

Abstract
Ischaemia/reperfusion causes intracellular calcium overloading in cardiac cells. Administration of calcium antagonists reduces myocardial infarct size. Recent in vitro studies have demonstrated that calcium plays a critical role in the signal transduction pathway leading to apoptosis. However, whether or not calcium antagonists may reduce myocardial apoptosis induced by ischaemia-reperfusion, and thus decrease myocardial infarction, has not been directly investigated.
The present study investigated the effects of benidipine, an L-type calcium channel blocker, on myocardial infarct size, apoptosis, necrosis and cardiac functional recovery in rabbits subjected to myocardial ischaemia/reperfusion (MI/R, 45 min/240 min). Ten minutes prior to coronary occlusion, rabbits were treated with vehicle or benidipine (10 μg kg−1 or 3 μg kg−1, i.v.).
In the vehicle-treated group, MI/R caused cardiomyocyte apoptosis as evidenced by DNA ladder formation and TUNEL positive nuclear staining (12.2±1.1%). Treatment with 10 μg kg−1 benidipine lowered blood pressure, decreased myocardial apoptosis (6.2±0.8%, P<0.01 vs vehicle) and necrosis, reduced infarct size (20±2.3% vs 49±2.6%, P<0.01), and improved cardiac functional recovery after reperfusion. Administering benidipine at 3 μg kg−1, a dose at which no haemodynamic effect was observed, also exerted significant anti-apoptosis effects, which were not significantly different from those observed with higher dose benidipine treatment. However, treatment with this low dose benidipine failed to reduce myocardial necrosis.
These results demonstrate that benidipine, a calcium antagonist, exerts significant anti-apoptosis effects, which are independent of haemodynamic changes. Administration of benidipine at a higher dose produced favourable haemodynamic effects and provided additional protection against myocardial necrotic injury and further improved cardiac functional recovery.
Keywords: Myocardial reperfusion, apoptosis, calcium antagonist
Introduction
Ischaemic heart disease is the single-most important cause of death in developed countries. Growing evidence from both animal experiments and clinical observations indicates that myocardial infarction after ischaemia and reperfusion is caused not only by necrosis, a traditional cell death pathway, but also by apoptosis, a gene controlled programme cell death (Gottlieb & Engler, 1999). We and others have previously demonstrated that the administration of antioxidants or agents that specifically block the signal transduction leading to apoptosis significantly improves cardiac function after myocardial ischaemia and reperfusion, suggesting that anti-apoptosis treatment may improve clinical outcomes in patients with ischaemic heart disease (Yue et al., 1998; Yaoita et al., 1998; Ma et al., 1999).
It has been long recognized that ischaemia, and ischaemia followed by reperfusion, cause intracellular calcium overloading in cardiac cells and that administration of calcium antagonists before ischaemia significantly reduces myocardial infarct size and improves myocardial functional recovery after reperfusion (Lefer et al., 1979). On the other hand, recent studies have demonstrated that calcium plays a key regulatory role in multiple sites of the signal transduction pathway leading to apoptosis in diverse cell types (McConkey & Orrenius, 1997). Calcium overloading results in overactivation of either those enzymes that directly cause DNA fragmentation, such as calcium- and magnesium-dependent endonuclease (DNase I), or proteins that regulate signal transduction leading to apoptosis, such as calcineurin, calpain and nuclear scaffold protease (Peitsch et al., 1993; Gottlieb & Engler, 1999; McConkey & Orrenius, 1997). Calcium overloading also increases free radical generation (Bagchi et al., 1997), a well-defined apoptosis-inducing factor (Das et al., 1999). Moreover, in cell culture systems, addition of dihydropyridine calcium channel blockers, amlodipine and nifedipine, reduces excessive apoptosis in ageing cerebellar granule cells (Mason et al., 1999) and in hypoxic neonatal rat cardiac myocytes (Chen et al., 1998). In animals subjected to renal ischaemia, administration of verapamil, an L-type calcium blocker, has been demonstrated to reduce tissue apoptosis (Raafat et al., 1997). These results strongly suggest that the calcium overloading occurring in ischaemia/reperfused cardiomyocytes may cause myocardial apoptosis, and that calcium antagonists may exert its infarct reduction effect partially through an anti-apoptotic effect. However, to date, the contribution of the potential anti-apoptotic effect of calcium antagonists on its infarct limitation and cardiac function improvement effects after myocardial ischaemia/reperfusion has not been directly investigated.
Accordingly, the aims of the present experiment were (a) to investigate whether administration of benidipine, a long-lasting vasodilatory calcium antagonist, may reduce myocardial apoptosis induced by ischaemia/reperfusion and thus contribute to its infarct reduction; and (b) to determine the influence of haemodynamic changes on anti-apoptosis effects associated with the administration of a calcium antagonist in the setting of myocardial ischaemia and reperfusion.
Methods
Materials
Benidipine, a long-acting 1,4-dihydropyriding calcium antagonist (Kitakaze et al., 1999), was provided by Kyowa Hakko Kogyo Co., LTD (Tokyo, Japan). Previous experiments using a dog model have demonstrated that after a single i.v. injection at 3 μg kg−1 and 10 μg kg−1, its effect lasts >2 h and >3 h, respectively (Karasawa et al., 1988). All other compounds were purchased from Sigma Chemicals Co. (St. Louis, MO, U.S.A.). A total of 71 adult male New Zealand white rabbits (2.8 – 3.5 kg) were used. Data from seven rabbits (three from vehicle group, two from low dose and two from high dose benidipine groups) were excluded from the final analysis for one or both of the following reasons: (a) sustained hypotension during ischaemic or reperfusion phases (⩽60 mmHg for ⩾20 min), and (b) greater than two episodes of ventricular fibrillation. The experiments were performed in adherence to NIH Guidelines on the Use of Laboratory Animals and were approved by the Thomas Jefferson University Committee on Animal Care.
