

Abstract
This study was undertaken to determine whether endothelium-dependent relaxations are altered in mesenteric arteries from young female rats during oestrus cycle and after castration.
The contractile response to phenylephrine (Phe) was significantly enhanced in arteries from rats subjected to ovariectomy than in those from sham-operated (control) rats. Treatment of ovariectomized rats with 17β-oestradiol returned the Phe response to the control level. Arteries from rats at the diestrus stage also exhibited greater contraction in response to Phe. In the presence of 100 μM NG-nitro-L-arginine (L-NOARG), the enhancement of the Phe contractile response associated with oestrogen deficiency was not observed.
Endothelium-dependent relaxations elicited by acetylcholine (ACh) in arteries precontracted with Phe were significantly reduced in ovariectomized and diestrus rats regardless of whether endothelium-derived nitric oxide (NO) was blocked with L-NOARG. Treatment with 17β-oestradiol prevented the reduced vascular relaxant response to ACh in ovariectomized rats. The reduction in the ACh responses observed in ovariectomized and diestrus rats was eliminated when 500 nM apamin and 100 nM charybdotoxin were present.
ACh-induced endothelium-dependent hyperpolarizations were depressed in arteries from ovariectomized and diestrus rats. The hyperpolarizing response to ACh was significantly improved when ovariectomized rats were treated with 17β-oestradiol. The resting membrane potentials and pinacidil-induced hyperpolarizations were unaffected by ovariectomy or the diestrus stage.
These results suggest that oestrogen-deficient states of both short and long duration reduce the basal release of NO from the endothelium and specifically attenuate endothelium-dependent hyperpolarization and relaxation transduced by endothelium-derived hyperpolarizing factor.
Keywords: Oestrogen, oestrus cycle, acetylcholine, endothelium-dependent relaxation, endothelium-derived hyperpolarizing factor, mesenteric artery
Introduction
Epidemiological investigations have demonstrated that cardiovascular disease is a major cause of morbidity and mortality in men and postmenopausal women. It has been reported that oestrogen replacement therapy in postmenopausal women reduces the risk of cardiovascular diseases (Lerner & Kannel, 1986; Barrett-Connor & Bush, 1991), although a recent study has cast some doubt on the validity of this notion (Hulley et al., 1998). In addition, women who underwent early menopause, either surgically or naturally, are at increased risk of developing coronary heart disease in comparison with premenopausal women of the same age (Colditz et al., 1987). Therefore, oestrogen deficiency, independently of aging, may be responsible for the enhanced risk of cardiovascular diseases.
Although there is accumulating evidence for the protection by oestrogen replacement therapy against cardiovascular diseases, the exact mechanism of the favourable role of oestrogen in cardiovascular diseases still remains unsettled. Several studies suggest that oestrogen may cause a decrease in low-density lipoprotein and an increase in high-density lipoprotein, and thereby improve the lipid metabolism (Bush et al., 1987; Barrett-Connor & Bush, 1991). In addition to the favourable changes in lipid metabolism, there is evidence that oestrogen has a direct beneficial effect on cells/structures within the arterial wall independently of its lipoprotein effects (Sudhir et al., 1995; Holm et al., 1997).
Alterations in endothelial functions, especially changes in nitric oxide (NO) production, have been suggested to be one of the cellular mechanisms regulated by oestrogen in its preventive effect on the development of cardiovascular diseases (Hishikawa et al., 1995; Caulin-Glaser et al., 1997). Ovariectomy-induced impairment of endothelium-dependent relaxation (Gisclard et al., 1988) further supports the notion that oestrogen exerts its favourable effects on the vascular system through regulating endothelial functions.
Endothelium-dependent relaxation, one of the important endothelial functions, is known to be mediated by three different endothelium-dependent relaxing factors: NO, endothelium-derived hyperpolarizing factor (EDHF) and prostacyclin (Vanhoutte et al., 1986). A number of reports implicate NO production in the endothelium as an important target of exogenous as well as endogenous oestrogen (Hayashi et al., 1992; Bell et al., 1995). However, the influence of the oestrogen status on EDHF-mediated endothelium-dependent relaxation has not been fully investigated, although a recent work suggests that EDHF-mediated vasodilation is greatly affected by oestrus cycle and pregnancy (Dalle Lucca et al., 2000). Therefore, the aim of the present study was to examine how oestrogen status affects endothelium-dependent vascular relaxations mediated by NO and EDHF in young female rats. In addition to the oestrogen depletion after ovariectomy, we examined endothelium-dependent relaxation of mesenteric arteries obtained from female rats during oestrus cycle, i.e., the proestrous to oestrus stage and the diestrus stage, since the non-physiological effects associated with ovariectomy, such as depletion of circulating progesterone, are avoided.
Methods
Animal preparation
All procedures were performed in accordance with the guidelines of the Hokkaido University School of Medicine Animal Care and Use Committee. Female 8-week-old Wistar rats (150–160 g) were divided randomly in three groups, sham-operated, ovariectomized, and treated with 17β-oestradiol after ovariectomy. Rats were anaesthetized with ketamine (10 mg 100 g−1, i.p.) under aseptic conditions. Sham-operated (control) group received only laparotomy. Ovariectomy was carried out according to the methods by Nekooeian et al. (1998), in which the bilateral ovaries were ligated and then removed. For oestrogen replacement therapy, an osmotic pump producing sustained release of 17β-oestradiol at 10 μg day−1 was implanted subcutaneously at the back of ovariectomized rats. Each group of rats was caged separately and housed under controlled environmental conditions of temperature (24°C) and humidity (40–60%) on a 12 h light/dark cycle. These animals were given the standard chow and water ad libitum. After 4 weeks, the animals were anaesthetized with diethyl ether, and blood samples were collected from the inferior cava for measurement of oestradiol. After centrifugation of the blood for 10 min at 3000 r.p.m., plasma was collected and frozen at −20°C for later measurement of oestradiol. Only rats staying in the proestrus to oestrus stage at the day when the experiments were carried out were used as control.
In a series of experiments investigating endothelium-dependent relaxation of female rats at the diestrus stage, vaginal smears were taken at 1000 h from female rats aged 12 weeks, which had been confirmed previously to show regular oestrus cycles of 4 or 5 days, and those which were at the stage of diestrus under the microscopic examination of the vaginal smears stained with methylene blue (Carswell et al., 2000) were selected and used for the experiments at the same day.
Measurement of the relaxant responses of mesenteric arteries
Rats were killed by exsanguination under anaesthesia with gaseous diethyl ether. The level of anaesthesia was such as to maintain deep surgical anaesthesia but not to cause respiratory and circulatory collapse. After collecting the blood samples, the main branch of mesenteric arteries was carefully excised and placed on a dish containing oxygenated physiological salt solution (PSS) at room temperature. The arteries were then cleaned of adherent connective tissues, cut into rings of 3 mm length. Each ring was suspended by a pair of stainless steel pins in a water-jacketed bath filled with 6 ml of normal PSS. The composition of PSS was as follows (in mM); NaCl 118.2, KCl 4.7, CaCl2 2.5, MgCl2 1.2, KH2PO4 1.2, NaHCO3 25.0 and glucose 10.0. The solution in the bath was gassed with 95% O2 and 5% CO2 (pH 7.4) and its temperature was maintained at 37°C. The rings were stretched until a resting tension of 1 g was loaded and then allowed to equilibrate for at least 60 min before the start of experiments. At the resting tension of 1 g the rings were found to develop maximal active tension to stimulation with 80 mM K+. Isometric tension was monitored with isometric transducer (Nihon Kohden, AP-621G, Tokyo, Japan) and recorded on a pen recorder (Rikadenki, R-64, Tokyo, Japan). The rings were repeatedly challenged with 80 mM K+ until the contractions reached a constant level. High-K+ PSS was made by substituting NaCl with equimolar KCl.
