

Abstract
Previous data have shown that activation of β3-adrenoceptors stimulates vascular L-type Ca2+ channels through a Gαs-induced stimulation of the cyclic AMP/PKA pathway. The present study investigated whether β-adrenergic stimulation also uses the Gβγ/PI3K/PKC pathway to modulate L-type Ca2+ channels in rat portal vein myocytes.
Peak Ba2+ current (IBa) measured using the whole-cell patch clamp method was maximally increased by application of 10 μM isoprenaline after blockade of β3-adrenoceptors by 1 μM SR59230A. Under these conditions, the isoprenaline-induced stimulation of IBa was reversed by ICI-118551 (a specific β2-adrenoceptor antagonist) but not by atenolol (a specific β1-adrenoceptor antagonist). The β2-adrenoceptor agonist salbutamol increased IBa, an effect which was reversed by ICI-118551 whereas the β1-adrenoceptor agonist dobutamine had no effect on IBa.
Application of PKA inhibitors (H-89 and Rp 8-Br-cyclic AMPs) or a PKC inhibitor (calphostin C) alone did not affect the β2-adrenergic stimulation of IBa whereas simultaneous application of both PKA and PKC inhibitors completely blocked this stimulation.
The β2-adrenergic stimulation of L-type Ca2+ channels was blocked by a pre-treatment with cholera toxin and by intracellular application of an anti-Gαs antibody (directed against the carboxyl terminus of Gαs). In the presence of H-89, intracellular infusion of an anti-Gβcom antibody or a βARK1 peptide as well as a pre-treatment with wortmannin (a PI3K inhibitor) blocked the β2-adrenergic stimulation of IBa.
These results suggest that the β2-adrenergic stimulation of vascular L-type Ca2+ channels involves both Gαs and Gβγ subunits which exert their stimulatory effects through PKA and PI3K/PKC pathways, respectively.
Keywords: β-adrenoceptors, Ca2+ channels, G protein, protein kinase A, protein kinase C, phosphoinositide 3-kinase, smooth muscle
Introduction
In vascular and visceral smooth muscles, a number of studies have shown that β-adrenergic receptor stimulation leads to enhancement of L-type Ca2+ channel activity via the cyclic AMP/protein kinase A (PKA) transduction pathway (Ruiz-Velasco et al., 1998; Viard et al., 2000). After binding with agonist, the β-adrenergic receptor catalyzes the exchange of GDP for GTP on the α subunit of the heterotrimeric G proteins, resulting in dissociation of the α subunit from the βγ dimers. It is now well documented that both the Gαs subunit and the Gβγ dimers are able to transduce signals to effector molecules (Clapham & Neer, 1997). Previous studies from our laboratory have shown that muscarinic m3- and endothelin ETA-induced Ca2+ release in vascular myocytes is selectively transduced by Gαq and Gα11 subunits, respectively (Morel et al., 1997; Macrez et al., 1999). In contrast, angiotensin AT1 receptors transduce the signal to L-type Ca2+ channels by Gβγ dimer (Macrez-Leprêtre et al., 1997). In the absence of receptor activation, infusion of Gβγ subunits in the myocytes leads to stimulation of L-type Ca2+ channels (Viard et al., 1999; Zhong et al., 1999).
On the basis of RT – PCR experiments, three β-adrenoceptor subtypes may be expressed in rat portal vein myocytes (Viard et al., 2000). The transduction pathway activated by β3-adrenoceptors is selectively mediated through a Gαs-induced stimulation of the cyclic AMP/PKA pathway (Viard et al., 2000). However, the possible role of endogenous Gβγ subunits to the β-adrenergic stimulation of L-type Ca2+ channels is still unknown. No informations are available concerning the potential role of Gβγ subunits as possible contributors to stimulation of L-type Ca2+ channels in response to activation of β-adrenoceptor subtypes.
In the present study, we investigated the transduction pathways activated by non β3-adrenoceptors (i.e. β1- and/or β2-adrenoceptors) in the modulation of L-type Ca2+ channels in rat portal vein myocytes. We combined the use of inhibitors of PKA and PKC to determine whether one or both of these kinases contribute to the response and specific β-adrenoceptor agonists and antagonists to characterize the receptor subtypes involved. We show that Gαs and Gβγ subunits participate in the β2-adrenergic stimulation of L-type Ca2+ channels through activation of PKA and PKC, respectively.
Methods
The investigation conforms with the European Community guiding principles in the care and use of animals (86/609/CEE, CE Off J no. L358, 18 December 1986) and the French decree no. 87/748 of October 19, 1987 (J Off République Française, 20 October 1987, pp. 12245 – 12248). Authorizations to perform animal experiments according to this decree were obtained from the French Ministère de l'Agriculture et de la Pêche.
Cell preparation
Isolated myocytes from rat portal vein were obtained by enzymatic dispersion, as described previously (Leprêtre et al., 1994). Cells were seeded at density of ∼103 cells mm−2 on glass slides and maintained in short-term primary culture (2 – 36 h) in M199 containing 5% foetal calf serum, 2 mM glutamine, 1 mM pyruvate 20 U ml−1 penicillin and 20 μg ml−1 streptomycin.
Membrane current measurement
Voltage-clamp and membrane current recordings were performed with a standard patch-clamp technique using an EPC-7 amplifier (List, Darmstadt-Eberstadt, Germany). Whole-cell recordings were performed with patch pipettes having resistances of 2 – 4 MΩ. Membrane potential and membrane currents were stored and analysed using a PC computer (P-clamp system, Axon Instruments, Foster City, CA, U.S.A.). Ba2+ currents were digitally corrected for leakage currents. In some experiments, Ba2+ current density is expressed as peak current amplitude per capacitance unit (in pA/pF). All experiments were performed at 30±1°C. Results are expressed as means±s.e. mean. Significance was tested by Student's t-test. P values <0.05 were considered as significant.
Solutions
The physiological solution used to record Ba2+ currents contained (in mM): NaCl 130, KCl 5.6, MgCl2 1, BaCl2 5, glucose 11, HEPES 10, pH 7.4 with NaOH. The basic pipette solution contained (in mM): CsCl 130, EGTA 10, ATPNa2 5, GTP 0.1, MgCl2 2, HEPES, 10 pH 7.3, with CsOH. Gβγ proteins were stored in a solution containing 20 mM Tris, 1 mM EDTA, 11 mM CHAPS, and 20 mM β-mercaptoethanol. At the concentration of Gβγ used in the experiments, the final concentration of detergent was 100 μM CHAPS, which alone had no effects on Ba2+ current density (Viard et al., 1999). β-adrenergic agonists, phorbol ester and 8-Br-cAMP were extracellularly applied to the recorded cell by pressure ejection from a glass pipette.