Experimental preparation
Rabbits were anaesthetized with sodium pentobarbitone (30 mg kg−1, i.v.) and ventilated with a Harvard small animal respirator. A polyethylene catheter was inserted into the right external jugular vein for supplemental pentobarbitone injection and for administration of test compounds. The arterial blood pressure was measured via a polyethylene catheter cannulated to the right femoral artery, and the left ventricular pressure (LVP) was measured via a Millar Mikro-tip catheter transducer that was inserted into the left ventricular cavity through the left carotid artery.
Following midline thoracotomy, a 4-0 silk ligature was placed around the major marginal branch of the left circumflex coronary, 10 – 12 mm from its origin. After a 20 min stabilization period, myocardial ischaemia (MI) was initiated by complete ligation of the marginal coronary artery. After 45 min of ischaemia, the ligature was untied and the ischaemic myocardium was reperfused (R) for 4 h. Sham MI/R rabbits were subjected to the same surgical procedures performed on MI/R rabbits, except that the suture was left untied. The rabbits were randomly assigned to one of the following groups: (1) sham MI/R+benidipine (3, 5, 10 μg kg−1, n=4 for each dose); (2) MI/R+vehicle (0.15% Tween 80 in saline, n=12); (3) MI/R+benidipine (3 μg kg−1, n=11); (4) MI/R+benidipine (10 μg kg−1, n=11). Each drug or vehicle was given intravenously over 1 min 10 min prior to ischaemia.
Myocardial functional injury
MI/R-induced cardiac dysfunction was continuously monitored during the entire MI/R period. The arterial blood pressure and LVP were sampled at 250 Hz and digitally processed via a haemodynamic analysing system (Po-Ne-Mah Physiology Platform P3 Plus, Gould Instrument Systems, Inc., Valley View, OH, U.S.A.). Mean arterial blood pressure (MABP), heart rate (HR), left ventricular end diastolic pressure (LVEDP), maximal positive and negative values of the instantaneous first derivative of LVP (+dP/dtmax and −dP/dtmax) and cardiac contractile index (CI=+dP/dtmax/p) were derived by computer algorithms. The pressure-rate-index (PRI), calculated as the product of MABP and HR divided by 1000, was employed as an approximation of myocardial oxygen demand.
Plasma creatine kinase accumulation
Arterial blood samples (1 ml) were drawn immediately before ligation (0 min), 45 min after ischaemia and hourly thereafter. Plasma creatine kinase (CK) activity was measured in a blinded manner using a Sigma kit and expressed as IU per g of protein.
Myocardial infarct
At the end of the 4 h reperfusion period, the ligature around the marginal coronary artery was retied and 20 ml of 2% Evans blue dye was injected into the left ventricular cavity. The heart was quickly excised, and the atria, right ventricle and fatty tissues were removed from the heart. The unstained portion of the left ventricular myocardium (i.e. the area-at-risk, AAR) was separated from the Evans blue-stained portion of the myocardium (i.e. the area-not-at-risk, ANAR). One half of the AAR tissue was immediately frozen in liquid nitrogen for detection of DNA fragmentation as described below. The other half of the AAR was sliced into 1 mm thick sections and incubated at 37°C in 0.1% solution of nitro blue tetrazolium (NBT) in phosphate buffer for 15 min. At the end of this time, the unstained infarct tissue was separated from the stained non-infarct viable tissue in a blinded manner. Samples from all three portions of the left ventricular cardiac tissue were weighed. The AAR as a percentage of the total left ventricle mass, the infarcted myocardial tissue as a percentage of AAR, and the infarcted myocardial tissue as a percentage of total left ventricle mass were calculated.
Myocardial apoptosis
Myocardial apoptosis was qualitatively analysed by detection of DNA fragmentation (DNA ladders). The frozen heart tissues (stored in liquid nitrogen) were minced while thawing in 600 μl of lysis buffer (Puregene DNA Isolation Kit) and quickly homogenized using 30 – 50 strokes with a microfuge tube pestle on ice. The tissue was digested with proteinase K (100 μg ml−1) at 56°C overnight and then incubated with DNA-free RNase at 37°C for an additional 1 h. Digested tissues were precipitated with protein precipitation solution with protein precipitated solution and centrifuged at 13,000×g for 5 min. Supernatants containing DNA were precipitated and centrifuged again. The resulting DNA pellets were washed with 75% ethanol and dissolved in DNA hydration solution. Ten μg of DNA was loaded into 1.8% agarose gel containing 0.5 μg ml−1 ethidium bromide. DNA electrophoresis was carried out at 60 V for 1 – 2 h. DNA ladder formation, a ‘hallmark' of tissue apoptosis, was visualized under ultraviolet light and photographed for a permanent record.
To determine myocardial apoptosis in a quantitative manner, four rabbits in each group were studied in an additional experiment. After MI/R, the hearts were perfused first with 0.9% NaCl for 5 min and then with 4% paraformaldehyde in PBS (pH 7.4) for 20 min. Four longitudinal sections from ischaemic regions were cut and further fixed in 4% paraformaldehyde in PBS for 24 h at room temperature. Fixed tissues were then embedded in a paraffin block and two slides at 4 – 5 μm thickness were cut from each tissue block. Immunohistochemical procedures for detecting apoptotic cardiomyocytes were performed by using an apoptosis detection kit (Boehringer Mannheim, Ridgefield, CT, U.S.A.) according to the manufacturer's instructions. The digoxigenin-conjugated dUTP was incorporated to the ends of DNA fragments by terminal deoxynucleotidyl transferase (TdT). The signal of TdT-mediated dUTP nick end labelling was then detected by an anti-fluorescein antibody conjugated with alkaline phosphatase, a reporter enzyme that catalytically generates a red-coloured product from Vector red substrate. For each slide, 10 fields were randomly chosen, and using a defined rectangular field area (20×objective), a total of 100 cells per field were counted. The index of apoptosis was determined (i.e., number of positively stained apoptotic myocytes/total number of myocytes counted×100) from a total of 80 fields per heart. Assays were performed in a blinded manner.