Relaxation to acetylcholine (ACh) (1 nM–100 μM) of the ring preparations was usually determined after precontraction with 10 μM phenylephrine (Phe). There was no significant difference in the magnitude of contractions produced by Phe at this concentration among the groups (see Results). The relaxant response of the artery to ACh was determined first without any treatment, then the relaxant response to ACh was tested in the presence of 100 μM L-NOARG and 10 μM indomethacin in order to exclude the involvement of endothelium-derived NO and prostanoids. Because incubation with L-NOARG markedly enhanced Phe-induced contractions, care was taken to match the contractions in the absence and presence of L-NOARG. Thus, in the experiments with L-NOARG, the tissue was precontracted with 300 nM Phe. We defined the relaxant responses to ACh obtained in the presence of L-NOARG and indomethacin as EDHF-mediated relaxations, since it was confirmed that the remaining endothelium-dependent relaxations to ACh, i.e, the L-NOARG- and indomethacin-resistant relaxations, were completely eliminated by 30 mM K+ PSS or by the combination of apamin and charybdotoxin (ChTX) as previously reported (Fukao et al., 1997; Doughty et al., 1999). The relaxant responses to ACh were also tested in the presence of 10 μM indomethacin, 100 nM ChTX and 500 nM apamin in order to exclude the involement of prostanoids and EDHF. We defined the ACh responses obtained in the presence of indomethacin, ChTX and apamin as NO-mediated relaxations, since the remaining relaxant responses to ACh were abolished after treatment with 100 μM L-NOARG. The degree of relaxations was expressed as a percentage of the height of contraction induced by Phe. As the preliminary experiments revealed that endothelium-dependent relaxations to ACh in mesenteric arteries from control and ovariectomized rats were marginally affected by indomethacin alone, we focused to examine alterations in the NO- and EDHF-mediated responses.
Recording of the membrane potentials from smooth muscle cells of mesenteric arteries
Rat mesenteric arterial rings were prepared as described above. The arterial rings were opened longitudinally. Care was taken to ensure that the endothelial layer was not damaged during processing of the tissue preparation. Where indicated, the endothelial layer was removed by gently rubbing the intimal surface of the vessel with a moistened cotton ball. The tissue was pinned down, the intimal side upward, on the bottom of an organ chamber (capacity 3 ml), and superfused at a constant flow rate of 7 ml min−1 with PSS aerated with 95% O2 and 5% CO2. The temperature of perfusate was kept constant at 37°C. After the preparations had been equilibrated for 60 min, glass microeletrodes filled with 3 M KCl (tip resistance 40–80 MΩ) were inserted into the smooth muscle cells from the intimal side. Successful impalements were signalled by sudden negative drop in potential from the baseline (zero potential reference). Following a first drop in voltage, the microelectrode was further advanced into the arterial wall to ensure that a smooth muscle cell was impaled. Following a stable recording of membrane potential for at least 3 min, ACh or pinacidil was applied to the preparations through the superfusing fluid, and the changes in the membrane potentials were measured through a high-impedance amplifier (Nihon Kohden, MEZ-8201). When L-NOARG or indomethacin was used, these agents were applied 15 min before the addition of ACh. Electrical signals were continuously monitored on an oscilloscope (Nihon Kohden, VC-10) and recorded on a chart recorder (Watanabe Sokki, WR310, Tokyo, Japan). For data analysis we used only the results of the experiments through which a single impalement was maintained.
Measurement of blood plasma 17β-oestradiol
Blood plasma concentrations of the oestradiol levels were measured by a standard procedure using the IMMUNOTECH OESTRADIOL assay kit (Réf. 2464; TFB, Tokyo, Japan).
Drugs
The following drugs were used: ACh chloride (Wako, Osaka, Japan), ketamine hydrochloride (Sankyo, Tokyo, Japan), pinacidil (Shionogi, Osaka, Japan), L-phenylephrine hydrochloride, L-NOARG, indomethacin, 17β-oestadiol, ChTX and apamin (Sigma, St. Louis, MO, U.S.A.). Pinacidil and L-NOARG were prepared in 0.2 N HCl. Other drugs were dissolved in distilled water. PSS was used for further dilution to the proper concentrations.
Statistical analysis
All data are expressed as means±s.e.mean. Two-way analysis of ANOVA was used to compare the concentration-responses curves for Phe-induced contraction or ACh-induced relaxation between the groups, followed by the Scheffé's multiple comparison test. Other variables were compared by the use of paired and unpaired Student's t-test. The analyses were carried out using the software Stat View (Abacus Concepts, Berkeley, CA, U.S.A.). Values of P<0.05 were accepted as indicating a significant difference. The half maximal concentration (EC50) of Phe or ACh was determined from log-probit plots of the individual response vs concentrations, and data are shown as the average of the individual value.
Results
Plasma oestradiol levels
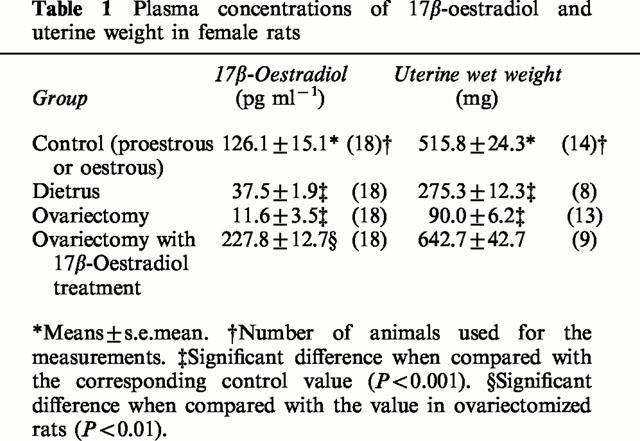
The plasma oestrogen levels estimated in female rats used for this study are shown with their uterine weights in Table 1. Because of the rapid shift of proestrus to oestrus stage, it was difficult to select only female rats staying at the proestrus stage based on the vaginal smear findings. Thus, rats included in the control group were either at the proestrus or at the oestrus stage. Reflecting this fact, the plasma levels of oestradiol were scattered from 40–200 pg ml−1. The plasma oestrogen levels at 4 weeks after ovariectomy were markedly reduced. However, eight of 18 ovariectomized rats showed the oestradiol levels of 10–50 pg ml−1, which were comparable to those of diestrus rats. This may result from compensatory secretion from adrenal cortex in ovariectomized animals. The other 10 rats revealed the level of oestradiol less than the detectable range. Supplementation with 17β-oestradiol to ovariectomized female rats maintained the high level of plasma oestradiol and prevented shrinkage of the uterus. The wet uterine weight was correlated with the plasma estradiol levels.
