

Abstract
Previous studies indicate that 3-nitropropionic acid (3-NPA) neurotoxicity involves the excitotoxic activation of N-methyl-D-aspartate (NMDA) receptors. Thus, we examined the effect of orphenadrine (an anticholinergic drug with NMDA receptor antagonist properties) on 3-NPA neurotoxicity in both cultured rat cerebellar granule cells (CGCs) and in rats.
Orphenadrine protected CGCs from 3-NPA-induced mortality, as assessed by both the neutral red viability assay and laser scanning cytometry, using propidium iodide staining.
For rats, two indirect markers of neuronal damage were used: the binding of [3H]-PK 11195 to the peripheral-type benzodiazepine receptor (PBR), a microglial marker, and expression of the 27 kD heat-shock protein (HSP27), a marker of activated astroglia. Systemic administration of 3-NPA (30 mg kg−1 per day for 3 days) induced a 170% increase in [3H]-PK 11195 binding, and expression of HSP27.
Both the increase in [3H]-PK 11195 and HSP 27 expression were prevented by previous administration of 30 mg kg−1 per day of orphenadrine for 3 days. Lower doses (10 and 20 mg kg−1) had no protective effect. Orphenadrine also reduced 3-NPA-induced mortality in a dose-dependent manner.
We propose that orphenadrine or orphenadrine-like drugs could be used to treat neurodegenerative disorders mediated by overactivation of NMDA receptors.
Keywords: Orphenadrine, HSP27, peripheral benzodiazepine receptor, N-methyl-D-aspartate, microgliosis, neurodegeneration, laser scanning cytometry
Introduction
Administration of 3-nitropropionic acid (3-NPA) to rats induces neuronal damage that mainly affects the striatum and mimics many of the histological and neurochemical features of Huntington's disease (HD) (Wüllner et al., 1994; Borlongan et al., 1995; Dawson et al., 1995; Kodsi & Swerlow, 1997; Sato et al., 1997; Brouillet et al., 1999). 3-NPA inhibits irreversibly succinate dehydrogenase, localized in the mitochondrial inner membrane, and complexes II and III of the respiratory chain and the tricarboxylic acid cycle (Alexi et al., 1998). These effects decrease ATP levels, leading to changes in the neuronal membrane potential. Mg2+, which normally blocks the N-methyl-D-aspartate (NMDA) receptor-coupled channel, is released. Glutamate then activates NMDA receptors and increases intracellular calcium, which initiates an excitotoxic cascade (Zeewalk & Nicklas, 1992; Beal et al., 1993). Studies of experimental models of Huntington's disease in mice (Bogdanow et al., 1998; Levine et al., 1999) and rats (Wüllner et al., 1994) report an increase in the response of striatal neurons to NMDA activation.
Several drugs such as antioxidants, lamotrigine and coenzyme Q10 (Schulz et al., 1996a,1996b) have been tested to prevent the neuronal damage induced by 3-NPA. Since a link has been suggested between mitochondrial dysfunction, striatal neurodegeneration and NMDA receptor activation, the neurodegenerative process in HD or 3-NPA-induced neurotoxicity could be prevented by blockade of NMDA receptors. MK-801 (dizocilpine), a non-competitive antagonist that blocks the NMDA receptor by binding to a site within its coupled ion channel (the phencyclidine site), protects against 3-NPA-induced neurotoxicity in rat brain striatum (Lee et al., 2000). However, the use of NMDA receptor antagonists in vivo causes serious side effects such as hallucinations and psychotomimetic behaviour, mainly due to a complete blockade of the NMDA receptor function.
Orphenadrine is a central nervous system (CNS)-acting antimuscarinic drug used in the treatment of Parkinson's disease that is devoid of psychotomimetic side effects. In addition, orphenadrine acts as a ‘mild' non-competitive antagonist of NMDA receptors by binding to the phencyclidine site (Kornhuber et al., 1995). It prevents the neurotoxicity induced by kainic acid in rats, and glutamate-induced neuronal cell death in cerebellar granule cells (CGCs) (Sureda et al., 1999). Moreover, some low-affinity channel-blocking antagonists at the NMDA receptor, such as memantine and amantadine, which possess fast strongly voltage-dependent open channel kinetics, are also neuroprotective and well tolerated (Parsons et al., 1999).
After brain damage, proliferation of microglia occurs (Streit et al., 1999). In the brain, peripheral-type benzodiazepine receptors (PBR) are mainly localized in microglia (Stephenson et al., 1995; Kuhlmann & Guilarte, 2000). Therefore, microgliosis is accompanied by an increase in the number of PBRs in the affected area (Benavides et al., 1990; Stephenson et al., 1995) and PBR is thus a suitable indirect indicator of neuronal damage (Benavides et al., 1987). In addition, the 27 kD heat-shock protein (HSP27) is expressed in astroglia after brain damage (Renkawek et al., 1993), and is used as a marker of astroglial stress. We hypothesize that orphenadrine may be suitable for the treatment of neurodegenerative disorders involving excitotoxicity. To test this, we evaluated the protective effect of orphenadrine against 3-NPA-induced neurotoxicity in both CGCs and in rat brain by measuring, respectively, its effect on cell viability and the increase of PBR density and HSP27 expression.
Methods
Drugs
3-NPA, orphenadrine, neutral red and cell culture salts and enzymes were obtained from Sigma Chemical Co. (St. Louis, MO, U.S.A.). Ro 5-4864 was purchased from Fluka Chemie AG (Germany). [3H]-PK 11195 (86 Ci mmol−1) was from New England Nuclear (Wilmington, DE, U.S.A.). Cell culture media were obtained from GIBCO (Life Technologies, Paisley, U.K.). Other chemical reagents were of analytical quality.
Cerebellar granule cell cultures
Primary cultures of CGCs were prepared from 7-day-old Sprague Dawley rat pups using the method of Nicoletti et al. (1986). Briefly, cerebella were quickly removed and cleaned of meninges, followed by manual slicing with a sterile blade, dissociation with trypsin and treatment with DNase. Cells were adjusted at 8×105 cells ml−1 and were plated on both poly-L-lysine-coated 24-well plates and glass coverslips at a density of 320,000 cells cm−2. Cultures were grown in Eagle's medium (BME basal medium) containing 10% FCS, 2 mM L-glutamine, 0.1 mg ml−1 gentamicin and 25 mM of KCl. Cytosine arabinoside (10 μM) was added 16 – 18 h after plating, in order to inhibit the growth of non-neuronal cells. Cultures prepared by this method were more than 95% enriched in granule neurons.
Treatment of CGCs and survival assay
CGCs were used after 7 – 10 days in culture. 3-NPA was dissolved in culture medium and neutralized with NaOH to pH 7.4 before being added to the cells at different concentrations. To investigate the neuroprotective effect of orphenadrine, different concentrations of this drug were added to the medium 30 min before 3-NPA. Cell death was determined 24 h after 3-NPA addition using the neutral red assay and laser scanning cytometry.
Laser scanning cytometry
CGCs grown on glass coverslips were stained for at least 10 min with propidium iodide at a final concentration of 10 μg ml−1 (Darzynkiewicz et al., 1999). Propidium iodide crosses disrupted cell membranes and stains the DNA of dead cells. Cell viability was measured using a laser scanning cytometer (LSC) (CompuCyte, Cambridge, MA, U.S.A.) with a red fluorescence detector. The laser scanned the coverslip and the cytometer stored the position, scatter and fluorescence of each cell, so that they could be easily localized and observed through the coupled microscope, equipped with a CCD camera. A total number of 2500 cells were analysed per coverslip and representative images were taken with a Leica DMRB fluo microscope using the 20× object lens.
