

Abstract
In this study, we tested the effects of the stilbene disulphonates DIDS and SITS on three different types of cloned KATP channel (Kir6.2/SUR1, Kir6.2/SUR2A and Kir6.2ΔC) heterologously expressed in Xenopus oocytes, with the aim of identifying the part of the channel which is involved in mediating disulphonate inhibition.
We found that the inhibitory site(s) for these drugs lies within the Kir6.2 subunit of the channel, although its properties are further modulated by the sulphonylurea (SUR) subunit. In particular, SUR2A reduces both the rate and extent of block, by impairing the ability of DIDS binding to produce channel closure.
The disulphonate-binding site interacts with the ATP inhibitory site on Kir6.2 because ATP is able to protect against irreversible channel inhibition by disulphonates. This effect is not mimicked by tolbutamide (at a concentration that interacts with Kir6.2) and is abolished by mutations that render the channel ATP insensitive.
A number of point mutations in both the N and C termini of Kir6.2 reduced the extent and reversibility of channel inhibition by SITS. The results are consistent with the idea that residue C42 of Kir6.2 is likely to be involved in covalently linking of SITS to the channel.
Other types of Kir channel (Kir1.1, Kir2.1 and Kir4.1) were also irreversibly blocked by DIDS, suggesting that these channels may share common binding sites for these stilbene disulphonates.
Keywords: ATP-sensitive K+ channel, stilbene disulphonates, potassium channel, inwardly rectifying potassium channel, ATP-binding site
Introduction
In a variety of tissues, ATP-sensitive K-channels (KATP channels) serve to couple electrical activity to the metabolic state of the cell (Ashcroft & Gribble, 1998; 1999; Aguilar-Bryan & Bryan, 1999). In addition to metabolism, KATP channels are regulated by sulphonylurea drugs, which inhibit channel activity and are used to enhance insulin secretion in type-2 diabetes mellitus, and by KATP channel openers which stimulate channel activity.
The KATP channel is an octameric (4 : 4) complex of two structurally distinct proteins: Kir6.2, and SUR (Clement et al., 1997; Inagaki et al., 1997; Shyng & Nichols, 1997). Kir6.2 acts as a common ATP-sensitive pore while SUR is a regulatory subunit that endows Kir6.2 with sensitivity to metabolism, sulphonylureas and KATP-channel openers (Nichols et al., 1996; Inagaki et al., 1996; Shyng et al., 1997; Tucker et al., 1997). KATP channels in pancreatic β-cells comprise Kir6.2 and SUR1 subunits (Inagaki et al., 1995; Sakura et al., 1995), while those of cardiac and skeletal muscle are composed of Kir6.2 and SUR2A (Inagaki et al., 1996). Although wild-type KATP channels require both Kir6.2 and SUR subunits for functional activity, a mutant form of Kir6.2 with a C-terminal truncation of 26 or 36 amino acids (Kir6.2ΔC) is capable of independent expression (Tucker et al., 1997).
Stilbene disulphonates are widely used as anion transport inhibitors and anion channel blockers. These compounds share structural similarities with sulphonylureas in that both types of drug have benzene rings and sulphonic acid residues. The latter observation led Furukawa et al. (1993) to test the effect of stilbene disulphonates on KATP channels in ventricular myocytes. They found that these drugs inhibited KATP channels when applied to the intracellular side of the membrane with an order of potency DIDS (IC50 71 μM)> SITS∼DNDS>DADS. The actions of DIDS and SITS were irreversible while those of DADS and DNDS were reversible. The Hill coefficient was around unity suggesting that despite the tetrameric nature of the KATP channel the interaction of a single stilbene disulphonate molecule was sufficient to cause channel inhibition. The compounds inhibited KATP channel activity without an obvious effect on the single-channel current amplitude. Analysis of the single-channel kinetics revealed that DIDS reduced the duration of the burst of openings characteristic of KATP channels and increased the long interburst closed time, consistent with the idea that the compound can interact with both the open state and the long closed state of the channel. The block was voltage independent and did not require divalent cations.
Stilbene disulphonates possessing SCN groups, like DIDS and SITS, are known to react covalently with lysine or cysteine residues (Gatto et al., 1997). The ability of DIDS to bind covalently to these residues is much greater than that of SITS, while DNDS and DADS do not have an SCN group that is able to bind covalently. The properties of stilbene disulphonate block of KATP channels reported by Furukawa et al. (1993) are consistent with the idea that irreversible block of the channel by DIDS (or SITS) is produced by a covalent reaction between the SCN group on the drug and the KATP channel. Inhibition of the KATP channel by stilbene disulphonates is thus likely to involve at least two steps: a reversible binding of the drug to the channel, followed by a covalent reaction that irreversibly attaches the stilbene disulphonate.