Chemicals and drugs
Isoprenaline, salbutamol, propranolol, prazosin, rauwolscine, dobutamine and wortmannin were from Sigma (St Quentin Fallavier, France). 8-Br-cAMP, Rp 8-Br-cAMPs, H-89, calphostin C and cholera toxin (CTX) were from Calbiochem (Meudon, France). Atenolol, ICI-118551, phorbol 12-myristate 13-acetate and 4α-phorbol 12-myristate 13-acetate were from RBI (Natick, MA, U.S.A.). SR59230A (3-(2-ethylphenoxy)-1[(1S)-1,2,3,4-tetrahydronapth-1-ylaminol]-(2S)-propranolol-oxalate) was from Sanofi (Milano, Italy). M199 medium was from Flow Laboratories (Puteaux, France). Streptomycin, penicillin, glutamine and pyruvate were from Life Technology (Paisley, U.K.). Rabbit anti-Gαs subunit antibody (371732) raised to the carboxyl-terminal amino acids, RMHLRQYELL, of Gαs was from Calbiochem. Rabbit anti-Gβcom antibody (SC-378) raised to the carboxyl-terminal amino acids, TDDGMAVATG SWDSFLKIWN, of Gβ1 subunit was from Santa-Cruz Biotechnology (Santa Cruz, CA, U.S.A.). Peptides corresponding to the Gβγ binding domain of β-adrenergic receptor kinase-1 (WKKELRDAYREAQQLVQRVPKMKNKPRS) or to a region outside the Gβγ binding site (AETDRLEARKKTKNKQLGHEEDY) were synthetized by Genosys (Cambridge, U.K.). Gβγ subunits purified from bovine brain Gi/Go proteins were a gift from B. Nürnberg (University of Berlin).
Results
β-adrenergic stimulation of L-type Ca2+ channels in vascular myocytes
We have previously shown that a combination of propranolol (a non-selective β1- and β2-adrenoceptor antagonist) and SR59230A (a β3-adrenoceptor antagonist) was required to inhibit the isoprenaline-induced maximal stimulation of L-type Ca2+ channels. Studied in isolation, the β3-adrenoceptor-induced transduction pathway involves the Gαs-induced stimulation of the cyclic AMP/PKA (Viard et al., 2000). Here, we combined the application of both inhibitors of PKA and PKC and antagonists of β-adrenoceptor subtypes to further investigate the transduction pathways activated by β1- and/or β2-adrenoceptors leading to stimulation of Ca2+ channels. Ba2+ currents through L-type Ca2+ channels were elicited every 20 s in single vascular myocytes bathed in 5 mM BaCl2 solution by 200 ms depolarizations to +10 mV from a holding potential of −40 mV. As previously reported (Viard et al., 1999), inward current progressively increased in amplitude over 2 – 3 min in almost all cells due to the diffusion of the pipette solution into the cytoplasm (Figure 1A). After steady-state was reached, application of isoprenaline (10 μM) resulted in an increase in Ba2+ current which reached a peak within 2 – 3 min (Figure 1B) without any variation in holding current. Removal of isoprenaline resulted in a progressive and slow return to basal Ba2+ current within 5 – 7 min. All the experiments reported in the present paper were carried out in the continuous presence of 1 μM SR59230A to inhibit β3-adrenoceptors and stimulation of Ba2+ current was measured a time to peak effect during 3 min-lasting applications of stimulating substances. Neither time to peak current nor inactivation kinetics were modified in the presence of isoprenaline (Figure 1B).
Figure 1.
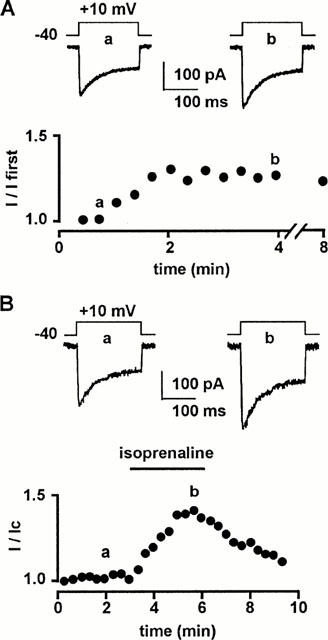
Effects of isoprenaline on L-type Ca2+ channels in rat portal myocytes. (A) Time course of peak Ba2+ current (expressed as a fraction of the first current obtained just after breakthrough into the whole-cell recording mode) in cells dialyzed with a control pipette solution. Inset: typical Ba2+ currents (a,b) evoked by a depolarization to +10 mV from a holding potential of −40 mV corresponding to lowercase letters on the curve. (B) Time course of peak Ba2+ current during the application of 10 μM isoprenaline, 3 min after reaching steady-state current. Inset: typical Ba2+ currents (a,b) corresponding to lowercase letters on the curve. External solution contained 5 mM Ba2+, 10 nM prazosin and 10 nM rauwolscine (to inhibit α1- and α2-adrenoceptors) and 1 μM SR59230A (to inhibit β3-adrenoceptors).
To identify the β-adrenergic subtypes responsible for the non-β3-adrenoceptor-induced stimulation of L-type Ca2+ channels, we tested the effects of isoprenaline and of selective β1-and β2-adrenergic agonists and antagonists. In control conditions, the mean Ba2+ current density was 5.8±0.4 pA/pF (n=45). Salbutamol (10 μM), a potent β2-adrenergic agonist (Skeberdis et al., 1997), increased the Ba2+ current density by 45±7% (Figure 2A) whereas dobutamine (10 μM), a potent β1-adrenergic agonist (Doggrell, 1990), had no detectable effect on the Ba2+ current density (Figure 2A). Isoprenaline (10 μM) increased the Ba2+ current density by 41±5% (Figure 2A), a value not significantly different from that obtained with salbutamol. The agonist-induced increases in Ba2+ current density were not different from the increase in peak current expressed as a fraction of control current (Figure 2B), indicating that this method was adequate for measuring the stimulatory effect of externally-applied agonists. The stimulatory effect of salbutamol was abolished by application of 1 μM ICI-118551, a selective antagonist of β2-adrenoceptor (Bilski et al., 1983) or 1 μM propranolol (not shown), whereas application of 1 μM atenolol, a selective antagonist of β1-adrenoceptors (Satake et al., 1996) was ineffective (Figure 2B). As the stimulatory effect of isoprenaline was abolished by application of 1 μM ICI-118551 (Figure 2B) or 1 μM propranolol (not shown) but not significantly affected by application of 1 μM atenolol (Figure 2B), it is likely that the β2-adrenoceptors are the predominant receptor subtype underlying non β3-adrenoceptor-induced stimulation of Ba2+ current.
Figure 2.