Neutral red viability assay
Twenty-four hours after 3-NPA addition, 10 μl of a neutral red (NR) solution was added to each culture well to give a final concentration of 40 μg ml−1 (Babich & Borenfreund, 1991). The NR solution had been preincubated overnight at 37°C and centrifuged (1500×g for 10 min) prior to use to remove fine precipitates of dye crystals. The assay plate was then returned to the incubator for another 3 h to allow for uptake of the supravital dye into the viable CGCs. Thereafter, the media was removed and the cells were rapidly washed with PBS followed by the addition of 0.2 ml of a solution of 1% acetic acid-50% ethanol to extract the dye from the cells. After 10 min at room temperature and gentle shaking, the absorbance of the extracted dye was measured in a microplate spectrophotometer (BioRad 550) equipped with a 540 nm filter. Surviving cells are given as percentage compared with untreated cells (controls). The EC50 value for 3-NPA was calculated by non-linear regression using GraphPAD Prism software (GraphPAD Software, San Diego, CA, U.S.A.).
In vivo drug treatments
The ethics committee of the Universitat de Barcelona, according to the European Community guidelines, approved the protocol concerning the use of experimental animals carried out in this study. Adult male Sprague Dawley rats (275 – 300 g) were obtained from Harlan Ibérica (Spain). They were housed at 21±1°C and maintained under a 12 h light/dark cycle. On day 1, 3-NPA was dissolved in saline, adjusted to pH 7.4 with NaOH and administered to a group of 16 rats (3-NPA group) at a dose of 30 mg kg−1 i.p. once a day for 3 days. With this dosing regimen the animals displayed the symptoms of 3-NPA toxicity after 3 – 4 days. Three groups of 10 animals received, respectively, a single dose of 10, 20 or 30 mg kg−1 i.p. of orphenadrine (dissolved in saline) 30 min before 3-NPA (30 mg kg−1, i.p.), on each day of treatment. Another group of 10 animals were injected with saline (control group) and three groups of three animals per group received orphenadrine alone at 10, 20 or 30 mg kg−1, i.p., respectively (orphenadrine controls). The animals were weighed on days 1 – 4 and 8. On day 8, surviving rats were killed by decapitation, and striatum, hippocampus and cerebral cortex were dissected out, frozen on dry ice and stored at −80°C.
Tissue homogenate preparation
Tissue samples were thawed on ice, weighed and homogenized in 10 volumes of cold homogenization buffer (5 mM Tris-HCl, 320 mM sucrose, 4.5 μg μl−1 aprotinin, 10−4 M phenylmethylsulphonyl fluoride (PMSF) and 10−3 M sodium orthovanadate, pH 7.4) with a Potter-Elvehjem Teflon-glass homogenizer. The homogenates were centrifuged at 15,000×g for 30 min at 4°C. The pellets were resuspended in buffer, washed twice and re-centrifuged at 15,000×g for 30 min. The final pellets were resuspended in Tris-HCl 50 mM buffer (pH 7.4) containing protease inhibitors (4.5 μg μl−1 aprotinin, 10−4 M PMSF and 10−3 M sodium orthovanadate). Striatum preparations from five saline-treated (controls) and four 3-NPA-treated rats were pooled and reserved for saturation binding experiments. The remaining homogenates were aliquoted separately for both binding and Western blot assays. The homogenates were stored at −80°C until use. Protein content was determined by the Bradford method (Bradford, 1976), using bovine serum albumin as the standard.
Radioligand binding assays
Equilibrium binding assays were performed at 0 – 4°C using [3H]-PK 11195. Assays were done in a final volume of 0.25 ml (Tris-HCl 50 mM, pH 7.4), which contained 125 μl of [3H]-PK 11195 (in a concentration range of 0.5 – 15 nM) and 125 μl of tissue homogenate preparation (100 μg of protein per assay). Specific binding was defined as the difference between the radioactivity bound in the absence (total binding) and in the presence (non-specific binding) of 10 μM of unlabelled Ro 5-4864.
After incubation for 120 min, samples were filtered under vacuum on to Whatman GF/B glass fibre filters pre-soaked in 0.5% polyethyleneimine. Filters were rapidly washed three times in 4 ml of ice-cold Tris-HCl (50 mM) and placed in vials containing 10 ml of Cocktail Scharlau Biogreen 1 (FEROSA, Spain). Radioactivity was measured by liquid scintillation spectroscopy in a Beckman LS-1800 with an efficiency of 40%. The specific binding in these conditions was about 90%.
Control 3-NPA and orphenadrine treated animals were compared by measuring specific [3H]-PK 11195 binding at a concentration of 2 nM in the conditions described above.
Data analyses and statistics
Analysis of saturation isotherms (Kd, dissociation constant; Bmax, maximum density of binding sites) was determined by computer-assisted non-linear regression, using Radlig 4.0 software (Biosoft, Elsevier). Saturation data are expressed as the best fit value±standard error of the mean (s.e.mean) obtained from three separate experiments carried out in duplicate. Other data are expressed as mean±s.e.mean from n determinations carried out in duplicate. Multiple mean comparisons were done by one-way analysis of variance (ANOVA) followed by Tukey's test. Differences were considered to be significant at the P<0.05 level.
Western blot analysis
Aliquots of tissue homogenate, containing 30 μg of protein per sample, were placed in sample buffer (0.5 M Tris-HCl pH 6.8, 10% glycerol, 2% (w v−1) SDS, 5% (v v−1) 2-β-mercaptoethanol, 0.05% bromophenol blue) and denaturalized by boiling at 95 – 100°C for 5 min.
Samples were separated by electrophoresis on 10% acrylamide gels. Subsequently, proteins were transferred to polyvinylidene fluoride sheets (Immobilon™-P, Millipore Corp., Bedford, MA, U.S.A.) using a transblot apparatus (Bio-Rad Laboratories, Hercules, CA, U.S.A.). Membranes were blocked overnight with 5% non-fat milk dissolved in TBS-T buffer (Tris 50 mM; NaCl 1.5%; Tween 20, 0.05%, pH 7.5). Membranes were then incubated with a primary rabbit polyclonal antibody anti-HSP27 (1 : 5000, SPA-801, StressGen Biotechnologies Corp., Victoria, BC, Canada). After 90 min, blots were washed thoroughly in TBS-T buffer and incubated for 1 h with peroxidase-conjugated anti rabbit IgG antibody (Amersham Corp., Arlington Heights, IL, U.S.A.). Immunoreactive protein was visualized using a chemiluminescence-based detection kit according to the manufacturer's protocol (ECL kit; Amersham Corp., Arlington Heights, IL, U.S.A.).
Results
Neuroprotective effects of orphenadrine against 3-NPA-induced neurotoxicity in cerebellar granule cell cultures
Twenty-four hours exposure to 3-NPA (1 – 1000 μM) caused a concentration-dependent neuronal mortality, as assessed by a decrease in neutral red accumulation (Figure 1) and an increase in propidium iodide staining (Figure 2). The EC50 value for 3-NPA was 144±3 μM (n=5). When CGCs were exposed to 100 μM 3-NPA in the presence of different concentrations of orphenadrine, they were protected from death (Figures 2 and 3). The highest cytoprotective effect of orphenadrine was at a concentration of 6 μM (Figure 3). Doses of orphenadrine above 24 μM failed to prevent 3-NPA toxicity and concentrations above 48 μM potentiated cell death. Orphenadrine alone, at a concentration of 12 μM, had no effect either on cell viability or on morphology but, at a concentration of 48 μM, induced a rapid necrotic death (data not shown). Visually, 3-NPA induced cellular aggregation, volume diminution and neurite fragmentation that were prevented by orphenadrine pre-treatment (Figure 4).