In this study, we have tested the effects of the stilbene disulphonates DIDS and SITS on different types of cloned KATP channels heterologously expressed in Xenopus oocytes, with the aim of identifying which part of the channel is involved in mediating disulphonate inhibition. We examined whether the stilbene disulphonate-binding site is located on the Kir6.2 or on the SUR subunit. We explored the relationship of the stilbene disulphonate-binding site to the sulphonylurea- and ATP-binding sites. And we investigated the effect of a number of point lysine and cysteine mutations in the N and C termini of the channel on the properties of stilbene disulphonate block.
Methods
Molecular biology
Mouse Kir6.2 (Genbank D50581; Inagaki et al., 1995; Sakura et al., 1995), rat SUR1 (Genbank L40624; Aguilar-Bryan et al., 1995) and SUR2A (Genbank D83598; Inagaki et al., 1996) cDNAs were cloned in the pBF vector. A truncated form of Kir6.2 (Kir6.2ΔC), which lacks the C-terminal 36 amino acids and forms functional channels in the absence of SUR, was prepared as described previously (Tucker et al., 1997). Site-directed mutations in Kir6.2ΔC were made using the ‘Quikchange' site-directed mutagenesis method (Stratagene, La Jolla, CA, U.S.A.). Capped mRNA was prepared using the mMESSAGE mMACHINE large scale in vitro transcription kit (Ambion, Austin, TX, U.S.A.), as previously described (Gribble et al., 1997a).
Oocyte collection
Female Xenopus laevis were anaesthetized with MS222 (2 g l−1 added to the water). One ovary was removed via a mini-laparotomy, the incision sutured and the animal allowed to recover. Immature stage V-VI oocytes were incubated for 60 min with 1.0 mg mg−1 collagenase (Sigma, type V) and manually defolliculated. Oocytes were either injected with ∼1 ng Kir6.2ΔC mRNA (wild-type or mutant) or coinjected with ∼0.1 ng Kir6.2 or Kir6.2ΔC mRNA (wild-type or mutant) and ∼2 ng of mRNA encoding either SUR1 or SUR2A. The final injection volume was 50 nl per oocyte. Isolated oocytes were maintained in Barth's solution and studied 1–4 days after injection (Gribble et al., 1997a).
Electrophysiology
Patch pipettes were pulled from borosilicate glass and had resistances of 250–500 kΩ when filled with pipette solution. Macroscopic currents were recorded from giant excised inside-out patches at a holding potential of −60 mV using an Axopatch 200B patch-clamp amplifier (Axon Instruments, Burlingame, U.S.A.). They were filtered at 0.2 kHz, digitized at 0.4 kHz using a Digidata 1200 Interface and analysed using pClamp software (Axon Instruments). Single-channel currents were recorded at −60 mV, filtered at 5 kHz and sampled at 20 Hz.
The pipette (external) solution contained (mM): 140 KCl, 1.2 MgCl2, 2.6 CaCl2, 10 HEPES (pH 7.4 with KOH). The intracellular (bath) solution contained (mM): 135 KCl, 1 K2SO4, 1 EGTA, 10 HEPES (pH 7.2 with KOH; final [K+]∼140 mM). A Mg2+-free solution was used to minimize rundown (Kozlowski & Ashford, 1990): it has been shown previously that this does not affect DIDS inhibition (Furukawa et al., 1993). DIDS and SITS (Sigma) were dissolved directly in the solution prior to the experiment and used the same day. Experiments were carried out at 20–24°C. Rapid exchange of solutions was achieved by positioning the patch in the mouth of one of a series of adjacent inflow pipes placed in the bath. Patches were exposed to 3 mM ATP at intervals throughout the experiment, to monitor the zero current level and the patch accessibility.
Data analysis
Data are presented as mean±standard error of the mean. The time course of DIDS (or SITS) block was fit with one or more exponentials, using Microcal Origin. To evaluate the extent of stilbene disulphonate block, or the degree of recovery from block, we measured the steady-state current during, or following, DIDS application. In most cases, steady-state block was achieved within 1 min. Where currents showed substantial rundown (e.g. Figure 6C), the peak value was used (both in control and test solution).
Figure 6.
