., KONDO Y. Phospholipase C activation and Ca2+ mobilization by cloned human somatostatin receptor subtypes 1-5, in transfected COS-7 cells. FEBS Lett. 1994;348:192–196. doi: 10.1016/0014-5793(94)00603-2. [DOI] [PubMed] [Google Scholar]
- ALBINI A., FLORIO T., GIUNCIUGLIO D., MASIELLO L., CARLONE S., CORSARO A., THELLUNG S., CAI T., NOONAN D.M., SCHETTINI G. Somatostatin controls Kaposi's sarcoma tumor growth through inhibition of angiogenesis. FASEB J. 1999;13:647–655. doi: 10.1096/fasebj.13.6.647. [DOI] [PubMed] [Google Scholar]
- BERG K.A., MAAYANI S., GOLDFARB J., SCARAMELLINI C., LEFF P., CLARKE W.P. Effector pathway-dependent relative efficacy at serotonin type 2A and 2C receptors: evidence for agonist-directed trafficking of receptor stimulus. Mol. Pharmacol. 1998;54:94–104. [PubMed] [Google Scholar]
- BITO H., MORI M., SAKANAKA C., TAKANO T., HONDA Z., GOTOH Y., NISHIDA E., SHIMIZU T. Functional coupling of SSTR4, a major hippocampal somatostatin receptor, to adenylate cyclase inhibition, arachidonate release, and activation of the mitogen-activated protein kinase cascade. J. Biol. Chem. 1994;269:12722–12730. [PubMed] [Google Scholar]
- BUSCAIL L., DELESQUE N., ESTÉVE J., SAINT-LAURENT N., PRATS H., CLERC P., ROBBERECHT P., BELL G.I., LIEBOW C., SCHALLY A.V., VAYSSE N., SUSINI C. Stimulation of tyrosine phosphatase and inhibition of cell proliferation by somatostatin analogues: mediation by human somatostatin receptor subtypes SSTR1 and SSTR2. Proc. Natl. Acad. Sci. USA. 1994;91:2315–2319. doi: 10.1073/pnas.91.6.2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CARRUTHERS A.M., WARNER A.J., MICHEL A.D., FENIUK W., HUMPHREY P.P.A. Activation of adenylate cyclase by human recombinant sst5 receptors expressed in CHO-K1 cells and involvement of Gαs proteins. Br. J. Pharmacol. 1999;126:1221–1229. doi: 10.1038/sj.bjp.0702401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHILTON F.H. Potential phospholipid source(s) of arachidonate used for the synthesis of leukotrienes by the human neutrophil. Biochem. J. 1989;258:327–333. doi: 10.1042/bj2580327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DI MARZO V., PIOMELLI D. Participation of prostaglandin E2 in dopamine D2 receptor-dependent potentiation of arachidonic acid release. J. Neurochem. 1992;59:379–382. doi: 10.1111/j.1471-4159.1992.tb08915.x. [DOI] [PubMed] [Google Scholar]
- DUERSON K., WHITE R.E., JIANG F., SCHONBRUNN A., ARMSTRONG D.L. Somatostatin stimulates BKca channels in rat pituitary tumor cells through lipoxygenase metabolites of arachidonic acid. Neuropharmacology. 1996;35:949–961. doi: 10.1016/0028-3908(96)00131-1. [DOI] [PubMed] [Google Scholar]
- EMANUELSSON H., BEATT K.J., BAGGER J.P., BALCON R., HEIKKILA J., PIESSENS J., SCHAEFFER M., SURYAPRANATA H., FOEGH M. Long-term effects of angiopeptin treatment in coronary angioplasty. Reduction of clinical events but not angiographic restenosis. European Angiopeptin Study Group. Circulation. 1995;91:1689–1696. doi: 10.1161/01.cir.91.6.1689. [DOI] [PubMed] [Google Scholar]
- FLORIO T., RIM C., HERSHBERGER R.E., LODA M., STORK J.S. The somatostatin receptor SSTR1 is coupled to phosphotyrosine phosphatase activity in CHO-K1 cells. Mol. Endocrinol. 1994;8:1289–1297. doi: 10.1210/mend.8.10.7854346. [DOI] [PubMed] [Google Scholar]