Figure 1.
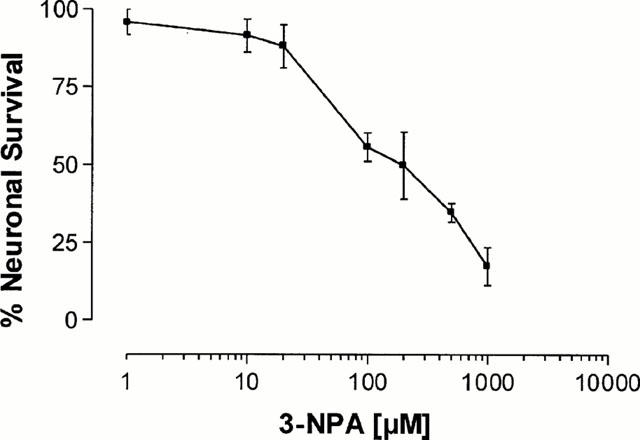
Cerebellar granule cell viability after treatment with 3-NPA. 7 – 10 days old CGCs were treated with a range of concentrations of 3-NPA (1 – 1000 μM). Cell viability was assessed 24 h after by neutral red assay. The results are presented as the means±s.e.mean from three to five separate experiments carried out in quadruplicate.
Figure 2.
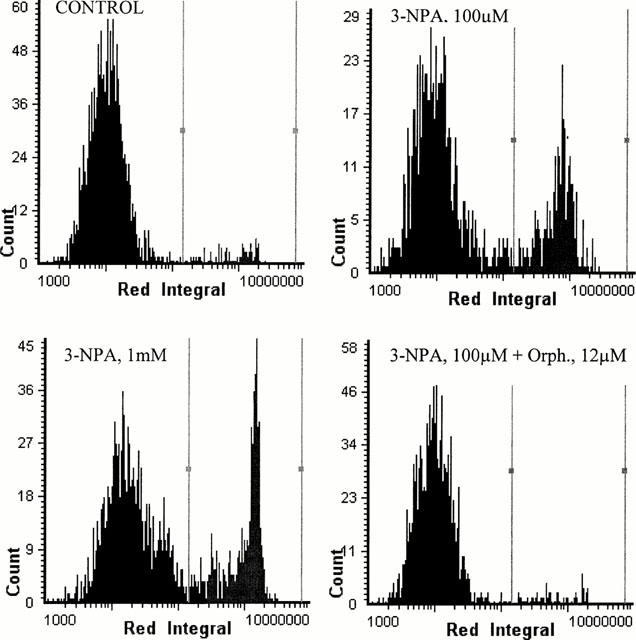
Laser scanning histograms showing the effect of orphenadrine against 3-NPA induced toxicity in CGCs. Pre-treatment of CGCs with orphenadrine induced a diminution of propidium iodide staining (cells between vertical bars), indicating a neuroprotective effect. The ordinate is the number of cells at each propidium iodide fluorescence intensity. Histograms are each one representative of those obtained from three different experiments carried out in triplicate.
Figure 3.
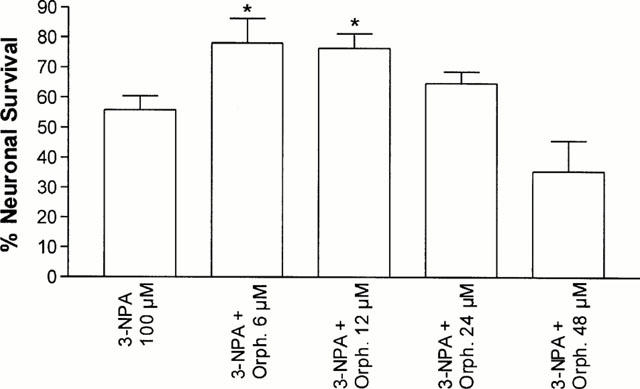
Orphenadrine protects cerebellar granule cells from 3-NPA neurotoxicity. Cerebellar granule cells were pre-treated with orphenadrine previously to 3-NPA (100 μM). Cell survival was assessed after 24 h by neutral red assay. The results are given as the percentage of absorbance of treated cells referred to that obtained in control cells. The results are presented as the means±s.e.mean from three to five separate experiments carried out in quadruplicate. Statistical analysis was performed using ANOVA. (*P<0.05 vs 3-NPA group).
Figure 4.
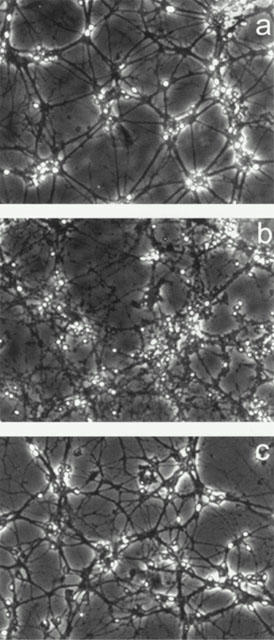
Phase-contrast micrographs of cerebellar granule cells injuried by 3-NPA and protected by orphenadrine. 3-NPA induced cellular aggregation, volume diminution and neurite fragmentation that were prevented by orphenadrine pre-treatment. Control cells (a), cells treated with 3-NPA 100 μM (b) and 3-NPA 100 μM plus 12 μM orphenadrine (c) are shown.
Effect on mortality and loss of body weight
3-NPA-treated animals showed general incoordination, drowsiness and general weakness. Some showed hindlimb paralysis without rigidity, recumbency and some of these died. Mortality in the groups 3-NPA and 3-NPA pre-treated with 10 mg kg−1 orphenadrine was 40%. The groups pre-treated with 20 and 30 mg kg−1 orphenadrine showed 30 and 10% mortality, respectively.
All the groups of 3-NPA treated rats suffered a significant loss of body weight (Figure 5), which continued until day 4, when it was maximum. After day 4, a partial recovery of body weight was observed, although control values were not reached. Although the differences were not statistically significant, rats receiving 30 mg kg−1 orphenadrine before 3-NPA showed a slightly faster recovery of body weight.
Figure 5.
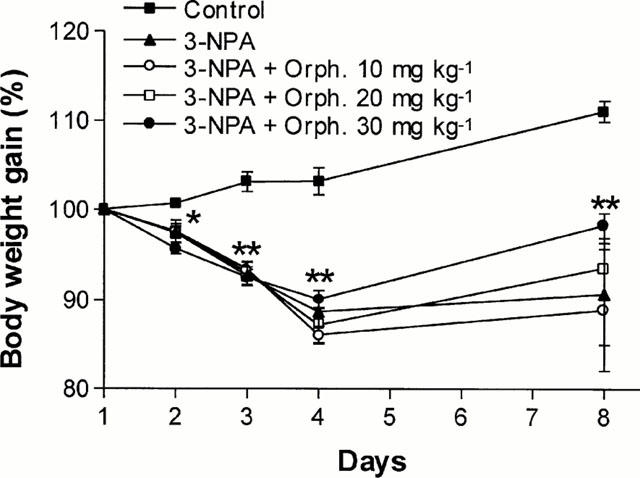
Temporal evolution of rats' body weight during (days 1 – 3) and after (days 4 – 8) treatment with 3-NPA or orphenadrine plus 3-NPA. Data are expressed as means±s.e.mean of the percentage of initial body weight (100%) for the surviving rats of each group (*P<0.05 and **P<0.01, for all the groups treated with 3-NPA vs control group).