Table 1.
Plasma concentrations of 17β-oestradiol and uterine weight in female rats
Phe-induced contraction of mesenteric arteries
Figure 1a depicts the concentration-response curves for Phe-induced contractions of mesenteric arteries from sham-operated (control) and ovariectomized rats. Arteries from ovariectomized animals exhibited higher sensitivity to Phe than those from controls, while there was no difference in the maximum response between the two groups of tissues. The EC50 value was 0.3±0.1 μM (n=6) for the ovarictomized group, which was significantly (P<0.05) smaller than the corresponding value of 1.5±0.4 μM (n=7) obtained in the control group. This resulted in significantly greater contraction in reponse to 1 μM Phe in the ovariectomized group than in the control group (Figure 1b). Treatment of ovariectomized rats with 17β-oestradiol reversed the response to 1 μM Phe to the control level. The contractile response to 1 μM Phe of arteries from diestrus rats was also greater than that of controls. There was no significant difference in contraction produced by 10 μM Phe among the experimental groups.
Figure 1.
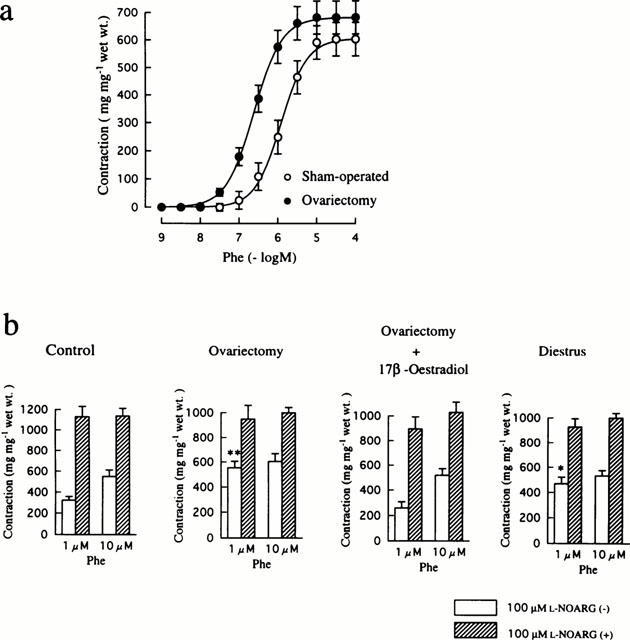
Influences of ovariectomy and oestrus cycle on Phe-induced contraction in rat mesenteric arteries. (a) Concentration-response curves for Phe-induced contraction in mesenteric arteries from sham-operated and ovariectomized rats. Phe was added to the bath cumulatively. (b) Effects of treatment with 100 μM L-NOARG on contractions induced by 1 and 10 μM Phe in mesenteric arteries from sham-operated, ovariectomized, ovariectomized and 17β-oestradiol-treated, and diestrus rats. The results are shown as means±s.e.mean of 6–10 experiments. *P<0.05 and **P<0.01 vs the corresponding value obtained in the control (sham-operated) group.
Incubation with 100 μM L-NOARG caused a significant increase in Phe-induced contractions in all groups (Figure 1b). This was especially evident in the contractile response to 1 μM Phe of arteries from control rats and rats that were treated with 17β-oestradiol after ovariectomy. As a result, the addition of L-NOARG abolished the difference in contraction induced by 1 μM Phe among the experimental groups. Mechanical removal of the endothelium resulted in the same effect on Phe-induced contractions as incubation with L-NOARG (data not shown). These findings indicate that NO released from the endothelium at the basal state actively modulates Phe-induced contractions, especially in the presence of endogenous oestrogen.
Decreased endothelium-dependent relaxation to ACh in mesenteric arteries from ovariectomized and diestrus rats
ACh produced endothelium-dependent relaxation in rat mesenteric arteries precontracted with 10 μM Phe in a concentration dependent manner. As shown in Figure 2a, ACh-induced relaxation was found to decrease in ovariectomized rats. The maximun relaxant response to ACh was not complete and significantly less than in the control group (88.0±1.3%, n=12 vs 99.8±0.2%, n=15, P<0.01). In addition, the EC50 value for ACh (589±161 nM, n=12) was significantly (P<0.001) greater than that in the control group (47±8 nM, n=15). Replacement therapy with 17β-oestradiol to ovariectomized rats caused a return of the maximum ACh response (99.0±0.4%, n=12). However, restoration of the rightward shift of the concentration-response curve for ACh was not complete (Figure 2b). Thus, the EC50 value for ACh was significantly changed to 116±25 nM (n=12), but was still greater than the control value.
Figure 2.
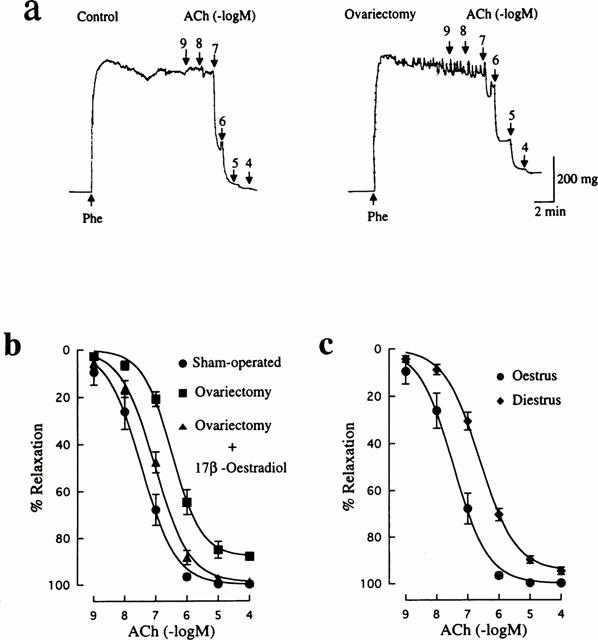
Influences of ovariectomy and oestrus cycle on endothelium-dependent relaxation induced by ACh in rat mesenteric arteries. (a) Typical traces illustrating the relaxant responses to ACh during 10 μM Phe-induced contraction in mesenteric arteries from sham-operated and ovariectomized rats. (b) Concentration-response curves for ACh-induced relaxation in mesenteric arteries from rats that were sham-operated, ovariectomized, and ovariectomized and 17β-oestradiol-treated. (c) Concentration-response curves for ACh-induced relaxation in mesenteric arteries from oestrus and diestrus rats. ACh was added to the bath cumulatively. The points are means of 6–8 experiments; vertical lines show s.e.mean. Responses are expressed as per cent relaxation of Phe-induced contraction.
ACh-induced relaxation in the diestrus group was also suppressed, as indicated by the rightward shift of the concentration-response curve for ACh (Figure 2c). The value of EC50 (288±45 nM, n=12) was significantly greater than the control value, although the maximum response (94.7±1.6%, n=12) was not significantly different from that in the control group.