Myocardial calcium content
The calcium content of the myocardial tissue was measured by atomic absorption spectrometry (Perkin-Elmer 460) as described previously (Murphy et al., 1996; Raffa et al., 1987). The calibration was performed using an atomic absorption standard calcium solution (Fisher Scientific) immediately before samples were assessed, and the results were expressed as μg g−1 dry weight (Karasawa & Kubo, 1990).
Statistical analysis
Time and group differences were determined by two-way analysis of variance for repeated measures, correcting for multiple comparisons by Holm's multiple rejective method as appropriate for an overall probability level of 0.05. Non-repetitive data, such as infarct size and apoptosis, was subjected to analysis of variance (ANOVA) followed by the Scheffe's correction for post-hoc t-test comparison. Probabilities of 0.05 or less were considered statistically significant.
Results
Heart rate, mean arterial blood pressure and pressure rate index
In sham MI/R rabbits, HR, MABP and PRI remained at a relatively stable level throughout the 4 h and 45 min observation period. Administration of benidipine in sham MI/R rabbit caused a dose-dependent decrease in MABP (−3.5±2.4, −11.8±2.3 and −29±2.6 mmHg at 3, 5 and 10 μg kg−1, respectively). Coronary occlusion caused a significant decline in HR, MABP and PRI in the first 10 min of ischaemia, followed by a partial recovery at the end of ischaemia. HR and MABP did not change significantly after reperfusion except that HR showed a transient decline in the first 20 min of reperfusion in the vehicle-treated group (Figure 1). Administration of benidipine at 10 μg kg−1 caused a significant decrease in both HR and MABP. PRI, an indirect measure of cardiac oxygen consumption, was markedly reduced and remained significantly low until 60 min after reperfusion (Figure 1). Administration of benidipine at 5 μg kg−1 also reduced PRI significantly, although to a lesser extent than 10 μg kg−1 benidipine (data not shown). In contrast, administration of 3 μg kg−1 benidipine exerted no significant effects on either HR or MABP and PRI remained at a level that was comparable to that seen with vehicle treatment (Figure 1). Therefore, it was unlikely that any protective effect demonstrated in this model by low dose benidipine (i.e., 3 μg kg−1) was the result of a reduction in myocardial oxygen demand.
Figure 1.
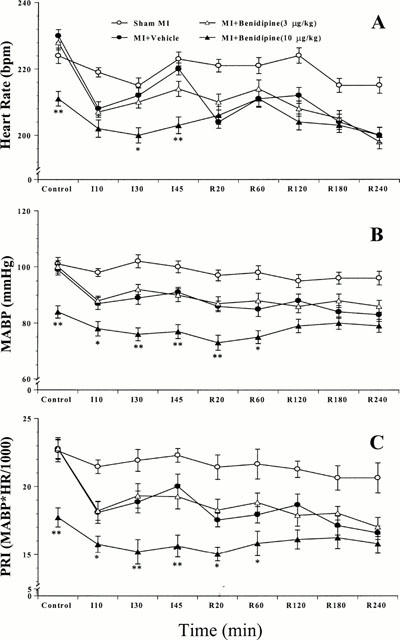
Haemodynamic changes in rabbits subjected to sham MI/R or MI/R. MI/R rabbits were treated with vehicle, 3 μg kg−1 benidipine, or 10 μg kg−1 benidipine. Depicted data are means±s.e.mean from 10 – 12 independent experiments. *P<0.05, **P<0.01 vs the vehicle-treated rabbits.
Effect of benidipine on myocardial infarct size
Forty-five min of ischaemia and 240 min of reperfusion resulted in significant myocardial infarction as evidenced by a large area of NBT negative staining. Treatment with benidipine reduced infarct size in a dose-dependent fashion. Thus, in the rabbits treated with 3 μg kg−1 benidipine, the NBT negative staining area was reduced to 32±2.5% of AAR. This value was further reduced to 20±2.3% when rabbits were treated with 10 μg kg−1 benidipine (P<0.01 vs vehicle and low dose benidipine treated group) (Figure 2).
Figure 2.
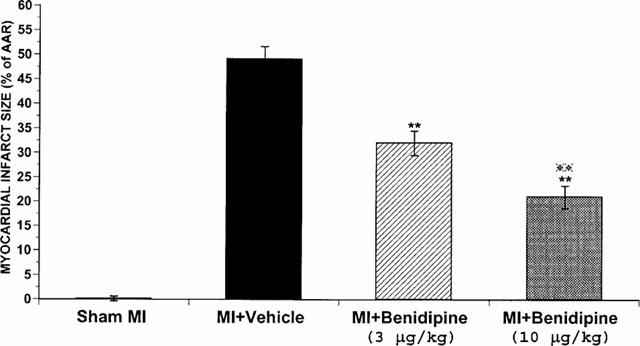
Tissue wet weight of the infarcted tissue as a percentage of area-at-risk (AAR). **P<0.01, vs the vehicle-treated rabbits.