- GARCIA M., SAKAMOTO K., SHIGEKAWA M., NAKANISHI S., ITO S. Multiple mechanisms of arachidonic acid release in Chinese hamster ovary cells transfected with cDNA of substance P receptor. Biochem. Pharmacol. 1994;48:1735–1741. doi: 10.1016/0006-2952(94)90459-6. [DOI] [PubMed] [Google Scholar]
- HERMANS E., CHALLISS R.A.J., NAHORSKI S.R. Effects of varying the expression level of recombinant human, Glu1α receptors on the pharmacological properties of agonists and antagonists. Br. J. Pharmacol. 1999;126:873–882. doi: 10.1038/sj.bjp.0702359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOYER D., BELL G.I., BERELOWITZ M., EPELBAUM J., FENIUK W., HUMPHREY P.P.A., O'CARROLL A.-M., PATEL Y.C., SCHONBRUN A., TAYLOR J.E., REISINE T. Classification and nomenclature of somatostatin receptors. Trends Pharmacol. Sci. 1995;16:86–88. doi: 10.1016/s0165-6147(00)88988-9. [DOI] [PubMed] [Google Scholar]
- HUMPHREY P.P.A., EPELBAUM J., FENIUK W., HOYER D., TAYLOR J.E., REISINE T.R. The IUPHAR Compendium of Receptor Characterization and Classification (International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification) IUPHAR Media, London; 1998. Somatostatin Receptors; pp. 246–255. [Google Scholar]
- KENAKIN T. The classification of seven transmembrane receptors in recombinant expression systems. Pharmacol. Rev. 1996;48:413–463. [PubMed] [Google Scholar]
- LAMMERS C.-H., SCHWEITZER P., FACCHINETTI P., ARRANG J.-M., MADAMBA S.G., SIGGINS G.R., PIOMELLI D. Arachidonate 5-lipoxygenase and its activating protein: prominent hippocampal expression and role in somatostatin signalling. J. Neurochem. 1996;66:146–152. doi: 10.1046/j.1471-4159.1996.66010147.x. [DOI] [PubMed] [Google Scholar]
- LAUDER H., SELLERS L.A., FAN T.-P.D., FENIUK W., HUMPHREY P.P.A. Somatostatin sst5 inhibition of receptor mediated regeneration of rat aortic vascular smooth muscle cells. Br. J. Pharmacol. 1997;122:663–670. doi: 10.1038/sj.bjp.0701443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NILSSON C.L., HELLSTRAND M., EKMAN A., ERIKSSON E. Direct dopamine D2-receptor-mediated modulation of arachidonic acid release in transfected CHO cells without the concomitant administration of a Ca2+-mobilizing agent. Br. J. Pharmacol. 1998;124:1651–1658. doi: 10.1038/sj.bjp.0702025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PATEL Y.C., SRIKANT C.B. Subtype selectivity of peptide analogs for all five cloned human somatostatin receptors (hsstr1-5) Endocrinology. 1994;135:2814–2817. doi: 10.1210/endo.135.6.7988476. [DOI] [PubMed] [Google Scholar]
- PATEL Y.C., GREENWOOD M.T., WARSZYNSKA A., PANETTA R., SRIKANT C.B. All five cloned human somatostatin receptors (hSSTR1-5) are functionally coupled to adenylyl cyclase. Biochem. Biophys. Res. Comm. 1994;198:605–612. doi: 10.1006/bbrc.1994.1088. [DOI] [PubMed] [Google Scholar]
- PIOMELLI D. Arachidonic acid in cell signalling. Curr. Opin. Cell Biol. 1993;5:274–280. doi: 10.1016/0955-0674(93)90116-8. [DOI] [PubMed] [Google Scholar]
- REARDON D.B., DENT P., WOOD S.L., KONG T., STURGILL T.N. Activation in vitro of somatostatin receptor subtypes 2, 3, or 4 stimulates protein tyrosine phosphatase activity in membranes from ras-transformed NIH3T3 cells: coexpression with catalytically inactive SHP-2 blocks responsiveness. Mol. Endocrinol. 1997;11:1062–1069. doi: 10.1210/mend.11.8.9960. [DOI] [PubMed] [Google Scholar]