Contribution of prostacyclin and NO to endothelium-dependent relaxation to ACh
In response to ACh, the endothelium produces three different kinds of relaxing factors: NO, prostacyclin and EDHF. The concentration-response curve for ACh-induced relaxation was unaffected by 10 μM indomethacin in either of the control or the ovariectomized group (data not shown), suggesting a minimal contribution of prostacyclin to the altered endothelium-dependent relaxant response to ACh of arteries from ovariectomized rats.
In order to examine the relative contribution of NO to the ACh relaxant responses, a manoeuvre to eliminate pharmacologically the involvement of EDHF and prostacyclin was applied to mesenteric arteries from each group. The concentration-response curves for ACh in the presence of 10 μM indomethacin, 500 nM apamin and 100 nM ChTX are shown in Figure 3b. The maximum relaxant response to ACh was not significantly reduced in the ovariectomized group. Although the EC50 value for ACh was greater in the ovariectomized group (368±99 nM, n=11) than that in the control group (128±32 nM, n=7), the difference did not reach a statistically significant level (P>0.05). The remaining relaxant response to ACh obtained in the presence of indomethacin, apamin and ChTX was completely suppressed by 100 μM L-NOARG (Figure 3a). Treatment with 17β-oestradiol of ovariectomized rats showed an EC50 value of 203±38 nM (n=7).
Figure 3.
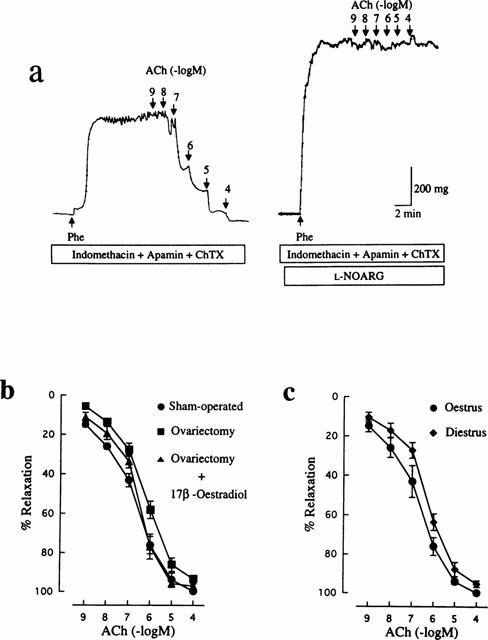
Influences of ovariectomy and oestrus cycle on endothelial NO-mediated relaxation induced by ACh in rat mesenteric arteries. (a) Typical traces illustrating the relaxant responses to ACh during 10 μM Phe-induced contraction in control (sham-operated) rat mesenteric arteries in the presence of 10 μM indomethacin, 500 nM apamin and 100 nM ChTX. Right panel shows that the remaining relaxation to ACh was abolished by further treatment with 100 μM L-NOARG. Indomethacin, apamin, ChTX and L-NOARG were added to the bath 15–20 min before application of Phe. (b) Concentration-response curves for ACh-induced relaxation in mesenteric arteries from rats that were sham-operated, ovariectomized, and ovariectomized and 17β-oestradiol-treated. (c) Concentration-response curves for ACh-induced relaxation in mesenteric arteries from oestrus and diestrus rats. All curves were constructed in the presence of indomethacin, apamin and ChTX. The points are means of 6–11 experiments; the vertical lines show s.e.mean. Responses are expressed as per cent relaxation of Phe-induced contraction. Statistically significant differences were not found between the concentration-response curves of the experimental groups.
The same result was obtained in arteries from diestrus rats (Figure 3c). In the presence of indomethacin, apamin and ChTX, the maximum response to ACh (95.1±1.6%, n=6) and the EC50 value (313±72 nM, n=6) were not statistically significantly different from the corresponding control values.
Contribution of EDHF to endothelium-dependent relaxation to ACh
ACh-induced relaxation in arteries precontracted by Phe in the presence of 100 μM L-NOARG and 10 μM indomethacin can be assumed to be caused by EDHF, based upon the finding that further treatment with the combination of 500 nM apamin and 100 nM ChTX abolished completely the ACh response (Figure 4a). As shown in Figure 4b, ACh-induced relaxation resistant to L-NOARG and indomethacin was depressed in arteries from ovariectomized rats compared with that in controls. The maximum relaxant response to ACh in the ovariectomized group (75.0±2.3%, n=13, P<0.001) was significantly less than that in the control group (98.3±0.8%, n=9), and the EC50 value for ACh was much greater in the ovariectomized group (1.2±0.2 μM, n=13, P<0.01) than the control value (0.2±0.1 μM, n=9). Treatment with 17β-oestradiol of ovariectomized rats prevented the down-regulation of the L-NOARG-resistant relaxant response to ACh (EC50: 0.4±0.1 μM, n=6) (Figure 4b).
Figure 4.
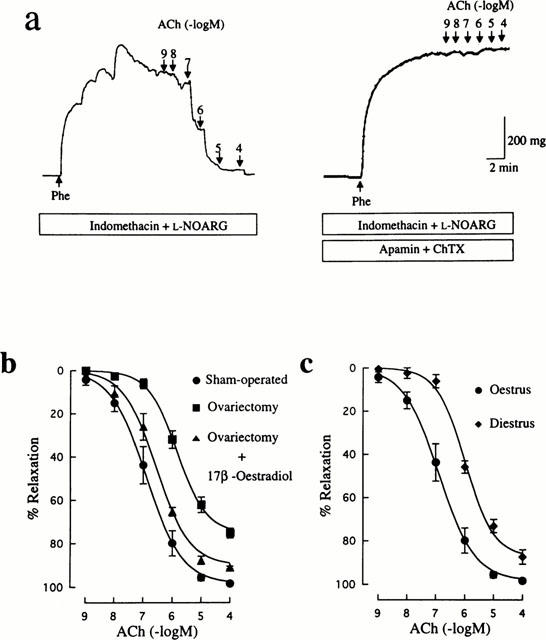
Influences of ovariectomy and oestrus cycle on EDHF-mediated relaxation induced by ACh in rat mesenteric arteries. (a) Typical traces illustrating the relaxant responses to ACh during 300 nM Phe-induced contraction in control (sham-operated) rat mesenteric arteries in the presence of 10 μM indomethacin and 100 μM L-NOARG. Right panel shows that the remaining relaxation to ACh was abolished by further treatment with 500 nM apamin and 100 nM ChTX. Indomethacin, L-NOARG, apamin and ChTX were added to the bath 15–20 min before application of Phe. (b) Concentration-response curves for ACh-induced relaxation in mesenteric arteries from rats that were sham-operated, ovariectomized, and ovariectomized and 17β-oesradiol-treated. (c) Concentration-response curves for ACh-induced relaxation in mesenteric arteries from oestrus and diestrus rats. All curves were constructed in the presence of indomethacin and L-NOARG. The points are means of 6–12 experiments; vertical lines show s.e.mean. Responses are expressed as per cent relaxation of Phe-induced contraction.
Arteries obtained from diestrus rats showed a significantly decreased relaxant response to ACh when compared with those from oestrus rats. The maximum response to ACh in the diestrus group (87.5±3.3%, n=8, P<0.05) was less than in the control group and the EC50 value for ACh (1.1±0.2 μM, n=8, P<0.01) was significantly greater than the control value.