Detection of DNA fragmentation (DNA ladder) and TUNEL staining in ischaemia/reperfused hearts and their inhibition by benidipine
It is now well accepted that DNA ladder formation is highly specific for apoptotic cell death, but lacks sensitivity and is difficult to quantify. In contrast, TUNEL staining of nuclei is extremely sensitive, but is less specific for apoptosis as some necrotic cells may stain positive. These two methods were thus used in combination to improve accuracy and reliability of our results. In myocardial tissue from sham MI hearts, no DNA ladder was detected (Figure 3, lane 0). In contrast, the formation of DNA nucleosome ladders was clearly detected in myocardial tissues obtained from I/R heart receiving only vehicle (Figure 3, lanes 1 – 3). In the 12 vehicle-treated rabbit hearts, clear DNA ladders were visualized in 10 heart samples (an incidence of 83.3%). In contrast, rabbits treated with either high dose (i.e., 10 μg kg−1) or low dose (i.e., 3 μg kg−1) benidipine exhibited markedly decreased DNA fragmentation. Specifically, in the 11 rabbits treated with 10 μg kg−1 benidipine, DNA ladder formation was absent in eight heart samples and significantly reduced in three other samples. In the 11 rabbits treated with 3 μg kg−1 benidipine, DNA ladder formation was absent in seven heart samples and significantly reduced in four other samples (Figure 3, lanes 4 – 6).
Figure 3.
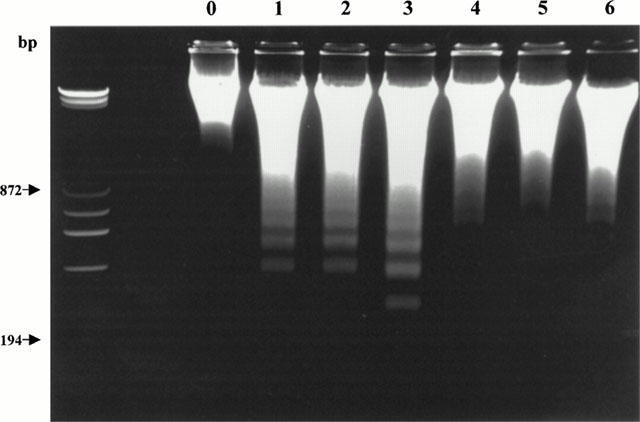
Representative photograph of electrophoretic analysis of internucleosomal DNA extracted from sham-operated control hearts (lane 0) or rabbit hearts exposed to I/R receiving either vehicle (lanes 1 – 3) or 3 μg kg−1 benidipine (lanes 4 – 6). The first lane is DNA cells size markers.
In sham MI hearts that lacked DNA ladders, few cells stained TUNEL positive (Figure 4A and 5). In contrast, TUNEL-positive nuclei were prevalent in tissues from ischaemic-reperfused hearts receiving only vehicle (Figure 4B and 5). Administration of benidipine at 10 μg kg−1 reduced the numbers of cells stained positive by TUNEL from 12.2±1.1 to 6.2±0.8% (Figure 5). Most interestingly, treatment with 3 μg kg−1 benidipine, a dose that exerted no significant effect on PRI, decreased apoptotic positive cells to 7.4±0.9%, a level that was not significantly different from that seen in the group treated with higher dose benidipine (Figure 4C and 5). Taken together, these results indicate that benidipine, an L-type calcium channel blocker, inhibited MI/R-induced myocardial apoptosis in a haemodynamically-independent manner.
Figure 4.
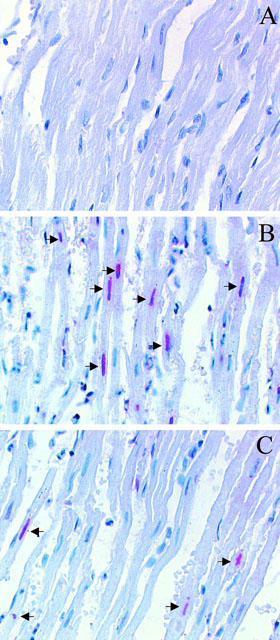
Representative photomicrographs of in situ detection of DNA fragments in heart tissue from rabbits subjected to sham MI/R (A), MI/R receiving vehicle (B), or MI/R treated with 3 μg kg−1 benidipine (C). The tissues were sectioned and stained using the TUNEL method, as described in ‘Methods'. Arrowheads signify positive nuclei.
Figure 5.

Percentage of nuclei staining positive for TUNEL in sham-operated control hearts or the rabbit hearts exposed to I/R receiving either vehicle or 3 μg kg−1 benidipine. Four longitudinal sections from each heart were cut and embedded in a paraffin block, and two slides per block were evaluated histologically. For each slide, a total of 10 fields were randomly chosen, a total of 100 cells per field were counted, and the percentage of myocytes exhibiting positive staining apoptosis was determined. **P<0.01 vs vehicle group.
In a separate series of study, six rabbits were subjected to MI/R as described above. Benidipine (3 μg kg−1) was administered 10 min before reperfusion (rather than 10 min before ischaemia) and effect of this treatment on the number of TUNEL positive cells in ischaemia-reperfused myocardial tissue was observed. The date from this experiment demonstrated that administration of benidipine shortly before reperfusion also significantly reduced apoptotic myocyte death (8.7±1.2%, P<0.05 vs vehicle group), although to a lesser extent than that exerted by benidipine given before ischaemia.
Effects of benidipine on myocardial necrotic injury after ischaemia and reperfusion
Plasma CK accumulation, an index of myocardial membrane damage and tissue necrosis, was measured during ischaemia and reperfusion. As illustrated in Figure 6, plasma CK activity did not change significantly during the entire observation period in sham ischaemic rabbits, indicating that the surgical operation alone did not result in significant myocardial necrotic injury. In the rabbits subjected to ischaemia and reperfusion receiving only vehicle, plasma CK activity was slightly increased during ischaemia and elevated substantially after reperfusion. This reperfusion-associated CK elevation was markedly blunted when rabbits were treated with 10 μg kg−1 benidipine (Figure 6). Interestingly, although administration of benidipine at 3 μg kg−1 inhibited apoptosis to a level that was comparable to that seen with 10 μg kg−1 benidipine treatment, administration of this low dose benidipine failed to attenuate plasma CK accumulation significantly (P>0.05 vs vehicle, P<0.05 vs high dose treatment).
Figure 6.