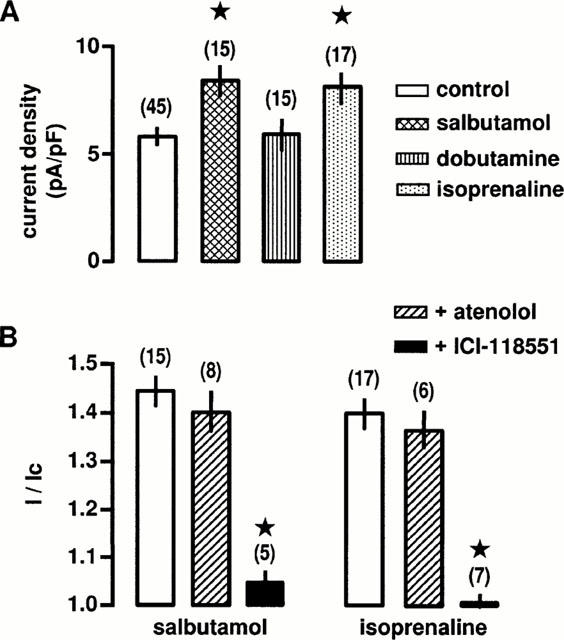
Effects of β-adrenoceptor agonists and antagonists on L-type Ca2+ channels. (A) Ba2+ current densities in control conditions and in the presence of 10 μM salbutamol (a β2-adrenoceptor agonist), 10 μM dobutamine (a β1-adrenoceptor agonist) or 10 μM isoprenaline. Current density was calculated at the time of peak effect. *Values significantly different from control (P<0.05). (B) Compiled data showing the effects of 1 μM ICI-118551 (a β2-adrenoceptor antagonist) and 1 μM atenolol (a β1-adrenoceptor antagonist) on the increase in peak Ba2+ current evoked by 10 μM salbutamol or 10 μM isoprenaline. Currents are expressed as a fraction of their values in the absence of agonists (I/Ic). *Values significantly different from those obtained in the presence of agonists (P<0.05). Data are means±s.e.mean with the number of cells tested indicated in parentheses. External solution contained 5 mM Ba2+, 10 nM prazosin, 10 nM rauwolscine and 1 μM SR59230A.
To evaluate whether PKA is involved in the intracellular pathway activated by β2-adrenoceptors, we tested the effects of PKA inhibitors (H-89 and Rp 8-Br-cyclic AMPs) on the stimulation of Ba2+ current evoked by isoprenaline (in the continuous presence of SR59203A). External application of H-89 (0.1 μM) or Rp 8-Br-cyclic AMPs (0.3 μM), applied 15 min before and throughout the current recording period did not change the peak Ba2+ current in control cells (data not shown) nor affect the stimulatory response of 0.1 μM PMA, an activator of PKC (Figure 3A,B). Increase in peak Ba2+ current evoked by 0.3 mM 8-Br-AMP was inhibited by H-89 and Rp 8-Br-cyclic AMPs, applied 15 min before and throughout the current recording period (Figure 3B). In contrast, application of H-89 or Rp 8-Br-cyclic AMPs alone had not significant effect on the isoprenaline-induced stimulation of peak Ba2+ current (Figure 3B). A similar absence of effect of PKA inhibitors was also observed on the salbutamol-induced stimulation of Ba2+ current (n=9). Because it has been recently suggested that PKC inhibitors (19 – 31 peptide or GF109203X) may act more selectively to some PKC isoforms (Wang et al., 1999), we tested calphostin C, a general inhibitor of PKCs, on the stimulation of Ba2+ currents involving activation of PKC or PKA. Superfusion of cells with 0.2 μM calphostin C for 10 – 15 min reduced the peak Ba2+ current in control cells by 25±5% (n=9). However, calphostin C-pretreatment did not affect the 8-Br-AMP-induced stimulation of L-type Ca2+ channels (Figure 4). Stimulation of peak Ba2+ current evoked by 0.1 μM PMA was blocked by 0.2 μM calphostin C (Figure 4A,B). In contrast, application of 0.2 μM calphostin C alone did not affect the isoprenaline-induced stimulation of peak Ba2+ current (Figure 4B). However, simultaneous applications of 0.2 μM calphostin C plus 0.1 μM H-89 completely inhibited the stimulatory effect of isoprenaline (Figure 4B). A similar inhibition was obtained when salbutamol was used to increase peak Ba2+ current (n=12). These results suggest that the β2-adrenergic stimulation of L-type Ca2+ channels is likely to involve both PKA and PKC.
Figure 3.
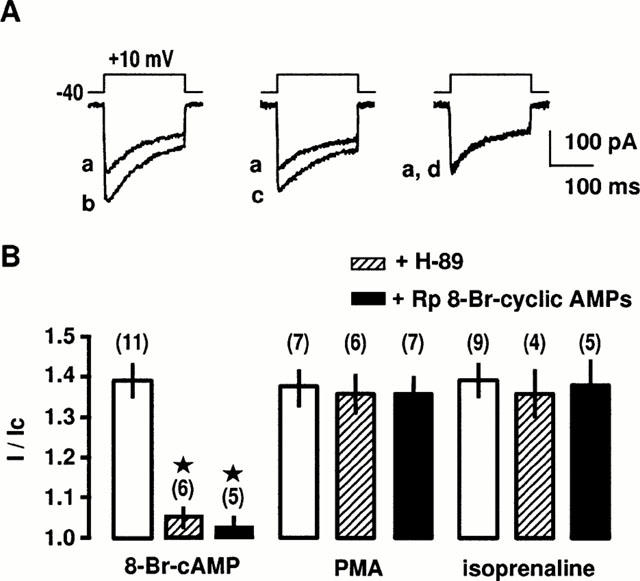
Effects of PKA inhibitors on β2-adrenergic stimulation of L-type Ca2+ channels. (A) Ba2+ currents evoked by a depolarization to +10 mV from a holding potential of −40 mV before (a) and during the application of 10 μM isoprenaline in control conditions (b), in the presence of 0.1 μM H-89 for 15 min (c) or 1 μM propranolol for 15 min (d). (B) Compiled data showing the effect of 0.1 μM H-89 and 3 μM Rp 8-Br-cyclic AMPs on the increase in Ba2+ current evoked by 0.3 mM 8-Br-cAMP, 0.1 μM phorbol 12-myristate 13-acetate (PMA) or 10 μM isoprenaline. Currents are expressed as a fraction of their values in the absence of the stimulating substances (I/Ic). Data are means±s.e.mean with the number of cells tested indicated in parentheses. *Values significantly different from those obtained in the presence of the stimulating substances (P<0.05). External solution contained 5 mM Ba2+, 10 nM prazosin, 10 nM rauwolscine and 1 μM SR59230A.
Figure 4.