[3H]-PK 11195 binding
Specific binding of [3H]-PK 11195 to homogenate preparations of striatum was saturable and of high affinity (Figure 6). Using non-linear regression analysis, our data were fitted by a single-site model, which yielded a Kd of 1.7±0.2 nM (n=3) for control and of 4.7±1 nM (n=3) for 3-NPA rats (P<0.01). Maximal binding site densities were respectively 400±37 fmol mg−1 protein and 843±45 fmol mg−1 protein (P<0.0001). All subsequent assays were performed using 2 nM [3H]-PK 11195.
Figure 6.
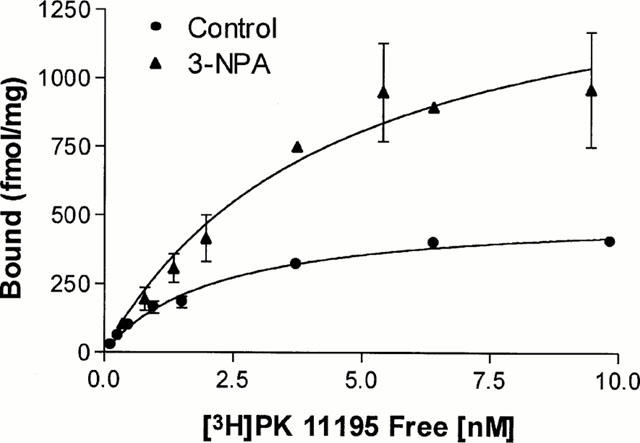
Saturation curves of [3H]-PK 11195 binding to striatum. Rats received daily 30 mg kg−1 3-NPA during 3 days and were sacrificed at day 8 from the start of treatment. Specific binding at increasing concentrations of [3H]-PK 11195 was measured to homogenates from control and 3-NPA-treated rats. Results are means±s.e.mean of data obtained from three separate experiments carried out in duplicate. Details of the binding procedure are described in Methods.
The density of [3H]-PK 11195 (2 nM) binding sites in striatum is given in Figure 7a. 3-NPA (30 mg kg−1 per day for 3 days) induced a significant 170% increase in PBR (171±16 fmol mg−1, n=5, for control compared with 463±57 fmol mg−1, n=6, for 3-NPA-treated rats, P<0.05). Pre-treatment with 30 mg kg−1 orphenadrine per day prevented the 3-NPA-induced PBR increase to a value not different from control values (244±9 fmol mg−1, n=9, P<0.05 vs 3-NPA group; P>0.05 vs control group). Lower doses of orphenadrine had no effect on the 3-NPA-induced increase in PBR. Orphenadrine alone had no effect on PBR levels (data not shown).
Figure 7.
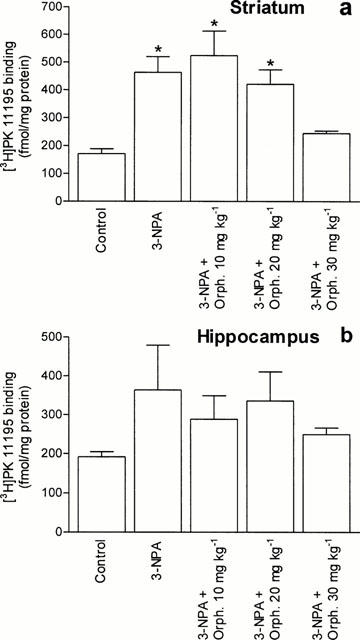
Effect of pre-treatment with orphenadrine on 3-NPA-induced increase in PBR density. [3H]-PK 11195 binding (2 nM) was performed to homogenate preparations of striatum (a) and hippocampus (b) of individual rats, as described in Methods. Values are expressed as means±s.e.mean of these obtained from five to nine animals. (*P<0.05 vs control).
3-NPA increased PBR levels in the hippocampus of three animals (193±13 fmol/mg for control, n=5, and 443±118 for 3-NPA treated, n=3, P<0.05). Figure 7b shows the mean PBR levels of all the treated animals. The PRB levels in the hippocampus from animals pre-treated with 30 mg kg−1 orphenadrine per day were not different from control (249±17 fmol mg−1, n=9, P>0.05 vs control; P<0.05 vs 3-NPA). No increase in PBR levels was found in cortex of 3-NPA rats (224±44 fmol mg−1, n=6, vs 205±29 fmol mg−1, n=5, control group, P>0.05).
Western blot analysis
The Western blot analysis of stress protein in the striatum showed a marked expression of HSP27 in 3-NPA rats, whereas no expression was detected either in control or in animals treated with orphenadrine alone. Pre-treatment with 30 mg kg−1 orphenadrine before 3-NPA administration inhibited HSP27 expression. Lower doses of orphenadrine had no effect on HSP27 expression after 3-NPA administration. Only three of the rats treated with 3-NPA alone expressed HSP27 in the hippocampus but none of the rats pre-treated with 30 mg kg−1 of orphenadrine expressed this protein in this area (n=6 for each group). Figure 8 shows representative Western blots of striatum and hippocampus originating from the same rats. The animals showing the highest levels of PBR also showed the highest HSP27 expression.
Figure 8.
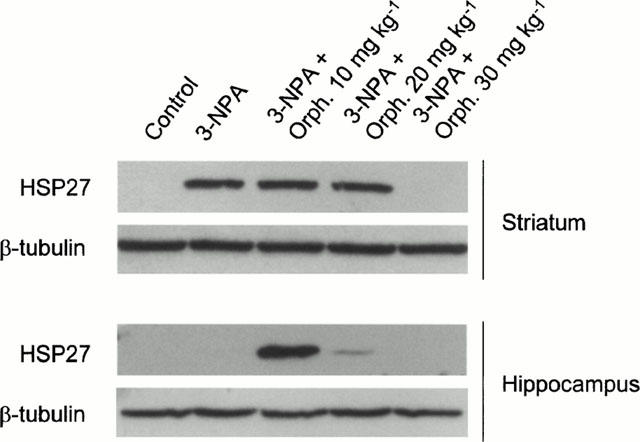
Representative Western blots showing HSP27 expression in striatum and hippocampus of the same rats after treatment with 3-NPA, alone or preceded by different doses of orphenadrine. Pre-treatment with 30 mg kg−1 orphenadrine prevented HSP27 expression induced by 3-NPA treatment in the striatum. Owing to the preference of 3-NPA lesions for the striatum, not all the rats treated with 3-NPA alone expressed this protein in the hippocampus (second lane). Western blots were performed in duplicate, with samples originating from at least four animals of each group of treatment. β-tubulin expression was used as a gel load control.
Discussion
This study demonstrates that orphenadrine protects in vitro and in vivo against 3-NPA-induced neuronal damage because it prevents cell mortality and the expression of two markers of neuronal damage (HSP27 and increase in PBR number) in the striatum of rats.
Histological and neurochemical analyses have demonstrated that systemic administration of 3-NPA to mature rats induces striatal damage (Beal et al., 1993; Miller & Zaborszky, 1997; Lee et al., 2000). Microgliosis accompanies the neuronal degeneration induced by several neurotoxins in rat brain. As in the brain PBRs are concentrated in the microglia, their increase is widely being used as an indirect marker of neuronal damage (Benavides et al., 1990; Stephenson et al., 1995). For example, density of PBR increases in brain regions injured by excitotoxicity (Shoemaker et al., 1982; Camins et al., 1998) or focal ischemia (Benavides et al., 1990). In our experiments, 3-NPA administration to rats increased both Bmax and Kd for [3H]-PK 11195 binding. Therefore, these results indicate that the damage induced by 3-NPA is also accompanied by an increase in [3H]-PK 11195 binding to striatum. This increase is prevented by orphenadrine at a dose of 30 mg kg−1 before 3-NPA administration, indicating a protective effect. The mitochondrial damage induced by this neurotoxin could lead to an alteration of mitochondrial membrane structure that could be responsible for the decrease of the [3H]-PK 11195 affinity for the peripheral benzodiazepine receptor in the damaged striatum.