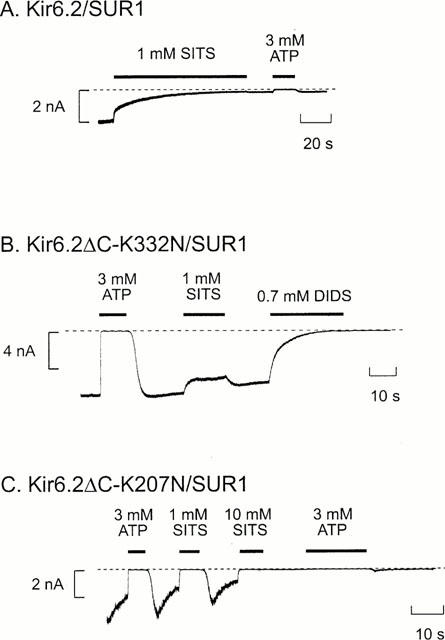
Effects of mutations on block by stilbene disulphonates. Macroscopic currents recorded at −60 mV in inside-out patches from oocytes coexpressing SUR1 and wild-type (A) or mutant (B,C) Kir6.2. ATP (3 mM), SITS (1 or 10 mM) or DIDS (0.7 mM) were added as indicated by the bars. The dashed line indicates the zero current level.
Results
Macroscopic currents were recorded in inside-out membrane patches from Xenopus oocytes expressing Kir6.2ΔC or coexpressing Kir6.2 and either SUR1 or SUR2A. In all cases, the currents were small in the cell-attached configuration but increased markedly when the patch was excised into nucleotide-free solution, consistent with the idea that the KATP channel is blocked in the intact oocyte by cytoplasmic nucleotides such as ATP.
Figure 1 shows that application of DIDS (0.7 mM) to the intracellular membrane surface irreversibly inhibited all three types of KATP channel. Kir6.2ΔC channels were blocked by 99±2% (n=6), indicating that DIDS interacts directly with the Kir6.2 subunit. Both Kir6.2/SUR1 and Kir6.2/SUR2A currents were also blocked by DIDS. The mean extent of steady-state block of Kir6.2/SUR1 currents was 99.6±0.3% (n=5) and that of Kir6.2/SUR2A currents was 77±5% (n=8). Thus the presence of SUR2A, but not SUR1, somehow decreases the extent of block. As in the case of DIDS, SITS (1 mM) completely and irreversibly blocked both Kir6.2ΔC and Kir6.2/SUR1 currents (by 98.8±0.1% (n=5) and 98.9±0.1% (n=6), respectively; (see Figures 4 and 6A).
Figure 1.
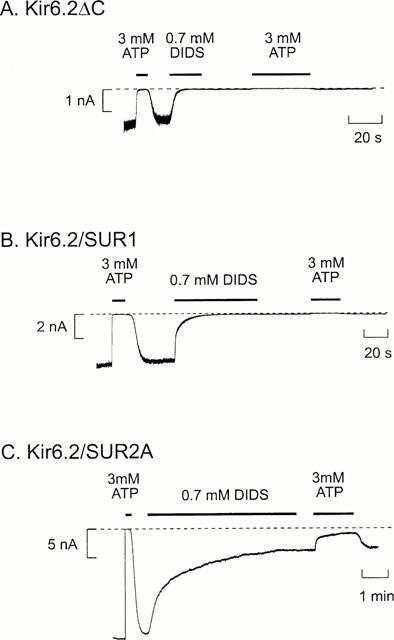
Inhibition of KATP currents by DIDS. Macroscopic currents recorded from inside-out patches at a holding potential of −60 mV from oocytes expressing Kir6.2ΔC (A) or coexpressing Kir6.2 and either SUR1 (B) or SUR2A (C). DIDS (0.7 mM) or ATP (3 mM) were added as indicated by the bars. The dashed line indicates the zero current level. Note the different time scale in C.
Figure 4.