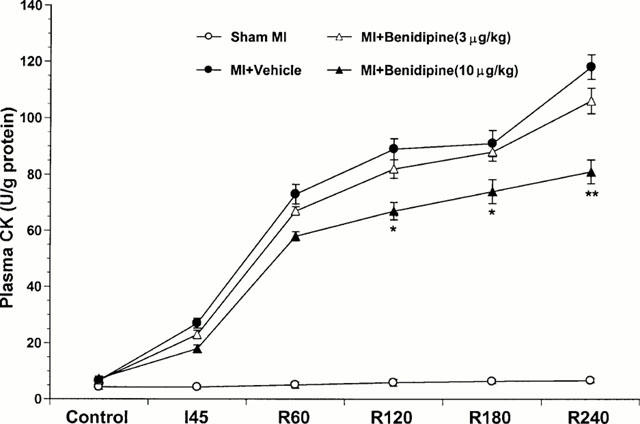
Plasma creatine kinase (CK) activity expressed as International Units per gram of protein measured before ischaemia (control), at the end of ischaemia (I-45) and hourly thereafter for the four study groups. *P<0.05; **P<0.01 vs the vehicle-treated group.
Effect of benidipine on cardiac function recovery after reperfusion
In sham I/R rabbits, LVEDP, CI and −dP/dtmax remained at a relatively stable level during the entire observation period, indicating that the surgical operation did not cause significant cardiac depression. Moreover, administration of benidipine at either dose did not alter any parameters observed, suggesting that benidipine had no significant effect on basal cardiac function. LVEDP was significantly elevated after ischaemia and reperfusion in the rabbits subjected to ischaemia and reperfusion receiving only vehicle. Administration of benidipine attenuated LVEDP elevation in a dose-dependent manner (Figure 7A). In the rabbits treated with high dose benidipine, LVEDP elevation was attenuated during the entire I/R period. At 120 min of reperfusion and thereafter, LVEDP in this high dose benidipine-treated group was not only significantly lower than LVEDP in vehicle-treated rabbits, but also significantly lower than LVEDP in those rabbits treated with low dose benidipine (Figure 7A). Treatment with low dose benidipine did not reduce LVEDP during ischaemia and during the early period of reperfusion. However, this treatment prevented the secondary elevation of LVEDP that occurred in the vehicle group after 60 min of reperfusion. Therefore, at 120 min of reperfusion and thereafter, LVEDP in the low dose benidipine group was also significantly reduced (Figure 7A).
Figure 7.
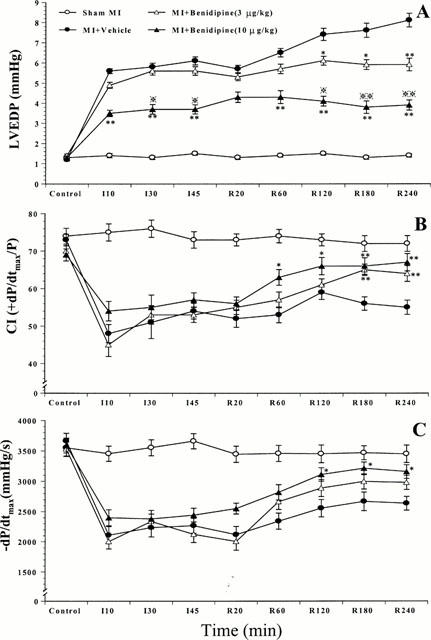
Effects of benidipine treatment on cardiac contractile function measured by LVEDP (A) and CI (B), and relaxation function as measured by −dP/dtmax (C). *P<0.05, **P<0.01 vs the vehicle-treated rabbits.
The maximal positive rate of the first derivative of left ventricular pressure (+dP/dtmax) is a commonly used index of myocardial contractility. However, +dP/dtmax is severely affected by alterations in LVP. To limit the influence of LVP on +dP/dtmax and to determine cardiac contractile function more precisely, we utilized the contractile index (CI), a calculated parameter derived from dP/dt and left ventricular pressure (CI=+dP/dtmax/LVP at dP/dtmax). In all three I/R groups, CI was markedly reduced during the ischaemic period. Administration of benidipine slightly decreased CI before ischaemia and moderately attenuated CI decline during ischaemia. However, these differences did not reach statistical significance. In vehicle-treated rabbits, CI initially improved in the first 120 min of reperfusion, but secondarily declined thereafter. Administration of benidipine at either dose significantly improved CI recovery after reperfusion. Although CI showed a faster and better recovery in the rabbits treated with high dose benidipine, the difference was not statistically significant when compared with low dose benidipine treatment (Figure 7B).
I/R caused not only a significant depression in cardiac contractile function, but also a marked impairment of cardiac diastolic function as measured by −dP/dtmax. Treatment with high dose, but not low dose of benidipine, significantly improved −dP/dtmax at the end of reperfusion (Figure 7C). Taken together, these results indicate that administration of benidipine at 10 μg kg−1, a dose that reduced myocardial apoptosis as well as necrosis, improved both cardiac contraction and relaxation function. Administration of benidipine at 3 μg kg−1, a sub-vasodilatory dose that inhibited myocardial apoptosis but had no significant effect on necrosis, significantly improved cardiac contractile functional recovery after reperfusion.
Effect of benidipine on calcium overloading in ischaemia-reperfused myocardial tissue
Benidipine exerted no significant effect on calcium content in myocardial tissue not exposed to ischaemia/reperfusion (187±10 μg g−1 vs 194±11 μg g−1 dry weight of myocardial tissue in vehicle-treated heart, P>0.05). In contrast, the administration of benidipine at 10 μg kg−1 markedly attenuated calcium overloading in ischaemic/reperfused tissue (467±42 μg g−1 vs 766±41 μg g−1 in vehicle-treated ischaemic/reperfused myocardial tissue, P<0.01). Treatment with low dose benidipine (i.e., 3 μg kg−1) also significantly reduced calcium overloading in ischaemic/reperfused myocardial tissue (624±38 μg g−1, P<0.05 vs vehicle-treated heart). However, the protective effect exerted by this low dose treatment was significantly less than benidipine at its higher dose (P<0.05).