Several reports show that acute intraperitoneal administration of 3-NPA can damage cerebral areas other than the striatum, such as the hippocampus and the cortex (Beal et al., 1993; Miller & Zaborsky, 1997; Brouillet et al., 1999; Lee et al., 2000). Our results show that, in some of the animals, 3-NPA can induce an increase of PBR levels in the hippocampus. This increase was prevented by pre-treatment with 30 mg kg−1 orphenadrine. As we did not detect a significant increase in [3H]-PK 11195 binding in the cortex, this corroborates the reported selectivity for 3-NPA-induced damage, which affects mainly the striatum and, in some cases, can spread to the hippocampus.
On the other hand orphenadrine, at a dose of 30 mg kg−1, prevented the expression of HSP27 in the striatum after 3-NPA administration. Kato et al. (1994; 1995) described that the distribution of HSP27 corresponds to reactive astroglial cells. Moreover, its expression is increased after ischemia (Kato et al., 1994; 1995), administration of kainic acid (Plumier et al., 1996) and in Alzheimer's disease (Renkawek et al., 1993). In addition, astrocytosis is prominent in both Huntington's disease (Vonsattel et al., 1985) and in chronic experimental 3-NPA intoxication (Beal et al., 1993). Therefore HSP27 expression is a useful parameter to measure the extent of the 3-NPA toxicity in the striatum and, as it was prevented by orphenadrine (30 mg kg−1), indicates a neuroprotective effect of this drug. Owing to the reported preference of 3-NPA-induced lesions for the striatum, not all the rats treated with 3-NPA alone expressed HSP27 in the hippocampus. This is in agreement with the findings obtained by Beal et al. (1993), who reported that only some of the animals that received 3-NPA showed gliosis and neuronal loss in the hippocampus, in addition to striatal lesions. Thus the expression of HSP27 in the hippocampus is not reliable for measuring neuroprotective effects against 3-NPA as it does not follow the same pattern than the striatum. However, the fact that none of the rats pre-treated with orphenadrine 30 mg kg−1 expressed HSP27 in the hippocampus gives support to the protective effects observed in striatum.
HSP27 has a key role in reactive astrocytes by regulating the dynamics of actin filaments, probably during structural remodelling after neuronal degeneration (Plumier et al., 1996). Transfection of HSP27 has an antiapoptotic effect in neurons through an increase in the levels of glutathione (Guénal et al., 1997; Mehlen et al., 1999; Wagstaff et al., 1999). It remains to be determined whether the increase in HSP27 expression in astrocytes in vivo is also neuroprotective. In this context, the increase in HSP27 expression found after 3-NPA administration could be a response to neural injury. Moreover, the prevention of 3-NPA-induced HSP27 expression by orphenadrine contributes to the neuroprotective effect of this drug in this experimental model of Huntington's disease.
Following 3-NPA administration, all groups of rats suffered a loss of body weight, which was slightly recovered after discontinuing 3-NPA. Both central and peripheral effects of 3-NPA could explain this effect. The striatal lesions and bradykinesia (Guyot et al., 1997) could be partly responsible for reducing rat appetite and food intake. However, peripheral effects such as the impairment of energetic metabolism, mobilization of energy stores and lipid peroxidation by nitroanions (Fu et al., 1995) would be the main cause of weight loss. The rats pre-treated with 30 mg kg−1 orphenadrine also suffered from loss of weight and, although they recovered slightly better than the other groups, they did not reach control levels. This observation supports that the protective effect of orphenadrine takes place mainly in the CNS, where NMDA receptors are located.
Several hypotheses have been proposed to explain the mechanism of neuronal damage induced by 3-NPA. Among them, indirect excitotoxic lesion by NMDA receptor activation is the most widely described (Wüllner et al., 1994) although currently it is more accepted that 3-NPA neurotoxicity is the result of cooperative pathways between excitotoxicity, metabolic compromise and oxidative stress (reviewed by Alexi et al., 1998). 3-NPA, by inhibiting succinate dehydrogenase, a key enzyme of the Kreb's cycle, causes an energetic impairment which depletes ATP. The lack of ATP causes the failure of ATP-dependent ion pumps and channels which results in depolarization of glutamaceptive neurons and relief of the Mg2+ block in the NMDA receptor channel. Then physiological concentrations of glutamate can activate the receptor channel, leading to Ca2+ influx. This Ca2+ influx cannot be reversed because of the depleted ATP, leading to Ca2+ overload and excitotoxicity. On the other hand, Ca2+ activates enzymes such as nitric oxide synthase, which produces NO and free radicals that induce oxidative stress together with those produced by impaired mitochondrial function. In addition, dopamine, which has been found to increase after 3-NPA systemic administration, can also lead to the production of free radicals (Nishino et al., 1997). Moreover, energy collapse can by itself cause cell death due to inhibition of vital protein synthesis, which depends on ATP.
In view of the several pathways implicated, several strategies have been adopted to prevent 3-NPA-induced neuronal damage. Schulz et al. (1996a,1996b) assayed in vivo several compounds acting at these different pathways, that is: free radical trappers (S-PBN), coenzyme Q10, which improves mitochondrial function, and compounds that inhibit glutamate release (lamotrigine) or block NMDA receptors (MK-801, memantine). All these compounds showed a protective effect that was potentiated when they were associated with another compound acting at a different point. The protective effects of MK-801 and lamotrigine have also been assessed in vivo through magnetic resonance imaging and proton magnetic resonance spectroscopy (Lee et al., 2000). On the other hand, the key role for free radicals in 3-NPA toxicity has been demonstrated in transgenic mice expressing increased free radical scavenging activity (Beal et al., 1995) as they were more resistant than wild-type to 3-NPA neurotoxicity. Fu et al. (1995) found an increase in cerebral activity of superoxide dismutase and content of malonaldehyde that indicates a defensive response to oxidative stress, as well as an increased lipid peroxidation in liver. A recent study (La Fontaine et al., 2000) has measured brain protein oxidation in rats after 3-NPA treatment and provides evidence that a widespread oxidative damage occurs prior to the appearance of morphologic lesions.
Such cooperative neurotoxic pathways have also been evidenced in vitro. It has been demonstrated that 3-NPA is toxic to CGCs through a mechanism involving NMDA receptors activation because both competitive (AP5) and non-competitive (MK-801) NMDA receptor antagonists prevent its neurotoxic effect (Weller & Paul, 1993). Similar results have been reported for hippocampal neurons (Pang & Geddes, 1997). But in addition to NMDA receptor activation there is an increased Ca2+ influx and formation of free radicals because free radical scavengers protect CGCs from 3-NPA-induced damage (Olsen et al., 1999). Also, there exists a synergism between the toxicities of 3-NPA and NMDA in striatal cultures (Greene et al., 1998), indicating that excitotoxicity and metabolic impairment act coordinately. In fact, energy-compromised neurons are more vulnerable to excitotoxicity of glutamate (Novelli et al., 1988). As far as the resulting cell death pathway is concerned, some authors (Weller & Paul, 1993; Pang & Geddes, 1997) reported that, in neuronal cultures, 3-NPA toxicity could be prevented after a short exposure (until 24 h) to this toxin with NMDA receptor antagonists but, after longer periods of exposure, these compounds were unable to prevent cell death. These observations pointed to the existence of an acute excitotoxic cell death and a slowly evolving apoptotic death that cannot be attenuated by NMDA receptor blockade, as a result of metabolic impairment. Our experiments were performed at 24 h exposure to 3-NPA and therefore the death was mostly excitotoxic. Under these circumstances, NMDA receptor blockade by orphenadrine could be protective, as shown by our results. We used CGCs after 7 – 10 days in culture, when the neurite networking was complete. Older cells were not assayed because an increased sensibility of CGCs to 3-NPA toxicity after day 13 in culture has been reported (Weller & Paul, 1993).