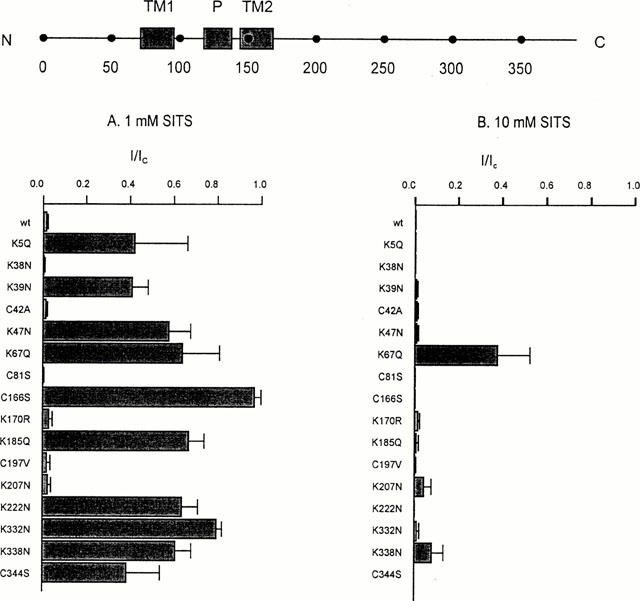
Effects of Kir6.2 mutations on the amplitude of SITS block. (Top) Schematic model of Kir6.2, with the positions of the C terminus (C), N terminus (N), transmembrane domains (TM1, TM2), pore loop (P) and various residues indicated. (Bottom) Mean KATP current (I), remaining in the presence of 1 mM SITS (left) and 10 mM SITS (right), expressed as a fraction of that in the absence of the compound (IC). Oocytes were coinjected with SUR1 and mutant Kir6.2, as indicated. The number of patches ranged between 3–6 for each mutant.
The time course of the onset of DIDS block of Kir6.2ΔC or Kir6.2/SUR1 currents could be reasonably well fitted by a single exponential with a time constant (τ) of 4.4±1.5 s (n=6) for Kir6.2ΔC and of 6.3±1.1 s (n=5) for Kir6.2/SUR1. These values are not significantly different. In the case of Kir6.2/SUR2A currents, the onset of inhibition did not always follow a single exponential (n=3 out of eight patches). We therefore fitted the time course of Kir6.2/SUR2A block in all patches with the sum of two exponentials: these had mean time constants of 12.9±3.1 s (τ1) and of 94.8±14.4 s (τ2) and mean amplitudes of 50±6% each (n=8). No recovery of Kir6.2/SUR2A currents was observed when DIDS was removed during the initial phase of block, at a time when inhibition was dominated by the fast exponential, indicating that both the fast and slow block represent irreversible reactions. These results indicate that whereas the presence of SUR1 does not alter the rate of DIDS block, SUR2A markedly slows the rate of inhibition.
When the DIDS concentration was reduced 10 fold (to 70 μM) both Kir6.2ΔC and Kir6.2/SUR1 currents were still fully blocked – by 98.3±1.5% (n=5) and 97.8±1.7% (n=5), respectively – consistent with the idea that DIDS interacts covalently with the KATP channel. However, the rate of block was slowed, the time constant being 77±12 s (n=5; P<0.001) for Kir6.2ΔC and 33.1±5.9 s (n=5; P<0.001) for Kir6.2/SUR1. A 10 fold reduction in DIDS concentration slightly decreased the extent of block of Kir6.2/SUR2A channels: from 77±5% (n=8) to 67±13% (n=5, n.s.). The fast time constant was also slowed (τ1=24±5 s, n=5; P<0.001 against 700 μM DIDS) but the slow time constant was unaffected (τ2=75±32 s, n=5).
As described above, DIDS did not completely block Kir6.2/SUR2A currents. The residual (unblocked) current showed a reduced sensitivity to ATP (Figure 1C), the inhibitory effect of 3 mM ATP being reduced from 99.4±0.5% (n=8) in the absence of DIDS to 30±24% (n=5) in its presence. It is clear from the large standard error that the extent of ATP inhibition after DIDS application was quite variable: indeed in two experiments, 3 mM ATP activated instead of inhibiting the channel. ATP has multiple effects on the KATP channel (Aguilar-Bryan & Bryan, 1999; Ashcroft & Gribble, 1999). It blocks the channel by interaction with a site on Kir6.2. It also activates the channel, by several mechanisms. These include interaction with the nucleotide-binding domains of SUR; and generation of phospholipids such as PIP2 that stimulate channel activity and reduce rundown. KATP channel activity in the presence of MgATP is determined by the balance between these stimulatory and inhibitory effects. Channel activation by ATP following DIDS application might therefore be due to a reduced ATP inhibition, or to enhanced stimulatory effects.
The onset of ATP block was also slower in the presence of DIDS (Figure 1C), the time constant increasing from 0.23±0.03 to 13.3±3.8 s (n=6; P<0.001). This suggests there may be an interaction between the ATP- and DIDS-binding sites.