Discussion
A growing body of evidence now indicates that ischaemia, and ischaemia followed by reperfusion, results in cardiomyocyte death with typical apoptotic features (Gottlieb & Engler, 1999). In human postmortem studies of myocardial infarction, apoptotic cardiomyocytes are clearly detected in the hypoperfused border zone between the central infarct area and noncompromised myocardial tissue (Saraste et al., 1997; Olivetti et al., 1996). Observations in animal models of myocardial infarction suggest that apoptosis may contribute substantially to cell death even within the central infarct area with 5 to 33% of the cardiomyocytes staining positive for DNA fragmentation (Kajstura et al., 1996; Bialik et al., 1997). Moreover, results from our laboratory and other investigators have demonstrated that treatment with free radical scavengers (Yue et al., 1998), caspase inhibitors (Yaoita et al., 1998), or p38 MAPK inhibitors (Ma et al., 1999) in animals subjected to myocardial ischaemia and reperfusion significantly reduces myocardial apoptosis and infarct size, and improves cardiac contractile function.
It has been long recognized that calcium overload in ischaemia/reperfused cardiomyocytes is one of the major factors that ultimately results in myocardial infarction (Bagchi et al., 1997). Marked elevations of intracellular calcium concentration result in overactivation of hydrolytic enzymes, lead to exaggerated energy expenditure, impair energy production, initiate cytoskeletal degradation, and ultimately result in cell membrane disruption and myocardial necrosis (Bagchi et al., 1997). Accumulating evidence now indicates that calcium overloading may also promote apoptotic cell death (McConkey & Orrenius, 1997). In vitro experiments have shown that addition of intracellular calcium buffering agents, or extracellular calcium chelators significantly inhibits caspase activation, DNA fragmentation and apoptotic cell death (McConkey & Orrenius, 1996; 1997). In contrast, agents that directly mobilize calcium, such as calcium ionophores, have been shown to trigger apoptosis in diverse cell types (Wyllie et al., 1984). Moreover, recent studies have demonstrated that addition of calcium blockers abrogates apoptosis in neuronal cells during the ageing process (Mason et al., 1999), and in cardiac myocytes subjected to chemical hypoxia (Chen et al., 1998), further supporting the claim that calcium influx plays a significant role in apoptotic cell death.
In the present experiments, we have demonstrated that treatment with benidipine, an L-type calcium channel blocker, significantly reduced MI/R-induced myocardial apoptosis. To our knowledge, this is the first study that directly demonstrated the anti-apoptosis property of a calcium antagonist contributes to its infarct reduction after myocardial ischaemia and reperfusion. Most importantly, we have demonstrated that treatment with benidipine decreased total infarct size and improved cardiac function by different mechanisms that are related to benidipine dose. Treatment with 10 μg kg−1 benidipine markedly reduced myocardial infarct size and significantly improved cardiac contractile as well as relaxation function. Decreased myocardial necrosis (as evidenced by reduced CK release) as well as apoptosis (as evidenced by less DNA ladder formation and reduced number of cells with TUNEL positive staining) both contribute to its profound cardioprotection at this high dose. In contrast, treatment with 3 μg kg−1 benidipine had no significant haemodynamic effect and did not significantly reduce cardiac oxygen demand. This treatment reduced myocardial infarct size primarily through an anti-apoptotic effect because treatment at this dose failed to reduce CK release significantly. Moreover, low dose benidipine treatment significantly improved cardiac contractile function, although to a lesser extent than high dose treatment. Interestingly, a recent study demonstrates that the degree of intracellular calcium increase is a critical determinant of apoptosis or necrosis (McConkey, 1998). A low to moderate (200 – 400 nM) increase in intracellular calcium causes cell death via apoptosis, whereas uncontrolled, massive calcium influx (to levels above 1 μM) is invariably associated with necrosis (McConkey, 1998). This may explain our present finding that a low dose benidipine treatment (which may protect those cells with a moderate calcium overloading) only decreased apoptosis, but failed to reduce necrosis (which was associated with a severe calcium overloading and would require a higher dose of calcium antagonist to block it) after myocardial ischaemia and reperfusion. Unfortunately, this hypothesis can not be directly proven using an in vitro model because measurement of tissue calcium content does not provide information on calcium content at the individual myocyte level.
The exact mechanisms by which benidipine may reduce MI/R-induced myocardial apoptosis could not be answered directly by the present study. However, based on previous in vivo and in vitro study findings, it can be reasonably speculated that benidipine may attenuate apoptosis via a number of mechanisms. Substantial evidence indicates that the mitochondria play a critical regulatory role in the signal transduction pathway leading to apoptosis (Gross et al., 1999). It is also well documented that calcium antagonists attenuate mitochondrial injury resulting from ischaemia and reperfusion and preserve mitochondrial function (Boraso et al., 1993). By this mechanism, calcium antagonist may prevent the formation of the permeability transition pore in the mitochondrial membrane, inhibit the release of pro-apoptosis molecules such as apaf-1 and apaf-2 (cytochrome c) from the mitochondria, and reduce myocardial apoptosis. Reduced calcium overloading achieved by calcium antagonist treatment may also attenuate myocardial apoptosis through prevention of the dephosphorylation of Bad, a pro-apoptosis protein of Bcl-2 family, by calcineurin (a calcium/calmodulin-dependent protein serine/threonine phosphatase). Preventing the activation of calcineurin keeps Bad in its phosphorylated state and inhibits its translocation to the mitochondrial surface, preventing subsequent cytochrome c release (Wang et al., 1999). Moreover, calcium overload has been demonstrated to directly activate calcium- and magnesium-dependent endonuclease (DNase I), thus resulting in DNA fragmentation and cell apoptosis (Gottlieb & Engler, 1999). Calcium antagonists may prevent this DNase activation and reduce myocardial apoptosis. In addition, previous studies have demonstrated that calcium overloading increases the generation of free radicals (Bagchi et al., 1997), a species that induces apoptotic cell death in various cell types (Das et al., 1999) and that calcium antagonists significantly reduce free radical generation in ischaemic/reperfused tissue. This anti-oxidant effect of calcium antagonists may thus also contribute to their anti-apoptosis effects in myocardial ischaemia and reperfusion.