Orphenadrine, in addition to its antimuscarinic properties, acts as a non-competitive NMDA receptor antagonist, and inhibits [3H]-MK-801 binding to the phencyclidine (PCP) site at the NMDA receptor complex (Kornhuber et al., 1995). Thus, protection by orphenadrine may be due to the blockade of NMDA receptors. Indeed, orphenadrine protects CGCs from glutamate neurotoxicity and protects rat brain from the excitotoxic lesion induced by kainic acid (Sureda et al., 1999). Similarly, in the present study we have found that orphenadrine protects CGCs from 3-NPA-induced toxicity.
High-affinity NMDA receptor channel blockers such as MK-801 and PCP produce severe psychotomimetic side effects owing to a persistent blockade of the receptors and cannot be used therapeutically (see Parsons et al., 1999 for a review). Their slow unblocking kinetics and low voltage dependency make them unable to leave the channel even when the neuron depolarizes after a physiological stimulus by glutamate (mM concentrations). Under pathological conditions, NMDA receptors are activated by lower (μM) concentrations of glutamate than under physiological conditions, but for much longer periods of time (Mitani et al., 1992). Thus, there would be of interest NMDA receptor channel blockers that could discriminate between physiological and pathological activation and would block only under pathological circumstances. Memantine and amantadine are compounds that block the NMDA receptor channel at low micromolar concentrations (Ki 0.5 and 10 μM, respectively, whereas MK-801 and PCP show nanomolar affinity) and show fast, strongly voltage-dependent open channel kinetics (Sobolevsky et al., 1998; Parsons et al., 1999). This means that, under resting conditions, both Mg2+ and these compounds occupy the channel. Likewise, both are able to leave the channel upon strong synaptic depolarization (physiological conditions) due to their pronounced voltage-dependency and rapid unblocking kinetics. However, memantine and amantadine do not leave the channel so easily as Mg2+ upon moderate prolonged depolarization (pathologic). These features make these compounds able to permit physiological glutamatergic transmission and to block excitotoxic stimulus.
Orphenadrine possesses similar NMDA receptor antagonistic properties to amantadine and memantine. This drug has low micromolar affinity for the PCP site (Ki about 6 μM) and similar fast unblocking kinetics as memantine, both with a Koff value about 0.2 s−1 (Kornhuber et al., 1995; Parsons et al., 1999). Therefore orphenadrine would be expected to have similar neuroprotective effects. In fact, compounds such as amantadine, memantine, budipine and orphenadrine, which are well tolerated clinically and have been used for their anti-parkinsonian effects, have been proposed as candidates for clinical trials on the neuroprotective efficacy of NMDA receptor antagonism (Lange et al., 1997).
Although orphenadrine showed some toxic effect on CGCs at concentrations above 24 μM, no toxicity was observed in vivo at the neuroprotective dose of 30 mg kg−1. The behaviour of the rats treated with orphenadrine alone did not show any apparent disturbance compared with saline-treated rats. In humans, micromolar serum concentrations are reached under therapeutic doses (Altamura et al., 1986; Contin et al., 1987), which are enough for NMDA receptor blockade. Although orphenadrine has complex pharmacokinetics and some cases of serious adverse effects have been reported in Parkinson's patients (Gjerden et al., 1998), this drug is being widely used for the treatment of parkinsonism and muscular distonias. The existing clinical experience with orphenadrine allows control and prevents the adverse effects. Therefore, while other safer open-channel kinetics NMDA receptor blockers are being developed and tested in clinical trials, orphenadrine could be considered as a preventive treatment for neurodegenerative disorders involving excitotoxicity.
Although these neuroprotective effects of orphenadrine are promising, more studies are necessary to evaluate its therapeutic safety in the treatment of neurodegenerative diseases.
Acknowledgments
We are grateful to Mr Jaume Comas, Ms Rosario González and Ms Susanna Castel, from the Scientific-Technical Services of the University of Barcelona, for their technical assistance in LSC and phase-contrast microscopy. This study was supported by a CICYT Grant PM98-0195 and Fundació Caixa Sabadell.
Abbreviations
- CGCs
cerebellar granule cells
- CNS
central nervous system
- HD
Huntington's disease
- HSP27
heat shock protein 27
- LSC
laser scanning cytometer
- NMDA
N-methyl-D-aspartate
- 3-NPA
3-nitropropionic acid
- NR
neutral red
- Orph.
orphenadrine
- PBR
peripheral-type benzodiazepine receptor
- PMSF
phenylmethylsulphonyl fluoride
References
- ALEXI T., HUGHES P.E., FAULL R.L.M., WILLIAMS C.E. 3-Nitropropionic acid's lethal triplet: cooperative pathways of neurodegeneration. Neuroreport. 1998;9:57–64. doi: 10.1097/00001756-199808030-00001. [DOI] [PubMed] [Google Scholar]
- ALTAMURA A.C., BUCCIO M., COLACURCIO F., COLOMBO G., TERZI A. Orphenadrine plasma levels and amelioration of extrapyramidal side effects in shizophrenic patients treated with haloperidol. Acta Neurol. Napoli. 1986;8:19–26. [PubMed] [Google Scholar]
- BABICH H., BORENFREUND E. Citotoxicity of T-2 toxin and its metabolites determined with the neutral red cell viability assay. Appl. Environ. Microbiol. 1991;57:2101–2103. doi: 10.1128/aem.57.7.2101-2103.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BEAL M.F., BROUILLET E., JENKINS B.G., FERRANTE R.J., KOWALL N.W., MILLER J.M., STOREY E., SRIVASTAVA R., ROSEN B.R., HYMAN B.T. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J. Neurosci. 1993;13:4181–4192. doi: 10.1523/JNEUROSCI.13-10-04181.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BEAL M.F., FERRANTE R.J., HENSHAW R., MATTHEWS R.T., CHAN P.H., KOWALL N.W., EPSTEIN C.J., SCHULZ J.B. 3-Nitropropionic acid neurotoxicity is attenuated in copper/zinc superoxide dismutase transgenic mice. J. Neurochem. 1995;65:919–922. doi: 10.1046/j.1471-4159.1995.65020919.x. [DOI] [PubMed] [Google Scholar]
- BENAVIDES J., FAGE D., CARTER C., MACKENZIE E.T., SCATTON B. Peripheral type benzodiazepine binding sites are a sensitive indirect index of neuronal damage. Brain Res. 1987;421:162–172. doi: 10.1016/0006-8993(87)91287-x. [DOI] [PubMed] [Google Scholar]