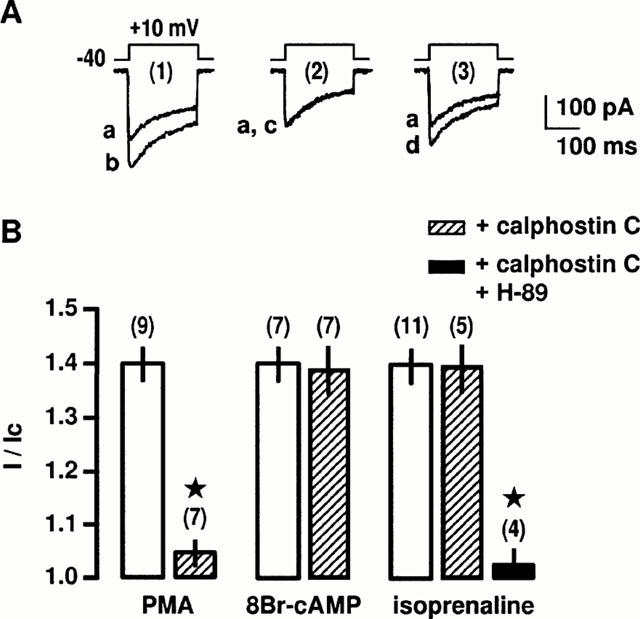
Effects of PKC and PKA inhibitors on β2-adrenergic stimulation of L-type Ca2+ channels. (A) Ba2+ currents evoked by a depolarization to +10 mV from a holding potential of −40 mV before (a) and during the application of 0.1 μM PMA for 5 min (b) in control conditions (1). In the presence of 0.2 μM calphostin C for 10 min (2 and 3), Ba2+ currents obtained before (a) and during the application of 0.1 μM PMA for 5 min (c) or 0.3 mM 8-Br-cAMP for 8 min (d). (B) Compiled data showing the effect of 0.2 μM calphostin C alone or associated with 0.1 μM H-89 on the increase in Ba2+ current evoked by 0.1 μM PMA, 0.3 mM 8-Br-cAMP and 10 μM isoprenaline. Currents are expressed as a fraction of their control values in the absence of stimulating substances (I/Ic). Data are means±s.e.mean with the number of cells tested indicated in parentheses. *Values significantly different from those obtained in the presence of stimulating substances (P<0.05). External solution contained 5 mM Ba2+, 10 nM prazosin, 10 nM rauwolscine and 1 μM SR59230A.
Simultaneous applications of 8-Br-AMP and PMA at concentrations producing a maximal stimulation of the Ba2+ current induced a slight further enhancement in Ba2+ current when compared to the application of one or the other activator alone. Thus, coapplication of both 8-Br-AMP and PMA did not result in a clear additive effect on L-type Ca2+ channel activity. However, when 8-Br-AMP and PMA were co-applied at submaximal concentrations, an additive stimulation of Ba2+ current was obtained which reached 49±5% (n=5); this stimulation was not different of that obtained with a maximal concentration of PKA or PKC activators.
Gαs- and Gβγ subunit-dependent pathways activated by β2-adrenergic stimulation
We have previously shown that intracellular application of purified Gβγ subunits from bovine brain to vascular myocytes stimulates L-type Ca2+ channels through a PI3K/PKC pathway (Viard et al., 1999). However, it is unclear whether a similar coupling is involved in the response to β-adrenergic stimulation. To identify the G protein involved in the β2-adrenergic stimulation, cells were infused with antibodies directed against the Gα-subunits. Antibodies directed against the carboxyl terminus of Gαs inhibit the interactions between the activated receptors and the Gs protein and therefore prevent the dissociation of the Gα- and Gβγ-subunits. Intracellular application of anti-Gαs antibody through the patch-clamp pipette for 5 min before application of isoprenaline inhibited in a concentration-dependent manner the isoprenaline-induced increase in Ba2+ current density (Figure 5A,B). Complete inhibition was obtained at 10 μg ml−1 anti-Gαs antibody. In contrast, intracellular application of 10 μg ml−1 anti-Gαq/11 antibody had no effect on the stimulation of the Ba2+ current evoked by isoprenaline (not shown). Furthermore, inactivated antibody obtained by heating at 95°C for 30 min had no effect on the β2-adrenergic stimulation (Figure 5A,B). Pre-treatment with 150 ng ml−1 CTX for 24 h increased slightly the Ba2+ current density by about 10% (from 6.3±0.3 pA/pF in control cells to 7.0±0.3 pA/pF in CTX-pretreated cells, n=24), suggesting that the permanent activation of Gs induced a desensitization of the transduction pathway. Under these conditions, β-agonists did not change significantly the Ba2+ current density (7.1±0.5 pA/pF, n=12). These results indicate that the Gs protein is involved in the transduction pathways leading to stimulation of L-type Ca2+ channels in response to stimulation of β2-adrenoceptors.
Figure 5.
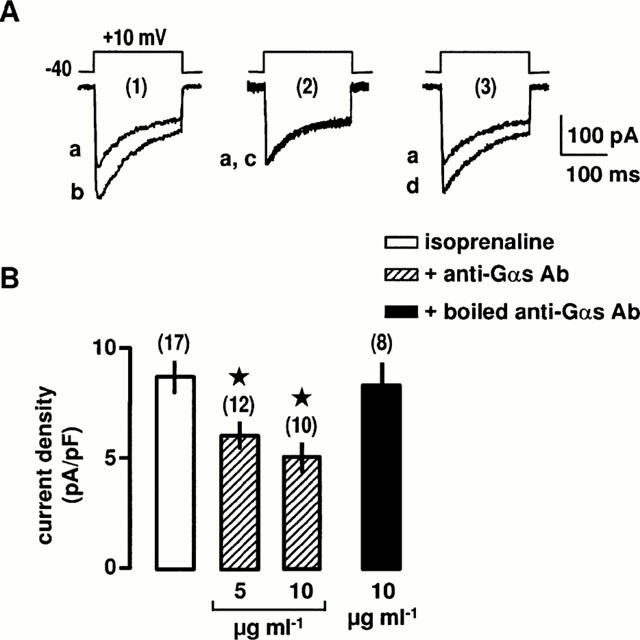
Effects of anti-Gαs antibody on the β2-adrenergic stimulation of L-type Ca2+ channels. (A) Ba2+ currents evoked by a depolarization to +10 mV from a holding potential of −40 mV before (a) and during the application of 10 μM isoprenaline for 5 min in control conditions (1b), in the intracellular presence of 10 μg ml−1 anti-Gαs antibody (2c) or 10 μg ml−1 boiled anti-Gαs antibody for 5 min (3d). (B) Compiled data showing the effects of anti-Gαs antibody on the increase in Ba2+ current density evoked by 10 μM isoprenaline. Data are means±s.e.mean with the number of cells tested indicated in parentheses. *Values significantly different from those obtained in the presence of isoprenaline (P<0.05). External solution contained 5 mM Ba2+, 10 nM prazosin, 10 nM rauwolscine and 1 μM SR59230A.