- SAKANAKA C., FERBY I., WAGA I., BITO H., SHIMIZU T. On the mechanism of cytosolic phospholipase A2 activation in CHO cells carrying somatostatin receptor: wortmannin-sensitive pathway to activate mitogen-activated protein kinase. Biochem. Biophys. Res. Comm. 1994;205:18–23. doi: 10.1006/bbrc.1994.2623. [DOI] [PubMed] [Google Scholar]
- SCHWEITZER P., MADAMBA S., SIGGINS G.R. Arachidonic acid metabolites as mediators of somatostatin-induced increase of neuronal M-current. Nature. 1990;346:464–467. doi: 10.1038/346464a0. [DOI] [PubMed] [Google Scholar]
- SHIMIZU T., WOLFE L.S. Arachidonic acid cascade and signal transduction. J. Neurochem. 1990;55:1–15. doi: 10.1111/j.1471-4159.1990.tb08813.x. [DOI] [PubMed] [Google Scholar]
- TAYLOR J.E., NELSON R., WOON C.-W. Real-time evaluation of somatostatin subtype 1 receptor activity employing the technique of cytosensor microphysiometry. Peptides. 1996;17:1257–1259. doi: 10.1016/s0196-9781(96)00189-1. [DOI] [PubMed] [Google Scholar]
- VON ESSEN R., OSTERMAIER R., GRUBE E., MÄURER W., TEBBE U., ERBEL R., ROTH M., OEL W., BROM J., WEIDINGER G. Effects of octreotide treatment on restenosis after coronary angioplasty. Circulation. 1997;96:1482–1487. doi: 10.1161/01.cir.96.5.1482. [DOI] [PubMed] [Google Scholar]
- WASHIZAKI K., SMITH Q.R., RAPOPORT S.I., PURDON A.D. Brain arachidonic acid incorporation and precursor pool specific activity during intravenous infusion of unesterified [3H]arachidonate in the anesthetized rat. J. Neurochem. 1994;63:727–736. doi: 10.1046/j.1471-4159.1994.63020727.x. [DOI] [PubMed] [Google Scholar]
- WILKINSON G.F., FENIUK W., HUMPHREY P.P.A. Characterization of human recombinant somatostatin sst5 receptors mediating activation of phosphoinositide metabolism. Br. J. Pharmacol. 1997a;121:91–96. doi: 10.1038/sj.bjp.0701116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WILKINSON G.F., FENIUK W., HUMPHREY P.P.A. Homologous and heterologous desensitisation of somatostatin-induced increases in intracellular Ca2+ and inositol 1,4,5-trisphosphate in CHO-K1 cells expressing human recombinant somatostatin sst5 receptors. Eur. J. Pharmacol. 1997b;340:277–285. doi: 10.1016/s0014-2999(97)01430-1. [DOI] [PubMed] [Google Scholar]
- WILKINSON G.F., THURLOW R.J., SELLERS L.A., COOTE J.E., FENIUK W., HUMPHREY P.P.A. Potent antagonism by BIM-23056 at the human recombinant somatostatin sst5 receptor. Br. J. Pharmacol. 1996;118:445–447. doi: 10.1111/j.1476-5381.1996.tb15423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WILLIAMS A.J., MICHEL A.D., FENIUK W., HUMPHREY P.P.A. Somatostatin5 receptor-mediated [35S]guanosine-5′-O-(3-thio)trisphosphate binding: agonist potencies and the influence of sodium chloride on intrinsic activity. Mol. Pharmacol. 1997;51:1060–1069. doi: 10.1124/mol.51.6.1060. [DOI] [PubMed] [Google Scholar]
- WOLTERING E.A., WATSON J.C., ALPERIN-LEA R.C., SHARMA C., KEENAN E., KUROZAWA D., BARRIE R. Somatostatin analogs: angiogenesis inhibitors with novel mechanism of action. Invest. New Drugs. 1997;15:77–86. doi: 10.1023/a:1005774713202. [DOI] [PubMed] [Google Scholar]
- XING M., INSEL P.A. Protein kinase C-dependent activation of cytosolic phospholipase A2 and mitogen-activated protein kinase by alpha1-adrenergic receptors in Madin-Darby canine kidney cells. J. Clin. Invest. 1996;97:1302–1310. doi: 10.1172/JCI118546. [DOI] [PMC free article] [PubMed] [Google Scholar]