In the summary, our present study demonstrated that benidipine, a long-lasting vasodilatory calcium antagonist, reduces MI/R-induced myocardial apoptosis in a haemodynamically-independent manner. We have provided the first direct evidence that the anti-apoptosis property of calcium antagonists significantly contributes to its overall infarct reduction and cardiac function improvement effects. This new observation may serve as a fundamental base for searching more effective treatments to limit MI/R-induced myocardial injury. In addition, previous clinical studies have demonstrated that in some patients, calcium antagonists are harmful, principally because they acutely lower blood pressure and result in a collateral steal phenomenon (Ehring & Heusch, 1997). Our present experimental results suggest that administration of a calcium antagonist at a sub-haemodynamic dose may therefore be preferable under pathologic conditions where myocardial apoptosis is predominant but blood pressure lowering effects need to be avoided (such as ischaemic heart failure).
Acknowledgments
The authors give special thanks to Dr C. Paul Bianchi, Department of Pharmacology at Thomas Jefferson University, for his expert assistance in the assay of tissue calcium content. This work is supported in part by the National Nature Science Foundation of China Grants 39970302 (to F. Gao) and 39925013, 39970807 (to X.L. Ma).
Abbreviations
- AAR
Area-at-risk
- ANAR
Area-not-at-risk
- CI
Cardiac contractile index
- CK
Creatine kinase
- HR
Heart rate
- LVEDP
Left ventricular end diastolic pressure
- LVP
Left ventricular pressure
- MABP
Mean arterial blood pressure
- MI/R
Myocardial ischaemia/reperfusion
- PRI
pressure-rate-index
References
- BAGCHI D., WETSCHER G.J., BAGCHI M., HINDER P.R., PERDIKIS G., STOHS S.J., HINDER R.A., DAS D.K. Interrelationship between cellular calcium homeostasis and free radical generation in myocardial reperfusion injury. Chem. Biol. Interact. 1997;104:65–85. doi: 10.1016/s0009-2797(97)03766-6. [DOI] [PubMed] [Google Scholar]
- BIALIK S., GEENEN D.L., SASSON I.E., CHENG R., HORNER J.W., EVANS S.M., LORD E.M., KOCH C.J., KITSIS R.N. Myocyte apoptosis during acute myocardial infarction in the mouse localizes to hypoxic regions but occurs independently of p53. J. Clin. Invest. 1997;100:1363–1372. doi: 10.1172/JCI119656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BORASO A., CARGNONI A., COMINI L., GAIA G., BERNOCCHI P., FERRARI R. Effect of lacidipine on ischaemic and reperfused isolated rabbit hearts. Mol. Cell Biochem. 1993;125:73–86. doi: 10.1007/BF00926837. [DOI] [PubMed] [Google Scholar]
- CHEN S.J., BRADLEY M.E., LEE T.C. Chemical hypoxia triggers apoptosis of cultured neonatal rat cardiac myocytes: modulation by calcium-regulated proteases and protein kinases. Mol. Cell Biochem. 1998;178:141–149. doi: 10.1023/a:1006893528428. [DOI] [PubMed] [Google Scholar]
- DAS D.K., ENGELMAN R.M., MAULIK N. Oxygen free radical signaling in ischemic preconditioning. Ann. N.Y. Acad. Sci. 1999;874:49–65. doi: 10.1111/j.1749-6632.1999.tb09224.x. [DOI] [PubMed] [Google Scholar]
- EHRING T., HEUSCH G. Dihydropyridine calcium antagonists: beneficial or adverse effects in the setting of myocardial ischaemia/reperfusion. Cardiology. 1997;88 Suppl 1:3–14. doi: 10.1159/000177451. [DOI] [PubMed] [Google Scholar]
- GOTTLIEB R.A., ENGLER R.L. Apoptosis in myocardial ischemia/reperfusion. Ann. N.Y. Acad. Sci. 1999;874:412–426. doi: 10.1111/j.1749-6632.1999.tb09255.x. [DOI] [PubMed] [Google Scholar]
- GROSS A., MCDONNELL J.M., KORSMEYER S.J. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- KAJSTURA J., CHENG W., REISS K., CLARK W.A., SONNENBLICK E.H., KRAJEWSKI S., REED J.C., OLIVETTI G., ANVERSA P. Apoptosis and necrotic myocyte cell deaths are independent contributing variables of infarct size in rats. Lab. Invest. 1996;74:86–107. [PubMed] [Google Scholar]
- KARASAWA A., KUBO K. Protection by benidipine hydrochloride (KW-3049), a calcium antagonist, of ischemic kidney in rats via inhibitions of Ca-overload, ATP-decline and lipid peroxidation. Jpn. J. Pharmacol. 1990;52:553–562. doi: 10.1254/jjp.52.553. [DOI] [PubMed] [Google Scholar]
- KARASAWA A., KUBO K., OKA T., NAKAMIZO N. Effects of the new calcium antagonist benidipine hydrochloride on cardiohemodynamics in anesthetized dogs. Arzneimmittelforschung. 1988;38:1713–1716. [PubMed] [Google Scholar]