ACh-induced hyperpolarization of vascular smooth muscle cells
As shown in Figure 5, ACh produced endothelium-dependent hyperpolarization of smooth muscle cells of mesenteric arteries. The hyperpolarizing response to ACh was not affected by treatment with either 100 μM L-NOARG alone or in combination with 10 μM indomethacin. However, combined treatment with 500 nM apamin and 100 nM ChTX significantly suppressed the hyperpolarizing response to 1 μM ACh from 14.7±0.3 mV to 2.3±0.6 mV (n=3).
Figure 5.
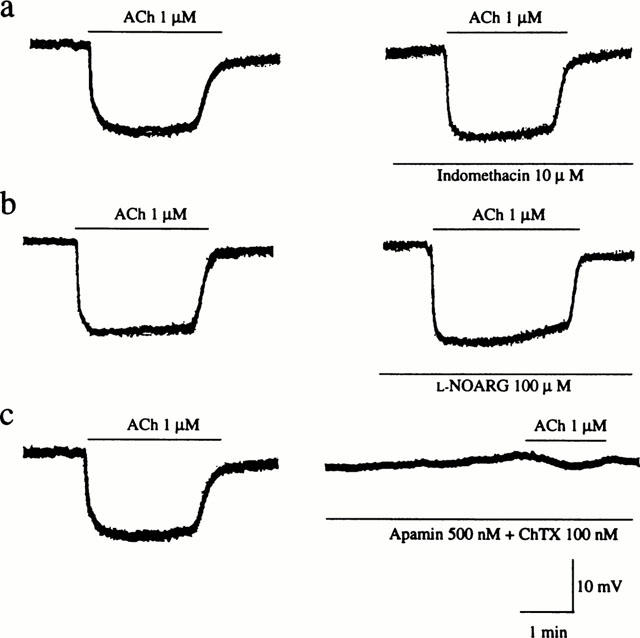
Effects of indomethacin, L-NOARG and apamin plus ChTX on membrane hyperpolarization induced by ACh in control (sham-operated) rat mesenteric arteries. Actual recordings of the hyperpolarizing responses to 1 μM ACh before (left panels) and after (right panels) treatment with 10 μM indomethacin (a), 100 μM L-NOARG (b) or 500 nM apamin plus 100 nM ChTX (c) are shown.
The average resting membrane potentials of smooth muscle cells of mesenteric arteries obtained from four groups of rats were not significantly different from each other (control: −52.5±0.6 mV, n=13; ovariectomized: −50.6±0.9 mV, n=13; ovariectomized and 17β-oestradiol-treated: −53.6±0.7 mV, n=13; diestrus: −50.5±0.9 mV, n=11). However, the amplitudes of hyperpolarization by 1 μM ACh were significantly diminished in the ovariectomized group (3.4±0.8 mV, n=13) and the diestrus group (7.2±0.4 mV, n=11) compared to that in the control group (15.3±0.5 mV, n=13), while oestrogen therapy restored the reduced hyperpolarizing response after ovariectomy (12.8±0.8 mV, n=13). Such original registrations are illustrated in Figure 6a–d. In control arteries, ACh produced concentration-dependent hyperpolarizations with an EC50 of 167±21 nM (n=8) (Figure 6e). Ovariectomy caused markedly a downward shift of the concentration-response curve for ACh-induced hyperpolarization, which was significantly prevented by treatment with 17β-oestradiol. As shown in Figure 6f, the concentration-response curve was also depressed in the diestrus state. The reduction in the maximal response was not so marked, but the EC50 value (552±107 nM, n=7) was significantly (P<0.01) greater than the control value.
Figure 6.
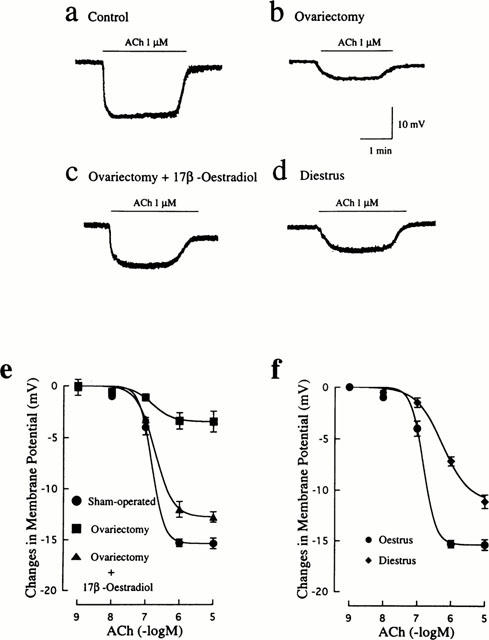
Influences of ovariectomy and oestrus cycle on endothelium-dependent hyperpolarization induced by ACh in rat mesenteric arteries. Actual recordings of the hyperpolarizing responses to 1 μM ACh in mesenteric arteries from sham-operated (a), ovariectomized (b), ovariectomized and 17β-oestradiol-treated (c), and diestrus (d) rats are shown. (e) Concentration-response curves for ACh-induced hyperpolarization in mesenteric arteries from rats that were sham-operated, ovariectomized, and ovariectomized and 17β-oestradiol-treated. (f) Concentration-response curves for ACh-induced hyperpolarization in mesenteric arteries from oestrus and diestrus rats. The points are means of 11–14 experiments; vertical lines show s.e.mean.
Pinacidil, an ATP-sensitive K+ channel opener, elicited endothelium-independent hyperpolarization of vascular smooth muscle cells. The hyperpolarizing responses to pinacidil (10 μM) were not significantly different among the experimental groups (Figure 7). Thus, the peak amplitudes of hyperpolarization induced by pinacidil in the control, ovariectomized, ovariectomized and 17β-oestradiol-treated, and diestrus groups were 25.5±0.9, 26.8±0.9, 24.8±1.3, and 25.3±0.9 mV (n=4–8), respectively.
Figure 7.
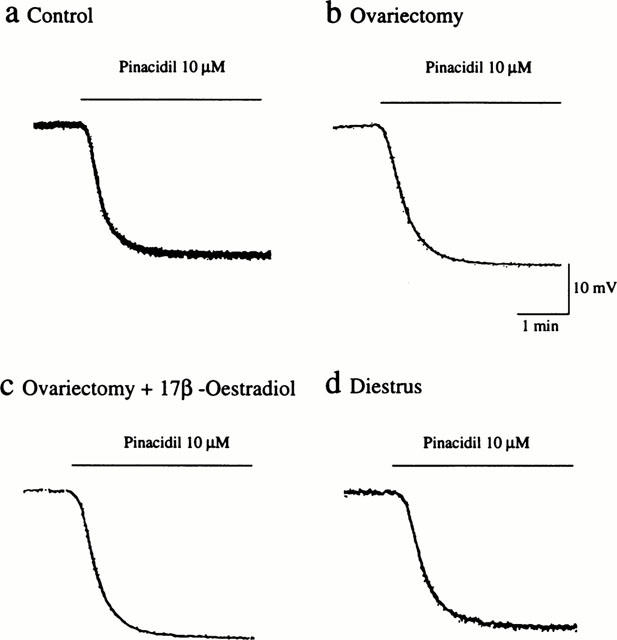
Influences of ovariectomy and oestrus cycle on endothelium-independent hyperpolarization induced by pinacidil in rat mesenteric arteries. Actual recordings of the hyperpolarizing responses to 10 μM pinacidil in mesenteric arteries from sham-operated (a), ovariectomized (b), ovariectomized and 17β-oestradiol-treated (c), and diestrus (d) rats are shown.