To demonstrate that the β2-adrenergic stimulation of L-type Ca2+ channels involves the Gβγ subunits, intracellular infusion of either an anti-Gβcom antibody or a peptide corresponding to the fragment of βARK1 was used to bind Gβγ subunits released after receptor activation and therefore to block activation of effectors. The following experiments were performed in the continuous presence of 0.1 μM H-89 to remove the PKA-activated pathway. Applications of βARK1 peptide and anti-Gβcom antibody had no significant effects on the Ba2+ current density in non-stimulated cells. The mean Ba2+ current density was 6.4±0.4 pA/pF in control conditions (n=11), 6.0±0.5 pA/pF in the presence of 10 μM βARK1 (n=10) and 6.1±0.5 pA/pF in the presence of 10 μg ml−1 anti-βcom antibody (n=9). As shown in Figure 6, anti-βcom antibody and βARK1 peptide (corresponding to the Gβγ binding domain of βARK1; Nair et al., 1995) inhibited in a concentration-dependent manner the β2-adrenergic increase in Ba2+ current density. In contrast, inactive βARK1 peptide and inactivated anti-βcom antibody (by heating at 95°C for 30 min) had no effect on the β2-adrenergic stimulation (not shown). It is noteworthy that in the absence of PKA inhibitors, maximal concentrations of anti-βcom antibody and βARK1 peptide alone were unable to inhibit the isoprenaline-induced stimulation of Ba2+ current (n=16). Taken together, these results suggest that both αs and βγ subunits of Gs proteins are involved in the β2-adrenergic stimulation of L-type Ca2+ channels.
Figure 6.
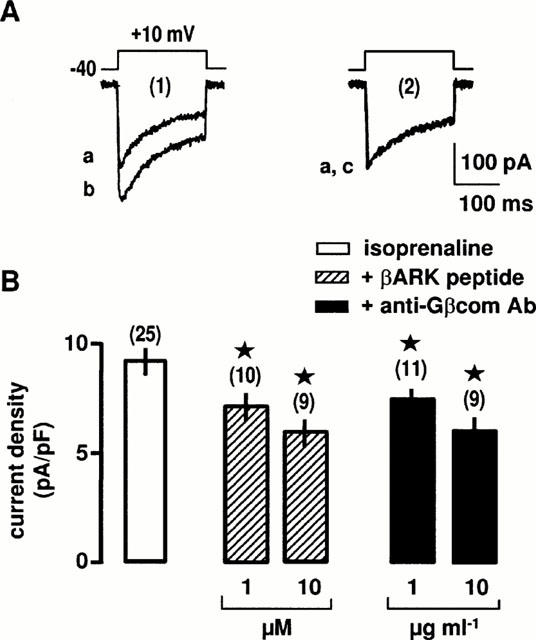
Effects of βARK1 peptide and anti-Gβcom antibody on the β2-adrenergic stimulation of L-type Ca2+ channels. (A) Ba2+ currents evoked by a membrane depolarization to +10 mV from a holding potential of −40 mV before (a) and during the application of 10 μM isoprenaline (b) in control conditions (1). In the intracellular presence of 10 μM βARK1 peptide (2), Ba2+ currents before (a) and during the application of 10 μM isoprenaline (c). (B) Compiled data showing the effects of βARK1 peptide and anti-Gβcom antibody on the increase in Ba2+ current density evoked by 10 μM isoprenaline. *Values significantly different from those obtained in the presence of isoprenaline alone (P<0.05). External solution contained 5 mM Ba2+, 10 nM prazosin, 10 nM rauwolscine, 1 μM SR59230A and 0.1 μM H-89.
Finally, we pre-treated the cells with the covalent membrane permeant-PI3K inhibitor wortmannin (Yano et al., 1993) to reveal the presence of PI3K in the transduction pathway activated by β2-adrenoceptors. As shown in Figure 7A,B, a pre-treatment for 30 min with increasing concentrations of wortmannin inhibited the Gβγ-induced increase in Ba2+ current density. Similarly, the isoprenaline-induced increase in Ba2+ current density was also completely blocked by a pre-treatment with 0.2 μM wortmannin (Figure 7A,B) in PKA inhibitor-containing solution to remove the PKA-activated pathway. No unspecific effect of wortmannin was detected and the same cells pre-treated with wortmannin were still responding to PMA by an increase in Ba2+ current (not shown). Taken together, these data suggest that after activation of β2-adrenoceptors, the Gβγ subunits released from the Gs proteins may stimulate the PI3K activity, leading to activation of PKC and stimulation of L-type Ca2+ channels.
Figure 7.
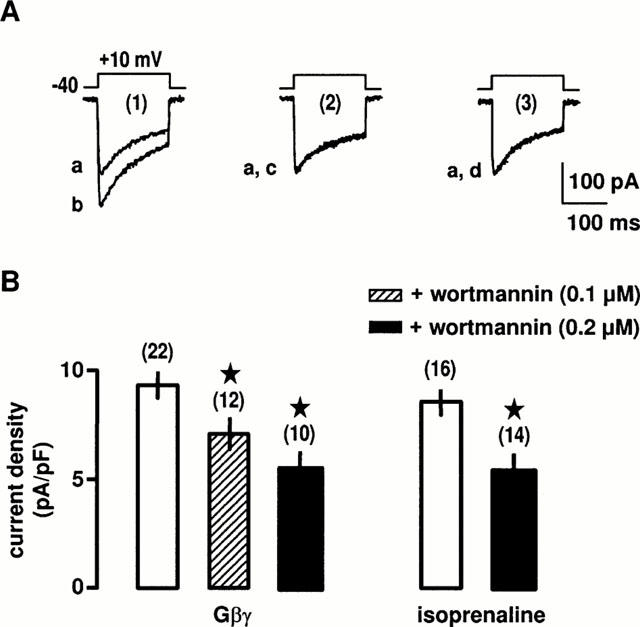
Effects of PI3K inhibitor on the β2-adrenergic stimulation of L-type Ca2+ channels. (A) Ba2+ currents evoked by a membrane depolarization to +10 mV from a holding potential of −40 mV before (a) and during the intracellular application of 0.2 μM Gβγ for 5 min (b) in control conditions (1). In the presence of 0.2 μM wortmannin for 30 min (2 and 3), Ba2+ currents before (a) and during the application of 0.2 μM Gβγ (c) or 10 μM isoprenaline for 5 min (d). (B) Compiled data showing the effects of wortmannin on the increase in Ba2+ current density evoked by 0.2 μM Gβγ and 10 μM isoprenaline. *Values significantly different from those obtained in the presence of Gβγ alone (P<0.05). External solution contained 5 mM Ba2+, 10 nM prazosin, 10 nM rauwolscine, 1 μM SR59230A and 0.1 μM H-89.
Discussion
The results of the present study indicate that in vascular myocytes the β-adrenergic stimulation of L-type Ca2+ channels depends on both β2- and β3-adrenoceptor-activated pathways. The β2-adrenergic pathway involves both the Gαs and the Gβγ subunits leading to activation of different downstream effectors, PKA and PI3K/PKC, respectively.