- BENAVIDES J., CAPDEVILLE C., DAUPHIN F., DUVOIS A., DUVERGER D., FAGE D., GOTTI B., MACKENZIE E.T., SCATTON B. The quantification of brain lesions with ω3 ligand: a critical analysis of animal models of cerebral ischemia and neurodegeneration. Brain Res. 1990;522:275–289. doi: 10.1016/0006-8993(90)91472-s. [DOI] [PubMed] [Google Scholar]
- BOGDANOV M.B., FERRANTE R.J., KUEMMERLE S., KLIVENYI P., BEAL M.F. Increased vulnerability to 3-Nitropropionic acid in an animal model of Huntington's disease. J. Neurochem. 1998;71:2642–2644. doi: 10.1046/j.1471-4159.1998.71062642.x. [DOI] [PubMed] [Google Scholar]
- BORLONGAN C.V., KOUTOUZIS T.K., FREEMAN T.B., CAHILL D.V., GANBERG P.R. Behavioral pathology induced by repeated systemic injections of 3-nitropropionic acid mimics the motoric symptoms of Huntington's disease. Brain Res. 1995;697:254–257. doi: 10.1016/0006-8993(95)00901-2. [DOI] [PubMed] [Google Scholar]
- BRADFORD M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- BROUILLET E., CONDE F., BEAL M.F., HANTRAYE P. Replicating Huntington's disease phenotype in experimental animals. Progr. Neurobiol. 1999;59:427–468. doi: 10.1016/s0301-0082(99)00005-2. [DOI] [PubMed] [Google Scholar]
- CAMINS A., GABRIEL C., AGUIRRE L., SUREDA F.X., PUBILL D., PALLÁS M., ESCUBEDO E., CAMARASA J. U-83836E prevents kainic acid-induced neuronal damage. Naunyn-Schmiedeberg's Arch. Pharmacol. 1998;357:413–418. doi: 10.1007/pl00005187. [DOI] [PubMed] [Google Scholar]
- CONTIN M., RIVA R., ALBANI F., BARUZZI A.. Simple and rapid GLC method for the determination of orphenadrine in human plasma. Biomed. Chromatogr. 1987;2:193–194. doi: 10.1002/bmc.1130020504. [DOI] [PubMed] [Google Scholar]
- DARZYNKIEWICZ Z., BEDNER E., LI X., GORCZYCA W., MELAMED M.R. Laser-scanning cytometry: a new instrumentation with many applications. Exp. Cell Res. 1999;249:1–12. doi: 10.1006/excr.1999.4477. [DOI] [PubMed] [Google Scholar]
- DAWSON R.D., BEAL M.F., BONDY S.C., DI MONTE D.A., ISOM G.E. Excitotoxins, aging, and environmental neurotoxins: implications for understanding human neurodegenerative diseases. Toxicol. Appl. Pharmacol. 1995;134:1–7. doi: 10.1006/taap.1995.1163. [DOI] [PubMed] [Google Scholar]
- FU Y.T., HE F.S., ZHANG S.L., ZHANG J.S. Lipid peroxidation in rats intoxicated with 3-nitropropionic acid. Toxicon. 1995;33:327–331. doi: 10.1016/0041-0101(94)00173-6. [DOI] [PubMed] [Google Scholar]
- GJERDEN P., ENGELSTAD K.S., PETTERSEN G., SLORDAL L. Fatalities caused by anticholinergic antiparkinsonian drugs. Analysis of findings in a 11-year national material. Tidsskr. Nor. Laegeforen. 1998;118:42–44. [PubMed] [Google Scholar]
- GREENE J.G., SHEU S.S., GROSS R.A., GREENAMYRE J.T. 3-Nitropropionic acid exacerbates N-methyl-D-aspartate toxicity in striatal culture by multiple mechanisms. Neuroscience. 1998;84:503–510. doi: 10.1016/s0306-4522(97)00389-8. [DOI] [PubMed] [Google Scholar]
- GUÉNAL I., SIDOTI-DE FRAISSE C., GAUMER S., MIGNOTTE B. Bcl-2 and HSP27 act at different levels to suppress programmed cell death. Oncogene. 1997;15:347–360. doi: 10.1038/sj.onc.1201182. [DOI] [PubMed] [Google Scholar]
- GUYOT M.C., HANTRAYE P., DOLAN R., PALFI S., MAZIÉRE M., BROUILLET E. Quantifiable bradykinesia, gait abnormalities and Huntington's disease-like striatal lesions in rats chronically treated with 3-nitropropionic acid. Neuroscience. 1997;79:45–56. doi: 10.1016/s0306-4522(96)00602-1. [DOI] [PubMed] [Google Scholar]
- KATO H., KOGURE K., LIU X.H., ARAKI T., KATO K., ITOYAMA Y. Immunohistochemical localization of the low molecular weight stress protein hsp27 following focal cerebral ischemia in the rat. Brain Res. 1995;679:1–7. doi: 10.1016/0006-8993(95)00198-y. [DOI] [PubMed] [Google Scholar]
- KATO H., LIU Y., KOGURE K., KATO K. Induction of 27-kDa heat shock protein following cerebral ischemia in a rat model of ischemic tolerance. Brain Res. 1994;634:235–244. doi: 10.1016/0006-8993(94)91926-7. [DOI] [PubMed] [Google Scholar]
- KODSI M.H., SWERLOW N.R. Mitochondrial toxin 3-nitropropionic acid produces startle reflex abnormalities and striatal damage in rats that model some features of Huntington's disease. Neurosci. Lett. 1997;231:103–107. doi: 10.1016/s0304-3940(97)00482-5. [DOI] [PubMed] [Google Scholar]
- KORNHUBER J., PARSONS C.G., HARTMANN S., RETZ W., KAMOLZ S., THOME J., RIEDERER P. Orphenadrine is an uncompetitive N-methyl-D-aspartate antagonist: binding and path clamp studies. J. Neural. Transm. 1995;102:237–246. doi: 10.1007/BF01281158. [DOI] [PubMed] [Google Scholar]
- KUHLMAN A.C., GUILARTE T.R. Cellular and subcellular localization of peripheral benzodiazepine receptors after trimethyltin neurotoxicity. J. Neurochem. 2000;74:1694–1704. doi: 10.1046/j.1471-4159.2000.0741694.x. [DOI] [PubMed] [Google Scholar]
- LA FONTAINE M.A., GEDDES J.W., BANKS A., BUTTERFIELD D.A. 3-Nitropropionic acid induced in vivo protein oxidation in striatal and cortical synaptosomes: insights into Huntington's disease. Brain Res. 2000;858:356–362. doi: 10.1016/s0006-8993(00)01948-x. [DOI] [PubMed] [Google Scholar]
- LANGE K.W., KORNHUBER J., RIEDERER P. Dopamine/glutamate interactions in Parkinson's disease. Neurosci. Biobehav. Revs. 1997;21:393–400. doi: 10.1016/s0149-7634(96)00043-7. [DOI] [PubMed] [Google Scholar]
- LEE W.T., SHEN Y.Z., CHANG C. Neuroprotective effect of lamotrigine and MK-801 on rat brain lesions induced by 3-nitropropionic acid: evaluation by magnetic resonance imaging and in vivo proton magnetic resonance spectroscopy. Neuroscience. 2000;95:89–95. doi: 10.1016/s0306-4522(99)00410-8. [DOI] [PubMed] [Google Scholar]
- LEVINE M.S., KLAPSTEIN G.J., KOPPEL A., GRUEN E., CEPEDA C., VARGAS M.E., JOKEL E.S., CARPENTER E.M., ZANJANI H., HURST R.S., EFSTRATIADIS A., ZEITLIN S., CHESSELET M.F. Enhanced sensitivity to N-methyl-D-aspartate receptor activation in transgenic and knockin mouse models of Huntington's disease. J. Neurosci. Res. 1999;58:515–532. [PubMed] [Google Scholar]
- MEHLEN P., CORONAS V., LJUBIC-THIBAL V., DUCASSE C., GRANGER L., JOURDAN F., ARRIGO A.P. Small stress protein HSP27 accumulation during dopamine-mediated differentiation of rat olfactory neurons counteracts apoptosis. Cell Death Differ. 1999;6:227–233. doi: 10.1038/sj.cdd.4400483. [DOI] [PubMed] [Google Scholar]