Discussion
The major findings of this study are that: (1) the endothelium-dependent relaxant response of female rat mesenteric artery to ACh was markedly depressed in the oestrogen-deficient states of either a long-term (ovariectomy) or a short-term (diestrus cycle); (2) the depressed endothelium-dependent relaxant response to ACh was mainly attributable to a diminished EDHF response; (3) the contractile response of mesenteric artery to Phe was significantly enhanced in oestrogen-deficient rats, but this enhancement was masked in the presence of L-NOARG; and (4) the changes in endothelium-dependent relaxations and contractions detected at the long-term oestrogen-deficient state were almost completely suppressed by oestrogen replacement therapy during the oestrogen-deficient period.
Matured female rats exhibit cyclical alternation in the plasma oestrogen level in 4 or 5 day cycles, which is sensitively reflected by the stage-dependent change in the uterine weights (Nequin et al., 1979). Thus, rats at the diestrus stage provide a good experimental model of short-term oestrogen deficiency. We found that ACh-induced endothelium-dependent relaxation was suppressed in mesenteric arteries from diestrus rats as detected in arteries after ovariectomy, although its extent was less severe in the former. The increased contractile response to Phe in diestrus rats was comparable to that in ovariectomized rats, and the enhancement of the Phe contraction amplitude by L-NOARG treatment was the same between arteries from ovariectomized and diestrus rats. These facts indicate that both vascular relaxation elicited by agonist-stimulated release of endothelium-derived relaxing factors and the basal release of NO from the endothelium depend on the cyclical alteration in the plasma oestrogen level, and provide evidence that alteration in progesterone in association with oestrogen deficiency, which was unavoidable in ovariectomy, does not affect the endothelium function in a direct way. It could be speculated that irreversible morphological derangements in endothelial cells might occur after long-term oestrogen deficiency, and be the underlying mechanism of the altered endothelium functions after ovariectomy. However, the reversible cyclical alterations in the endothelium functions, depending on the plasma level of oestrogen, shown in our study suggests that the endothelium-dependent vascular changes observed in ovariectomized rats are simplified to be functional, and are not caused by constitutional derangements.
Endothelium-dependent relaxation of blood vessels is dependent on the relaxing factors which endothelial cells produce in response to shear stress on the cells, wall stress, blood bearing vasoactive substances, autacoids or cytokines. Among different relaxing factors reported so far (Vanhoutte et al., 1986), EDHF is distinct from other two factors, NO and prostacyclin, since EDHF is not still defined as a substance. Recent studies raise a question on the presence of EDHF as a releasable chemical entity from endothelial cells (Edwards et al., 1998, 1999; Doughty et al., 1999).
Pharmacological analysis using indomethacin, which inhibits the production of prostanoids, revealed that vasorelaxing prostanoids such as prostacyclin play a negligible role in the agonist-induced endothelium-dependent relaxation in rat female mesenteric arteries irrespective of the oestrogen status.
However, the oestrogen status was found to definitely affect the vascular responses to vasoactive agents through its influence on endothelium-derived NO. Mesenteric arteries from control female rats exhibited the less contractile response to Phe and the more marked potentiation of the Phe response in the presence of L-NOARG when compared to arteries from ovariectomized or diestrus rats. This suggests that the basal release of NO from the endothelium, which hampers the contractile action of Phe, is dependent on the oestrogen level. Several observations have suggested that NO synthase may be regulated by sex hormone. Treatment with oestradiol (not progesterone) increased calcium-dependent NO synthase actively in uterine artery and other tissues in female and male guinea-pigs (Weiner et al., 1994). Hayashi et al. (1992) have reported that aortae of female rabbits have a higher basal release of NO than male rabbit aortae, and ovariectomy eliminates the difference. The present findings are not inconsistent with these studies.
Suppression of agonist-induced endothelium-dependent relaxations in arteries from ovariectomized and diestrus rats, which was clearly shown by the rightward shift of the concentration-response curves for ACh-induced relaxation and the reduced maximum response, seemingly appeared to be in line with the reduced basal NO release at the oestrogen-deficiency state. However, further examination to determine a relative contribution to NO and EDHF to the suppression of endothelium-dependent relaxation induced by ACh, revealed that the L-NOARG-sensitive fraction, i.e, endothelium-dependent relaxation mediated by NO, was not being significantly depressed in arteries obtained from rats with either long-term or short-term deficiency of oestrogen. This suggests that the state of oestrogen deficiency preserves relatively well the agonist-induced release of NO from the endothelium in contrast to the diminished basal release of NO. Therefore it is likely that suppression of the relaxant response to ACh obtained in oestrogen-deficiency is mainly attributed to a reduced response to EDHF. In fact, the relaxant response to ACh in the presence of L-NOARG and indomethacin, which could exclude involement of NO and prostanoids from the endothelium in the ACh response, was significantly impaired in arteries from oestrogen-deficient rats. This response to ACh was confirmed to be derived from EDHF by the finding that treatment with combination of apamin and ChTX, which can selectively block the EDHF response (Doughty et al., 1999), inhibited almost completely the remaining response to ACh in the presence of L-NOARG and indomethacin. In support of these findings, measurement of the membrane potentials of smooth muscle cells of mesenteric arteries showed that the oestrogen-deficiency states significantly diminished hyperpolarization induced by ACh in the initial rapid phase and the following sustained phase of hyperpolazation to the same extent.
The hyperpolarizing response to ACh of vascular smooth muscle cells is known to be dependent on the intracellular Ca2+ concentration in endothelial cells. Thus, the initial hyperpolarization is related to Ca2+ released from intracellular stores, and the sustained hyperpolarization is due to Ca2+ influx through Ca2+-permeable cation channels via an undefined mechanism (Chen & Suzuki, 1990; Fukao et al., 1995). The decreased hyperpolarizing response of both the initial and sustained phases suggests the altered signal transduction of the muscarinic receptor mechanism on the Ca2+ release from intracellular stores, resulting in the incomplete fillness of intracellular Ca2+ stores, or the impaired function or reduced expression of the channels.
The findings that EDHF is a dominant contributor in endothelial-dependent vasorelaxation induced by ACh in females rats is in accordance with the study reported by McCulloch & Randall (1998), in which they examined sex difference in the relative contribution of NO and EDHF to agonist-induced endothelium-dependent relaxation in mesenteric arterial bed from female and male rats and found a dominant role of EDHF in female animals. However, whether the dominance of EDHF in endothelium-dependent relaxation in female is generalized to human awaits further study.
The present study indicates clearly that the endothelium-dependent relaxant responses to vasoactive agonists can be regulated by the oestrogen states by dominantly affecting the EDHF response. The fact that even a short-term deficiency of oestrogen for a few days, such as in the diestrus stage, results in suppression of endothelium-dependent relaxation, suggests that reversible and functional alterations in cellular constituents, including receptors, ion channels, or essential molecules of signal transduction, may be involved in the mechanism(s) of the impairment of endothelium-dependent relaxation.