Although it is generally accepted that the α subunits of Gs proteins play an important role in regulation of L-type Ca2+ channels in cardiac and smooth muscles during β-adrenergic stimulation, the role of both endogenous Gαs and Gβγ subunits is not completely clear. Gαs is generally associated with activation of the adenylyl cyclase/PKA pathway and the subsequent phosphorylation of L-type Ca2+ channels. In addition, PKA phosphorylation sites have been identified on both the α and β subunits of L-type Ca2+ channels (Hofmann et al., 1994). Vascular L-type Ca2+ channels have been shown to be stimulated by isoprenaline, 8-Br-cAMP and forskolin (Loirand et al., 1992; Viard et al., 2000), as well as by intracellular applications of exogenous Gαs and the catalytic subunit of PKA (Ruiz-Velasco et al., 1998). The present study provides additional evidence that β-adrenergic stimulation via β2-adrenoceptors activates the Gαs/PKA pathway for regulation of vascular L-type Ca2+ channels. When the Gβγ pathway was eliminated by application of calphostin C or infusion with anti-Gβcom antibody or βARK1 peptide, the isoprenaline-induced stimulation of L-type Ca2+ channel was entirely abolished by PKA inhibitors (H-89 and Rp 8-Br-cyclic AMPs). A similar transduction pathway involving Gαs and PKA has been identified in response to activation of β3-adrenoceptors in the same myocytes (Viard et al., 2000). Although both PKA and PKC are involved in L-type Ca2+ channels stimulation in response to β2-adrenoceptor activation, removal of one transduction coupling does not affect the stimulation of Ca2+ channels by the other one, suggesting that there is no additivity and that each one of the PKA- and PKC-mediated mechanisms is able to produce the maximal stimulation of L-type Ca2+ channels. However, it has to be noted that concentrations producing maximal effects on Ca2+ channel activity have been used in order to identify the transduction couplings. Therefore, this does not exclude the additivity of these couplings on the same effector, i.e. the L-type Ca2+ channel if PKA and PKC are activated at lower levels during physiological stimulations.
Recently, modulation of Ca2+ channels by Gβγ subunits has received much more attention. In neurons, presynaptic Ca2+ channels have been reported to be inhibited by neuromediators via direct interaction between the Ca2+ channel complex and the Gβγ subunits released from the activated G protein heterotrimer (Ikeda, 1996; De Waard et al., 1997; Zamponi et al., 1997). In contrast, we have reported that the β1γ3 dimer of G13 transduces the angiotensin II-induced stimulation of L-type Ca2+ channels in rat portal vein myocytes (Macrez-Leprêtre et al., 1997) and that direct infusion of purified Gβγ subunits also increases the Ca2+ channel current (Viard et al., 1999). A similar stimulatory effect of purified Gβγ subunits has been reported in rabbit portal vein myocytes (Zhong et al., 1999). The present data provides evidence that the Gβγ pathway also contributes to β-adrenergic stimulation by showing that the PKA inhibitors-resistant stimulation of L-type Ca2+ channels induced by β2-adrenoceptors was inhibited by intracellular application of anti-Gβcom antibody and βARK1 peptide.
Previously, we have proposed that the action of Gβγ subunits on L-type Ca2+ channels was due to activation of PKC (Viard et al., 1999). Several pathways may lead to stimulation of PKC, including Gβγ-activated PLC, PLD and PLA2 (Clapham & Neer, 1997). In addition, we have shown recently that Gβγ may also activate PKC via PI3K; accordingly, infusion of cells with purified PI3Kγ also stimulates L-type Ca2+ channels (Viard et al., 1999). It is not yet clear what specific βγ combination of G protein is coupled to Gαs since the subunit composition of the Gs proteins that interact with the β2- and β3-adrenoceptors has not been identified. Different combinations of β and γ subunits (except β1γ1) have been reported to have similar actions on various effectors (Dolphin, 1998). However, recent data have shown that activation of mitogen-activated protein kinase/extracellular signal-regulated kinase and inhibition of adenylyl cyclases V and VI appear to be Gβ isoform specific (Gβ1 being more efficient than Gβ5; Zhang et al., 1996; Bayewitch et al., 1998). Recombinant mammalian Gβ1 – 3γ2 complexes stimulate PI3Kγ with similar potency and efficacity whereas Gβ5γ2 is not effective and appears to be unable to stimulate L-type Ca2+ channels in vascular myocytes, suggesting that signalling specificity may be encoded in the direct protein – protein interaction between Gβγ and PI3K (Maier et al., 2000). Obviously, selective protein – protein interactions represent the first step in signalling specificity and may be a possible explanation for the absence of Gβγ-activated pathway during β3-adrenoceptors activation (Viard et al., 1999). It can be postulated that the Gβγ dimers coupled to Gαs may be different in relation to the existence of two forms of Gαs, as previously suggested (Chaudhry & Granneman, 1991; Chaudhry et al., 1994). Additional factors such as cell compartmentation, spatial and temporal expression of transduction components may be also involved in signalling specificity, but they remain to be studied in more detail.
Although three β1 – 3-adrenergic subtypes have been identified in portal vein myocytes by RT – PCR, we found that only β2- and β3-adrenoceptors stimulated L-type Ca2+ channels. Mixed populations of β-adrenergic receptor subtypes have been previously reported in vascular and visceral smooth muscles, but predominant roles for β2- and β3-adrenoceptors in inducing relaxation are generally demonstrated (De Boer et al., 1993; Satake et al., 1996; Yamazaki et al., 1998; Roberts et al., 1999). Existence of several β-adrenoceptor subtypes producing the same physiological effect raises the question of the role of these receptors. Although a redundant function of these receptors cannot be discarded, specific modulations of β-adrenoceptor subtype have been previously described. For example, β3-adrenoceptors, unlike β1- and β2-subtypes, lack regulatory phosphorylation sites for G protein receptor kinases (Liggett et al., 1993) and could be relatively resistant to agonist-induced desensitization. Thus, the function of β3-adrenoceptors may become predominant after desensitization of the β2-adrenoceptors when the sympathetic nervous system is highly stimulated.
The mechanisms by which activation of β-adrenoceptors induce relaxation in smooth muscles are not fully understood although several hypotheses have been proposed including Ca2+-dependent and Ca2+-independent mechanisms.
The Ca2+-independent mechanism is mainly related to stimulation of voltage-dependent K+ channels (Aiello et al. 1998) and/or ATP-sensitive K+ channels (Randall & McCulloch, 1995) by isoprenaline and PKA which causes hyperpolarization of the membrane potential. Two Ca2+-dependent mechanisms have been suggested to underlie the isoprenaline-induced relaxation in vascular myocytes: (1) Full activation of Ca2+-sensitive large-conductance K+ channels by isoprenaline and PKA requires an increase in [Ca2+]i (Song & Simard, 1995) which can result from PKA- and PKC-mediated stimulation on L-type Ca2+ channels (Tewari & Simard, 1994; Viard et al., 1999 and the present study); (2) Alternately, PKA-induced stimulation of sarcoplasmic reticulum Ca2+-ATPases may increase the luminal Ca2+ concentration and consequently stimulate the frequency of spontaneous Ca2+ sparks (due to the opening of clustered ryanodine-sensitive Ca2+ release channels; Porter et al., 1998). Since Ca2+ sparks have been reported to activate Ca2+-sensitive K+ channels (Mironneau et al., 1996; Perez et al., 1999), their stimulation by β-agonists could also lead to membrane potential hyperpolarization, closing of voltage-dependent Ca2+ channels and relaxation.