- MILLER P.J., ZABORSKY L. 3-Nitropropionic acid neurotoxicity: visualization by silver staining and implications for use as an animal model of Huntington's disease. Exp. Neurol. 1997;146:212–229. doi: 10.1006/exnr.1997.6522. [DOI] [PubMed] [Google Scholar]
- MITANI A., ANDOU Y., KATAOKA K. Selective vulnerability of hippocampal CA1 neurons cannot be explained in terms of an increase in glutamate concentration during ischemia in the gerbil-brain microdialysis study. Neuroscience. 1992;48:307–313. doi: 10.1016/0306-4522(92)90492-k. [DOI] [PubMed] [Google Scholar]
- NICOLETTI F., WROBLEWSKI J.T., NOVELLI A., ALHO H., GUIDOTTI A., COSTA E. The activation of inositol phospholopid hydrolysis as a signal transducing system for excitatory amino acids in primary cultures of cerebellar granule cells. J. Neurosci. 1986;6:1905–1911. doi: 10.1523/JNEUROSCI.06-07-01905.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NISHINO H., KUMAZAKI M., FUKUDA A., FUJIMOTO I., SHIMANO Y., HIDA H., SAKURAI T., DESHPANDE S.B., SHIMIZU H., MORIKAWA S., INUBUSHI T. Acute 3-nitropropionic acid intoxication induces striatal astrocytic cell death and dysfunction of the blood-brain barrier: involvement of dopamine. Neurosci. Res. 1997;27:343–355. doi: 10.1016/s0168-0102(97)01170-x. [DOI] [PubMed] [Google Scholar]
- NOVELLI A., REILLY J.A., LYSKO P.G., HENNEBERRY R.C. Glutamate becomes neurotixic via the N-methyl-D-aspartate receptor when intracellular energy levels are reduced. Brain Res. 1988;451:205–212. doi: 10.1016/0006-8993(88)90765-2. [DOI] [PubMed] [Google Scholar]
- OLSEN C., RUSTAD A., FONNUM F., PAULSEN R.E., HASSEL B. 3-Nitropropionic acid: an astrocyte-sparing neurotoxin in vitro. Brain Res. 1999;850:144–149. doi: 10.1016/s0006-8993(99)02115-0. [DOI] [PubMed] [Google Scholar]
- PANG Z., GEDDES J.W. Mechanisms of cell death induced by the mitochondrial toxin 3-nitropropionic acid: acute excitotoxic necrosis and delayed apoptosis. J. Neurosci. 1997;17:3064–3073. doi: 10.1523/JNEUROSCI.17-09-03064.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PARSONS C.G., DANYSZ W., QUACK G. Memantine is a clinically well tolerated N-methyl-D-asparate (NMDA) receptor antagonist: A review of preclinical data. Neuropharmacology. 1999;38:735–767. doi: 10.1016/s0028-3908(99)00019-2. [DOI] [PubMed] [Google Scholar]
- PLUMIER J.C.L., ARMSTRONG J.N., LANDRY J., BABITY J.M., ROBERTSON H.A., CURRIE R.W. Expression of the 27,000 Mol. Wt heat shock protein following kainic acid-induced status epilepticus in the rat. Neuroscience. 1996;75:849–856. doi: 10.1016/0306-4522(96)00317-x. [DOI] [PubMed] [Google Scholar]
- RENKAWEK K., BOSMAN G.J., GAESTEL M. Increased expression of heat-shock protein 27 kDa in Alzheimer disease: a preliminary study. Neuroreport. 1993;5:14–16. doi: 10.1097/00001756-199310000-00003. [DOI] [PubMed] [Google Scholar]
- SATO S., GOBBEL G.T., HONKANIEMI J., LI Y., KONDO T., MURAKAMI K., SATO M., COPIN J.C., CHAN P.H. Apoptosis in the rat striatum of rats following intraperitoneal administration of 3-nitropropionic acid. Brain Res. 1997;745:343–347. doi: 10.1016/s0006-8993(96)01231-0. [DOI] [PubMed] [Google Scholar]
- SCHULZ J.B., HENSHAW D.R., MACGARVEY U., BEAL M.F. Involvement of oxidative stress in 3-nitropropionic acid neurotoxicity. Neuroscience. 1996a;29:167–171. doi: 10.1016/0197-0186(95)00122-0. [DOI] [PubMed] [Google Scholar]
- SCHULZ J.B., MATTHEWS R.T., HENSHAW D.R., BEAL M.F. Neuroprotective strategies for treatment of lesions produced by mitochondrial toxins: implications for neurodegenerative diseases. Neuroscience. 1996b;71:1043–1048. doi: 10.1016/0306-4522(95)00527-7. [DOI] [PubMed] [Google Scholar]
- SHOEMAKER H., MORELLI M., DESHMKH P., YAMAMURA H. [3H] Ro5-4864 benzodiazepine in the kainate lesioned striatum and Huntington's diseased basal ganglia. Brain Res. 1982;248:396–401. doi: 10.1016/0006-8993(82)90602-3. [DOI] [PubMed] [Google Scholar]
- SOBOLEVSKY A., KOSHELEV S.G., KHODOROV B.I. Interaction of memantine and amantidine with agonist-unbound NMDA receptor channels in acutely isolated rat hippocampal neurons. J. Physiol. 1998;512:47–60. doi: 10.1111/j.1469-7793.1998.047bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEPHENSON D.T., SCHOBER D.A., SMALSTIG E.B., MINCY R.E., GEHLER D.R., CLEMENS J.A. Peripheral benzodiazepine receptors are colocalized with activated microglia following transient global forebrain ischemia in the rat. J. Neurosci. 1995;15:5263–5274. doi: 10.1523/JNEUROSCI.15-07-05263.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STREIT W.J., WALTER S.A., PENNELL N.A. Reactive microgliosis. Progr. Neurobiol. 1999;57:563–581. doi: 10.1016/s0301-0082(98)00069-0. [DOI] [PubMed] [Google Scholar]
- SUREDA F.X., GABRIEL C., PALLÁS M., ADAN J., MARTINEZ J.M., ESCUBEDO E., CAMARASA J., CAMINS A. In vitro and in vivo protective effects of orphenadrine on glutamate neurotoxicity. Neuropharmacology. 1999;38:671–677. doi: 10.1016/s0028-3908(98)00228-7. [DOI] [PubMed] [Google Scholar]
- VONSATTEL J.P., MYERS R.H., STEVENS T.J., FERRANTE R.J., BIRD E.D., RICHARDSON E.P. Neuropathological classification of Huntington's disease. J. Neuropathol. Exp. Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- WAGSTAFF M.J.D., COLLAçO-MORAES Y., SMITH J., BELLEROCHE J.S., COFFIN R.S., LATCHMAN D.S. Protection of neuronal cells from apoptosis by HSP27 delivered with a herpes simplex virus-based vector. J. Biol. Chem. 1999;274:5061–5069. doi: 10.1074/jbc.274.8.5061. [DOI] [PubMed] [Google Scholar]
- WELLER M., PAUL S.M. 3-Nitropropionic acid is an indirect excitoxin to cultured cerebellar granule neurons. Eur. J. Pharmacol. 1993;248:223–228. doi: 10.1016/0926-6917(93)90048-u. [DOI] [PubMed] [Google Scholar]
- WÜLLNER U., YOUNG A.B., PENNEY J.B., BEAL M.F. 3-Nitropropionic acid toxicity in the striatum. J. Neurochem. 1994;63:1772–1781. doi: 10.1046/j.1471-4159.1994.63051772.x. [DOI] [PubMed] [Google Scholar]
- ZEEWALK G.D., NICKLAS W.J. Evidence that the loss of the voltage-dependent Mg++ block at the N-methyl-D-aspartate receptor underlies receptor activation during inhibition of neuronal metabolism. J. Pharmacol. Exp. Ther. 1992;259:1211–1220. doi: 10.1111/j.1471-4159.1992.tb08430.x. [DOI] [PubMed] [Google Scholar]