These results also support the notion that the altered lipid metabolism, such as an increase in low-density lipoprotein and decrease in high-density lipoprotein, often observed in long-term deficiency of oestrogen (Bush et al., 1987; Sack et al., 1994) seems unlikely to play a major role in impaired endothelium-dependent vasorelaxation in oestrogen deficiency, but quite possibly is involved as an exacerbating factor, being in keeping with the work of Nascimento et al. (1999). Several studies have demonstrated a direct vasodilating effect of 17β-oestradiol through its blocking action on Ca2+ channels (Adams et al., 1989, Nakajima et al., 1995). These effects seem likely to be non-genomic, even though oestradiol is reported to exert some action by interacting with cell surface receptors (Sudhir et al., 1995). The concentrations of oestrogen used in these studies are supraphysiological, and we found that direct application of 17β-oestradiol did not improve the diminished hyperpolarizing response to ACh in mesenteric arteries from ovariectomized rats (unpublished observations). Thus, the alterations in endothelium-dependent relaxation observed in ovariectomized and diestrus rats shown in this study, may result from the insufficiency of endogenous oestrogen to act as a hormone regulating internal homeostasis.
The chemical identity of EDHF is still not clear at the moment. However, several candidates have been proposed so far to include the epoxyeicosatrienoic acids, a metabolite of cytochrome P-450 monooxygenase, and endocannabinoids, although there are several reports providing evidence against those studies (Félétou & Vanhoutte, 1999). Recently, K+ has been proposed to be an EDHF (Edwards et al., 1998), but remains to be confirmed (Lacy et al., 2000). Furthermore, some reports suggest that EDHF is not a diffusible factor but an endothelium-derived hyperpolarizing current transmitted to smooth muscle through gap junctions between endothelial cells and vascular smooth muscle cells (Kuhberger et al., 1994; Edwards et al., 1999). Studies reporting that the expression of K+ channels in uterine smooth muscle varies during the oestrus cycle and can be stimulated by oestrogen treatment (Boyle et al., 1987; Pragnell et al., 1990) suggest the possibility that K+ channels in the endothelium and/or gap junctions between endothelial cells and vascular smooth muscle cells might be flexibly altered in their expression responding to the oestrogen level.
In conclusion, oestrogen deficiency, either in the short-term during oestrus cycle, or long-term by ovariectomy, altered the endothelium-dependent responses of mesenteric arteries in female rats. The basal release of NO was impaired and the endothelium-dependent relaxant response to ACh was suppressed. Among the endothelium-derived relaxing factors, prostanoids was found to play a minimal role in the altered vascular response to ACh. The EDHF-mediated component of endothelium-dependent relaxation was specifically impaired in the oestrogen-deficient state, while the NO-mediated component was well reserved.
Abbreviations
- ACh
acetylcholine
- ChTX
charybdotoxin
- EDHF
endothelium-derived hyperpolarizing factor
- L-NOARG
NG-nitro-L-arginine
- NO
nitric oxide
- Phe
phenylephrine
- PSS
physiological salt solution
References
- ADAMS D.J., BARAKEH J., LASKEY R., VAN BREEMEN C. Ion channels and regulation of intracellular calium in vascular endothelial cells. FASEB J. 1989;3:2389–2400. doi: 10.1096/fasebj.3.12.2477294. [DOI] [PubMed] [Google Scholar]
- BARRETT-CONNOR E., BUSH T.L. Estrogen and coronary heart disease in women. JAMA. 1991;265:1861–1867. [PubMed] [Google Scholar]
- BELL D.R., RENSBERGER H.J., KORINIK D.R., KOSHY A. Estrogen pretreatment directly potentiates endothelium-dependent vasorelaxation of porcine coronary arteries. Am. J. Physiol. 1995;268:H377–H383. doi: 10.1152/ajpheart.1995.268.1.H377. [DOI] [PubMed] [Google Scholar]
- BOYLE M.B., MACLUSKEY N.F., NAFTOLIN F., KAZCZMAREL L.K. Hormonal regulation of K+ channel messenger RNA in rat myometrium during oestrus cycle and in pregnancy. Nature. 1987;330:373–375. doi: 10.1038/330373a0. [DOI] [PubMed] [Google Scholar]
- BUSH T.L., BARRETT-CONNOR E., COWAN L.D., CRIQUI M.H., WALLACE R.B., SUNCHINDRAN C.M., TYROLER H.A., RIFKIND B.M. Cardiovascular mortality and noncontraceptive estrogen use in women: results from the Lipid Research Clinics Program Follow-up Study. Circulation. 1987;75:1102–1109. doi: 10.1161/01.cir.75.6.1102. [DOI] [PubMed] [Google Scholar]
- CARSWELL H.V.O., DOMINICZAK A.F., MACRAE I.M. Estrogen status affects sensitivity to focal cerebral ischemia in stroke-prone spontaneously hypertensive rats. Am. J. Physiol. 2000;278:H290–H294. doi: 10.1152/ajpheart.2000.278.1.H290. [DOI] [PubMed] [Google Scholar]
- CAULIN-GLASER T., GARCIA-CARDENA G., SARREL P., SESSA W.C., BENDER J.R. 17β-Estradiol regulation of human endothelial cell basal nitric oxide release, independent of cytosolic Ca2+ mobilization. Circ. Res. 1997;81:885–892. doi: 10.1161/01.res.81.5.885. [DOI] [PubMed] [Google Scholar]
- CHEN G., SUZUKI H. Calcium dependency of the endothelium-dependent hyperpolarization in smooth muscle cells of the rabbit carotid artery. J. Physiol. 1990;421:521–534. doi: 10.1113/jphysiol.1990.sp017959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COLDITZ G.A., WILLETT W.C., STAMPFER M.J., ROSNER B., SPEIZER F.E., HENNEKENS C.H. Menopause and the risk of coronary heart disease in women. N. Engl. J. Med. 1987;316:1105–1110. doi: 10.1056/NEJM198704303161801. [DOI] [PubMed] [Google Scholar]
- DALLE LUCCA J.J., ADEAGBO A.S., ALSIP N.L. Influence of oestrous cycle and pregnancy on the reactivity of the rat mesenteric vascular bed. Hum. Reprod. 2000;15:961–968. doi: 10.1093/humrep/15.4.961. [DOI] [PubMed] [Google Scholar]
- DOUGHTY J.M., PLANE F., LANGTON P.D. Charybdotoxin and apamin block EDHF in rat mesenteric artery if selectively applied to the endothelium. Am. J. Physiol. 1999;276:H1107–H1112. doi: 10.1152/ajpheart.1999.276.3.H1107. [DOI] [PubMed] [Google Scholar]