In conclusion, our results suggest that the β-adrenergic stimulation of vascular L-type Ca2+ channels involves both β2- and β3-adrenoceptors and that these receptors exert their effects through two different pathways involving only PKA in the case of β3-adrenoceptors or both PKA and PKC for β2-adrenoceptors. The reasons for this overlapping of couplings to L-type Ca2+ channels are not known but strengthen the role of β-adrenergic agonists in the modulation of vascular relaxation.
Acknowledgments
This work was supported by grants from Centre National de la Recherche Scientifique and Fondation pour la Recherche Médicale (to P. Viard), France. We thank N. Biendon for secretarial assistance.
Abbreviations
- IBa
Ba2+ current
- PI3K
phosphoinositide 3-kinase
- PKA
protein kinase A
- PKC
protein kinase C
References
- AIELLO E.A., MALCOLM A.T., WALSH M.P., COLE W.C. Beta-adrenoceptor activation and PKA regulate delayed rectifier K+ channels of vascular smooth muscle cells. Am. J. Physiol. 1998;275:H448–H459. doi: 10.1152/ajpheart.1998.275.2.H448. [DOI] [PubMed] [Google Scholar]
- BAYEWITCH M.L., AVIDOR-REISS T., LEVY R., PFEUFFER T., NEVO I., SIMONDS W.F., VOGEL Z. Inhibition of adenylyl cyclase isoforms V and VI by various Gβγ subunits. FASEB J. 1998;12:1019–1025. doi: 10.1096/fasebj.12.11.1019. [DOI] [PubMed] [Google Scholar]
- BILSKI A.J., HALLIDAY S.E., FITZGERALD J.D., WALE J.L. The pharmacology of a beta 2-selective adrenoceptor antagonist (ICI 118,551) J. Cardiovasc. Pharmacol. 1983;5:430–437. doi: 10.1097/00005344-198305000-00013. [DOI] [PubMed] [Google Scholar]
- CHAUDHRY A., GRANNEMAN J.G. Developmental changes in adenylyl cyclase and GTP binding proteins in brown fat. Am. J. Physiol. 1991;261:R403–R411. doi: 10.1152/ajpregu.1991.261.2.R403. [DOI] [PubMed] [Google Scholar]
- CHAUDHRY A., MCKENZIE R.G., GEORGIC L.M., GRANNEMAN J.G. Differential interaction of β1- and β3-adrenergic receptors with Gi in rat adipocytes. Cell. Signalling. 1994;6:457–465. doi: 10.1016/0898-6568(94)90093-0. [DOI] [PubMed] [Google Scholar]
- CLAPHAM D.E., NEER E.J. G protein βγ subunits. Annu. Rev. Pharmacol. Toxicol. 1997;37:167–203. doi: 10.1146/annurev.pharmtox.37.1.167. [DOI] [PubMed] [Google Scholar]
- DE BOER R.E., BROUWER F., ZAAGSMA J. The β-adrenoceptors mediating relaxation of rat oesophageal muscularis mucosae are predominantly of the β3-, but also of the β2-subtype. Br. J. Pharmacol. 1993;110:442–446. doi: 10.1111/j.1476-5381.1993.tb13830.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE WAARD M., LIU H., WALKER D., SCOTT V.E., GURNETT C.A., CAMPBELL K.P. Direct binding of G-protein βγ complex to voltage-dependent Ca2+ channels. Nature. 1997;385:446–450. doi: 10.1038/385446a0. [DOI] [PubMed] [Google Scholar]
- DOGGRELL S.A. Assessment of the β2-adrenoceptor and Ca2+ channel-blocking activity of drugs with the rat portal vein. J. Pharmacol. Methods. 1990;24:145–156. doi: 10.1016/0160-5402(90)90025-g. [DOI] [PubMed] [Google Scholar]
- DOLPHIN A.C. Mechanisms of modulation of voltage-dependent calcium channels by G proteins. J. Physiol. (Lond.) 1998;506:3–11. doi: 10.1111/j.1469-7793.1998.003bx.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOFMANN F., BIEL M., FLOCKERZI V. Molecular basis for calcium channel diversity. Annu. Rev. Neurosci. 1994;17:399–418. doi: 10.1146/annurev.ne.17.030194.002151. [DOI] [PubMed] [Google Scholar]
- IKEDA S.R. Voltage-dependent modulation of N-type calcium channels by G-protein βγ subunits. Nature. 1996;380:255–258. doi: 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- LEPRÊTRE N., MIRONNEAU J., MOREL J.L. Both α1A- and α2A-adrenoreceptor subtypes stimulate voltage-operated L-type Ca2+ channels in rat portal vein myocytes: evidence for two distinct transduction pathways. J. Biol. Chem. 1994;269:29546–29552. [PubMed] [Google Scholar]
- LIGGETT S.B., FREEDMAN N.J., SCHWINN D.A., LEFKOWITZ R.J. Structural basis for receptor subtype-specific regulation revealed by a chimeric β3-/β2-adrenergic receptor. Proc. Natl. Acad. Sci. USA. 1993;90:3665–3669. doi: 10.1073/pnas.90.8.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOIRAND G., FAIDERBE S., BARON A., GEFFARD M., MIRONNEAU J. Autoanti-phosphatidylinositide antibodies specifically inhibit noradrenaline effects on Ca2+ and Cl− channels in rat portal vein myocytes. J. Biol. Chem. 1992;267:4312–4316. [PubMed] [Google Scholar]
- MACREZ N., MOREL J.L., MIRONNEAU J. Specific Gα11β3γ5 protein involvement in endothelin receptor-induced phosphatidylinositol hydrolysis and Ca2+ release in rat portal vein myocytes. Mol. Pharmacol. 1999;55:684–692. [PubMed] [Google Scholar]
- MACREZ-LEPRÊTRE N., KALKBRENNER F., MOREL J.L., SCHULTZ G., MIRONNEAU J. G protein heterotrimer Gα13β1γ3 couples the angiotensin AT1A receptor to increase in cytoplasmic Ca2+ in rat portal vein myocytes. J. Biol. Chem. 1997;272:10095–10102. doi: 10.1074/jbc.272.15.10095. [DOI] [PubMed] [Google Scholar]
- MAIER U., BABICH A., MACREZ N., LEOPOLDT D., GIERSCHIK P., ILLENBERGER D., NURNBERG B. Gβ5γ2 is a highly selective activator of phospholipid-dependent enzymes. J. Biol. Chem. 2000;275:13746–13754. doi: 10.1074/jbc.275.18.13746. [DOI] [PubMed] [Google Scholar]