- KITAKAZE M., KARASAWA A., KOBAYASHI H., TANAKA H., KUZUYA T., HORI M. Benidipine: a new calcium channel blocker with a cardioprotective effect. Cardiovasc. Drug Rev. 1999;17:1–15. [Google Scholar]
- LEFER A.M., POLANSKY E.W., BIANCHI C.P., NARAYAN S. Influence of verapamil on cellular integrity and electrolyte concentrations of ischemic myocardial tissue in the cat. Basic Res. Cardiol. 1979;74:555–567. doi: 10.1007/BF01907648. [DOI] [PubMed] [Google Scholar]
- MA X.L., KUMAR S., GAO F., LOUDEN C.S., LOPEZ B.L., CHRISTOPHER T.A., WANG C.L., LEE J.C., FEUERSTEIN G.Z., YUE T.L. Inhibition of p38 mitogen-activated protein kinase decreases cardiomyocyte apoptosis and improves cardiac function after myocardial ischemia and reperfusion. Circulation. 1999;99:1685–1691. doi: 10.1161/01.cir.99.13.1685. [DOI] [PubMed] [Google Scholar]
- MASON R.P., LEEDS P.R., JACOB R.F., HOUGH C.J., ZHANG K.G., MASON P.E., CHUANG D.M. Inhibition of excessive neuronal apoptosis by the calcium antagonist amlodipine and antioxidants in cerebellar granule cells. J. Neurochem. 1999;72:1448–1456. doi: 10.1046/j.1471-4159.1999.721448.x. [DOI] [PubMed] [Google Scholar]
- MCCONKEY D.J. Biochemical determinants of apoptosis and necrosis. Toxicol. Lett. 1998;99:157–168. doi: 10.1016/s0378-4274(98)00155-6. [DOI] [PubMed] [Google Scholar]
- MCCONKEY D.J., ORRENIUS S. Signal transduction pathways in apoptosis. Stem Cells. 1996;14:619–631. doi: 10.1002/stem.140619. [DOI] [PubMed] [Google Scholar]
- MCCONKEY D.J., ORRENIUS S. The role of calcium in the regulation of apoptosis. Biochem. Biophys. Res. Commun. 1997;239:357–366. doi: 10.1006/bbrc.1997.7409. [DOI] [PubMed] [Google Scholar]
- MURPHY L., PROCOPIO J.M., BIANCHI C.P. Cisplatinum enhancement of myocardial Mg2+ transport. Life Sci. 1996;59:1739–1744. doi: 10.1016/s0024-3205(96)00511-5. [DOI] [PubMed] [Google Scholar]
- OLIVETTI G., QUAINI F., SALA R., LAGRASTA C., CORRADI D., BONACINA E., CIGOLA E., ANVERSA P. Acute myocardial infarction in humans is associated with activation of programmed myocyte cell death in the surviving portion of the heart. J. Mol. Cell Cardiol. 1996;28:2005–2016. doi: 10.1006/jmcc.1996.0193. [DOI] [PubMed] [Google Scholar]
- PEITSCH M.C., POLZAR B., STEPHAN H., CROMPTON T., MACDONALD H.R., MANNHERZ H.G., TSCHOPP J. Characterization of the endogenous deoxyribonuclease involved in nuclear DNA degradation during apoptosis (programmed cell death) EMBO J. 1993;12:371–377. doi: 10.1002/j.1460-2075.1993.tb05666.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAAFAT A.M., MURRAY M.T., MCGUIRE T., DEFRAIN M., FRANKO A.P., ZAFAR R.S., PALMER K., DIEBEL L., DULCHAVSKY S.A. Calcium blockade reduces renal apoptosis during ischemia reperfusion. Shock. 1997;8:186–192. doi: 10.1097/00024382-199709000-00006. [DOI] [PubMed] [Google Scholar]
- RAFFA R.B., BIANCHI C.P., NARAYAN S.R. Reversible inhibition of acetylcholine contracture of molluscan smooth muscle by heavy metals: correlation to Ca++ and metal content. J. Pharmacol. Exp. Ther. 1987;243:200–204. [PubMed] [Google Scholar]
- SARASTE A., PULKKI K., KALLAJOKI M., HENRIKSEN K., PARVINEN M., VOIPIO-PULKKI L.M. Apoptosis in human acute myocardial infarction. Circulation. 1997;95:320–323. doi: 10.1161/01.cir.95.2.320. [DOI] [PubMed] [Google Scholar]
- WANG H.G., PATHAN N., ETHELL I.M., KRAJEWSKI S., YAMAGUCHI Y., SHIBASAKI F., MCKEON F., BOBO T., FRANKE T.F., REED J.C. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science. 1999;284:339–343. doi: 10.1126/science.284.5412.339. [DOI] [PubMed] [Google Scholar]
- WYLLIE A.H., MORRIS R.G., SMITH A.L., DUNLOP D. Chromatin cleavage in apoptosis: association with condensed chromatin morphology and dependence on macromolecular synthesis. J. Pathol. 1984;142:67–77. doi: 10.1002/path.1711420112. [DOI] [PubMed] [Google Scholar]
- YAOITA H., OGAWA K., MAEHARA K., MARUYAMA Y. Attenuation of ischemia/reperfusion injury in rats by a caspase inhibitor. Circulation. 1998;97:276–281. doi: 10.1161/01.cir.97.3.276. [DOI] [PubMed] [Google Scholar]
- YUE T.L., MA X.L., WANG X.K., ROMANIC A.M., LIU G.L., LOUDEN C., GU J.L., KUMAR S., POSTE G., RUFFOLO RR., JR, FEUERSTEIN G.Z. Possible involvement of stress-activated protein kinase signaling pathway and Fas receptor expression in prevention of ischemia/reperfusion-induced cardiomyocyte apoptosis by carvedilol. Circ. Res. 1998;82:166–174. doi: 10.1161/01.res.82.2.166. [DOI] [PubMed] [Google Scholar]