- EDWARDS G., DORA K.A., GARDENER M.J., GARLAND C.J., WESTON A.H. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- EDWARDS G., FELETOU M., GARDENER M.J., THOLLON C., VANHOUTTE P.M., WESTON A.H. Role of gap junctions in the responses to EDHF in rat and guinea-pig small arteries. Br. J. Pharmacol. 1999;128:1788–1794. doi: 10.1038/sj.bjp.0703009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FÉLÉTOU M., VANHOUTTE P.M. Endothelial dysfunction: a novel therapeutic target. The alternative: EDHF. J. Mol. Cell. Cardiol. 1999;31:15–22. doi: 10.1006/jmcc.1998.0840. [DOI] [PubMed] [Google Scholar]
- FUKAO M., HOTTORI Y., KANNO M., SAKUMA I., KITABATAKE A. Thapsigargin- and cyclopiazonic acid-induced endothelium-dependent hyper-polarization in rat mesenteric artery. Br. J. Pharmacol. 1995;115:987–992. doi: 10.1111/j.1476-5381.1995.tb15908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FUKAO M., HOTTORI Y., KANNO M., SAKUMA I., KITABATAKE A. Alterations in endothelium-dependent hyperpolarization and relaxation in mesenteric arteries from streptozotocin-induced diabetic rats. Br. J. Pharmacol. 1997;121:1383–1391. doi: 10.1038/sj.bjp.0701258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GISCLARD V., MILLER V.M., VANHOUTTE P.M. Effect of 17β-estradiol on endothelium-dependent responses in the rabbit. J. Pharmacol. Exp. Ther. 1988;244:19–22. [PubMed] [Google Scholar]
- HAYASHI T., FUKUTO J.M., IGNARRO L.J., CHAUDHURI G. Basal release of nitric oxide from aortic rings is greater in female rabbits than in male rabbits. Proc. Natl. Acad. Sci. U.S.A. 1992;89:11259–11263. doi: 10.1073/pnas.89.23.11259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HISHIKAWA K., NAKAKI T., MARUMO T., SUZUKI H., KATO R., SARUTA T. Up-regulation of nitric oxide synthase by estradiol in human aortic endthelial cells. FEBS Lett. 1995;360:291–293. doi: 10.1016/0014-5793(95)00124-r. [DOI] [PubMed] [Google Scholar]
- HOLM P., KORSGAARD N., SHALMI M., ANDERSEN H.L., HOUGAARD P., SKOUBY S.O., STENDER S. Significant reduction of the antiatherogenic effect of estrogen by long-term inhibition of nitric oxide synthesis in cholesterol-clamped rabbits. J. Clin. Invest. 1997;100:821–828. doi: 10.1172/JCI119597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HULLEY S., GRADY D., BUSH T., FURBERG C., HERRINGTON D., RIGGS B., VITTINGHOFF E. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. JAMA. 1998;280:605–613. doi: 10.1001/jama.280.7.605. [DOI] [PubMed] [Google Scholar]
- KUHBERGER E., GROSCHNER K., KUKOVETZ W.R., BRUNNER F. The role of myoendothelial cell contact in non-nitric oxide-mediated, non-prostanoid-mediated endothelium-dependent relaxation of porcine coronary artery. Br. J. Pharmacol. 1994;113:1289–1294. doi: 10.1111/j.1476-5381.1994.tb17138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LACY P.S., PILKINGTON G., HANVESAKUL R., FISH H.J., BOYLE J.P., THURSTON H. Evidence against potassium as an endothelium-derived hyperpolarizing factor in rat mesenteric small arteries. Br. J. Pharamacol. 2000;129:605–611. doi: 10.1038/sj.bjp.0703076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LERNER D.J., KANNEL W.B. Patterns of coronary heart disease morbidity and mortality in the sexs: a 26-year follow-up of the Framingham population. Am. Heart J. 1986;111:383–390. doi: 10.1016/0002-8703(86)90155-9. [DOI] [PubMed] [Google Scholar]
- McCULLOCH A.I., RANDALL M.D. Sex differences in the relative contributions of nitric oxide and EDHF to agonist-stimulated endothelium-dependent relaxations in the rat isolated mesenteric arterial bed. Br. J. Pharmacol. 1998;123:1700–1706. doi: 10.1038/sj.bjp.0701781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAKAJIMA T., KITAZAWA T., HAMADA E., HAZAMA H., OMATA M., KURACHI Y. 17β-Oestradiol inhibits the voltage-dependent L-type Ca2+ currents in aortic smooth muscle cells. Eur. J. Pharmacol. 1995;294:625–635. doi: 10.1016/0014-2999(95)00602-8. [DOI] [PubMed] [Google Scholar]
- NASCIMENTO C.A.D., KAUSER K., RUBANYI G.M. Effect of 17β-estradiol in hypercholesterolemic rabbits with severe endothelial dysfunction. Am. J. Physiol. 1999;276:H1788–H1794. doi: 10.1152/ajpheart.1999.276.5.H1788. [DOI] [PubMed] [Google Scholar]
- NEKOOEIAN A.A., LIM S.L., MAN R.Y.K., PANG C.C.Y. Chronic 17β-estradiol augments relaxant role of basal nitric oxide in blood vessels from rats with heart failure. Naunyn-Schmiedeberg's Arch. Pharmacol. 1998;358:671–677. doi: 10.1007/pl00005310. [DOI] [PubMed] [Google Scholar]
- NEQUIN L.G., ALVAREZ J., SCHWARTZ N.B. Measurement of serum steroid and gonadotropin levels and uterine and ovarian variables throughout 4 day and 5 day estrous cycles in the rat. Biol. Reproduct. 1979;20:659–670. doi: 10.1095/biolreprod20.3.659. [DOI] [PubMed] [Google Scholar]
- PRAGNELL M., SANAY K.J., TRIMMER J.S., MACLUSKY N.J., NAFTOLIN F., KACZMARELK L.K., BOYLE M.B. Estrogen induction of a small, putative K+ channel mRNA in rat uterus. Neuron. 1990;4:807–812. doi: 10.1016/0896-6273(90)90207-v. [DOI] [PubMed] [Google Scholar]
- SACK M.N., RADER D.J., CANNON R.O. Oestrogen and inhibition of oxidation of low-density lipoproteins in postmenopausal women. Lancet. 1994;343:269–270. doi: 10.1016/s0140-6736(94)91117-7. [DOI] [PubMed] [Google Scholar]
- SUDHIR K., CHOU T.M., MULLEN W.L., HASUSMANN D., COLLINS P., YOCK P.G., CHATTERJEE K. Mechanisms of estrogen-induced vasodilation: in vivo studies in canine coronary conductance and resistance arteries. J. Am. Coll. Cardiol. 1995;26:807–814. doi: 10.1016/0735-1097(95)00248-3. [DOI] [PubMed] [Google Scholar]
- VANHOUTTE P.M., RUBANYI G.M., MILLER V.M., HOUSTON D.S. Modulation of vascular smooth muscle contraction by the endothelium. Annv. Rev. Physiol. 1986;48:307–320. doi: 10.1146/annurev.ph.48.030186.001515. [DOI] [PubMed] [Google Scholar]
- WEINER C.P., LIZASOAIN L., BAYLIS S.A., KNOWLES R.G., CHARLES I.G., MONCADA S. Induction of calcium-dependent nitric oxide synthase by sex hormones. Proc. Natl. Acad. Sci. U.S.A. 1994;91:5212–5216. doi: 10.1073/pnas.91.11.5212. [DOI] [PMC free article] [PubMed] [Google Scholar]