- MIRONNEAU J., ARNAUDEAU S., MACREZ-LEPRETRE N., BOITTIN F.X. Ca2+ sparks and Ca2+ waves activate different Ca2+-dependent ion channels in single myocytes from rat portal vein. Cell Calcium. 1996;20:153–160. doi: 10.1016/s0143-4160(96)90104-9. [DOI] [PubMed] [Google Scholar]
- MOREL J.L., MACREZ N., MIRONNEAU J. Specific Gq protein involvement in muscarinic M3 receptor-induced specific phosphatidylinositol hydrolysis and Ca2+ release in mouse duodenal myocytes. Br. J. Pharmacol. 1997;121:451–458. doi: 10.1038/sj.bjp.0701157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAIR L.A., INGLESE J., STOFFEL R., KOCH W.J., LEFKOWITZ R.J., KWATRA M.M., GRANT A.O. Cardiac muscarinic potassium channel activity is attenuated by inhibitors of Gβγ. Circ. Res. 1995;76:832–838. doi: 10.1161/01.res.76.5.832. [DOI] [PubMed] [Google Scholar]
- PEREZ G., BONEV A.D., PATLAK J.B., NELSON M.T. Functional coupling of ryanodine receptors to KCa channels in smooth muscle cells from rat cerebral arteries. J. Gen. Physiol. 1999;113:229–237. doi: 10.1085/jgp.113.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PORTER V.A., BONEV A.D., KNOT H.J., HEPPNER T.J., STEVENSON A.S., KLEPPISCH T., LEDERER W.J., NELSON M.T. Frequency modulation of Ca2+ sparks is involved in regulation of arterial diameter by cyclic nucleotides. Am. J. Physiol. 1998;274:C1346–C1355. doi: 10.1152/ajpcell.1998.274.5.C1346. [DOI] [PubMed] [Google Scholar]
- RANDALL M.D., MCCULLOCH A.I. The involvement of ATP-sensitive potassium channels in beta-adrenoceptor-mediated vasorelaxation in the rat isolated mesenteric arterial bed. Br. J. Pharmacol. 1995;115:607–612. doi: 10.1111/j.1476-5381.1995.tb14975.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROBERTS S.J., PAPAIOANNOU M., EVANS B.A., SUMMERS R.J. Characterization of β-adrenoceptor mediated smooth muscle relaxation and the detection of mRNA for β1-, β2- and β3-adrenoceptors in rat ileum. Br. J. Pharmacol. 1999;127:949–961. doi: 10.1038/sj.bjp.0702605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RUIZ-VELAZCO V., ZHONG J., HUME J.R., KEEF K.D. Modulation of Ca2+ channels by cyclic nucleotides cross activation of opposing protein kinases in rabbit portal vein. Circ. Res. 1998;82:557–565. doi: 10.1161/01.res.82.5.557. [DOI] [PubMed] [Google Scholar]
- SATAKE N., SHIBATA M., SHIBATA S. The inhibitory effects of iberiotoxin and 4-aminopyridine on the relaxation induced by β1- and β2-adrenoceptor activation in rat aortic rings. Br. J. Pharmacol. 1996;119:505–510. doi: 10.1111/j.1476-5381.1996.tb15700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SKEBERDIS V.A., JUREVICIUS J., FISCHMEISTER A.R. Pharmacological characterization of the receptors involved in the β-adrenoceptor-mediated stimulation of the L-type Ca2+ current in frog ventricular myocytes. Br. J. Pharmacol. 1997;121:1277–1286. doi: 10.1038/sj.bjp.0701268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SONG Y., SIMARD J.M. Beta-adrenoceptor stimuation activates large-conductance Ca2+-activated K+ channels in smooth muscle cells from basilar artery of guinea pig. Pflügers Arch. 1995;430:984–993. doi: 10.1007/BF01837413. [DOI] [PubMed] [Google Scholar]
- TEWARI K, SIMARD J.M. Protein kinase A increases availability of calcium channels in smooth muscle cells from guinea pig basilar artery. Pflügers Arch. 1994;428:9–16. doi: 10.1007/BF00374746. [DOI] [PubMed] [Google Scholar]
- VIARD P., EXNER T., MAIER U., MIRONNEAU J., NÜRNBERG B., MACREZ N. Gβγ dimers stimulate vascular L-type Ca2+ channels via phosphoinositide 3-kinase. FASEB J. 1999;13:685–694. doi: 10.1096/fasebj.13.6.685. [DOI] [PubMed] [Google Scholar]
- VIARD P., MACREZ N., COUSSIN F., MOREL J.L., MIRONNEAU J. Beta-3 adrenergic stimulation of L-type Ca2+ channels in rat portal vein myocytes. Br. J. Pharmacol. 2000;129:1497–1505. doi: 10.1038/sj.bjp.0703187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG Y.X., DHULIPALA P.D., LI L., BENOVIC J.L., KOTLIKOFF M.I. Coupling of M2 muscarinic receptors to membrane ion channels via phosphoinositide 3-kinase gamma and atypical protein kinase C. J. Biol. Chem. 1999;274:13859–13864. doi: 10.1074/jbc.274.20.13859. [DOI] [PubMed] [Google Scholar]
- YAMAZAKI Y., TAKEDA H., AKAHANE M., IGAWA Y., NISHIZAWA O., AJISAWA Y. Species differences in the distribution of β-adrenoceptor subtypes in bladder smooth muscles. Br. J. Pharmacol. 1998;124:593–599. doi: 10.1038/sj.bjp.0701870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANO H., NAKANISHI S., KIMURA K., HANAI N., SAITOH Y., FUKUI Y., NONOMURA Y., MATSUDA Y. Inhibition of histamine secretion by wortmannin through the blockade of phosphatidyl 3-kinase in RBL-2H3 cells. J. Biol. Chem. 1993;268:25846–25856. [PubMed] [Google Scholar]
- ZAMPONI G.W., BOURINET E., NELSON D., NARGEOT J., SNUTCH T.P. Crosstalk between G proteins and protein kinase C mediated by the calcium channel alpha1 subunit. Nature. 1997;385:442–446. doi: 10.1038/385442a0. [DOI] [PubMed] [Google Scholar]
- ZHANG S., COSO O., LEE C., GUTKIND J.S., SIMONDS W.F. Selective activation of effector pathways by brain-specific G protein β5. J. Biol. Chem. 1996;271:33575–33579. doi: 10.1074/jbc.271.52.33575. [DOI] [PubMed] [Google Scholar]
- ZHONG J., DESSAUER C.W., KEEF K.D., HUME J.R. Regulation of L-type Ca2+ channels in rabbit portal vein by G protein αs and βγ. J. Physiol. (Lond.) 1999;517:109–120. doi: 10.1111/j.1469-7793.1999.0109z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]