Interactions of the DIDS- and ATP-binding sites
To investigate this possibility further, we examined whether the presence of ATP was able to protect against DIDS block. We first applied 3 mM ATP and then added 0.7 mM DIDS; both agents were subsequently removed at the same time. We found that ATP prevented the irreversible block of both Kir6.2ΔC and Kir6.2/SUR1 currents by DIDS (Figure 2A,B). The percentage recovery following removal of ATP and DIDS was 26±12% (n=4) and 24±6% (n=4) for Kir6.2ΔC and Kir6.2/SUR1, respectively, compared with no recovery at all following DIDS removal when ATP was absent. The ability of ATP to protect against irreversible inhibition by DIDS suggests that ATP either physically impedes the access of DIDS to its binding site, electrostatically impedes its access, or allosterically induces a conformational change in Kir6.2 that renders the DIDS-binding site less accessible.
Figure 2.
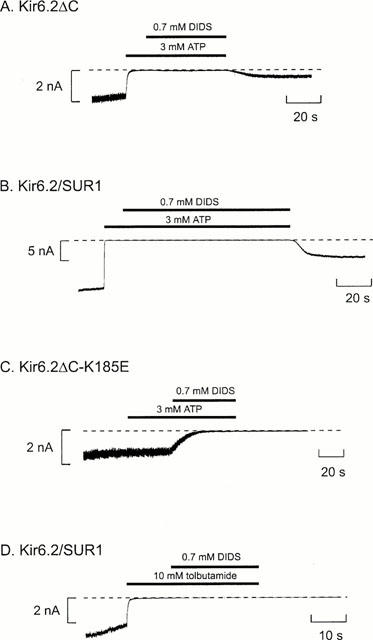
ATP protects against irreversible DIDS block. Macroscopic currents recorded at −60 mV in inside-out patches from oocytes expressing Kir6.2ΔC (A), or coexpressing SUR 1 and either wild-type (B,D) or mutant (C) Kir6.2. ATP (3 mM), DIDS (0.7 mM) or tolbutamide (10 mM) were added as indicated by the bars. The dashed line indicates the zero current level.
To confirm that the site at which ATP protects against DIDS inhibition is the same as that at which ATP mediates channel inhibition, we examined a mutant form of Kir6.2 (Kir6.2ΔC-K185E) that shows reduced ATP sensitivity (Reimann et al., 1999) and decreased ATP binding (Tanabe et al., 1999). In contrast to the wild-type channel, ATP did not protect against DIDS block of Kir6.2ΔC-K185E currents (Figure 2C). This adds further support to the idea that ATP interaction with the ATP-inhibitory site on Kir6.2 protects against DIDS block. The time constant of DIDS block was significantly (P<0.001) slowed: from 4.4±1.5 s (n=6) for the wild-type Kir6.2ΔC to 18.3±3.2 s (n=5) for the Kir6.2ΔC-K185E mutant. This result suggests that mutations affecting ATP sensitivity may also alter the properties of DIDS block. This is consistent with the idea that the two binding sites lie in close physical proximity.
In contrast to ATP, application of the inhibitory sulphonylurea tolbutamide (10 mM), did not prevent the irreversible block of Kir6.2/SUR1 currents by DIDS (percentage recovery, 0.4±0.3%; n=5), as shown in Figure 2D. The high tolbutamide concentration used in this experiment (10 mM) would be sufficient to interact with the low-affinity sulphonylurea site on Kir6.2 (Gribble et al., 1997b). Taken together with the effect of ATP on DIDS inhibition, therefore, this result suggests that the sulphonylurea- and disulphonate-binding sites on Kir6.2 are independent while the ATP- and disulphonate-binding sites can interact (either directly or indirectly). It also provides additional evidence that the ATP-binding site and the low-affinity sulphonylurea site on Kir6.2 are not identical. It further suggests that either the closed states produced by ATP and tolbutamide are different (so that the DIDS site is accessible when tolbutamide closes the channel but not when ATP does); or that bound ATP physically impedes access to the DIDS-binding site while tolbutamide does not.
Effects of point mutations in Kir6.2
We next mutated, one at a time, each of the lysine and cysteine residues in the N and C termini of Kir6.2 and analysed the effect of these mutations on the ability of DIDS and SITS to inhibit the channel. Because some mutations did not express sufficient current when mutant Kir6.2ΔC was expressed alone, most mutations were made in full length Kir6.2 which was then coexpressed with SUR1. This strategy ensured that only fully coupled octameric channels reached the surface membrane (Zerangue et al., 1999). In all but two cases (C166S, K67Q, see below), the properties of the DIDS inhibition were unaffected (Figure 3). In contrast, most point mutations reduced the extent of block by 1 mM SITS and/or enhanced its reversibility (Figures 4 and 5). For example, mutation of lysine 332 to asparagine (K332N) resulted in a much smaller block that was now almost fully reversible (Figures 4 and 6B), while the mutation K207N markedly increased the reversibility of block without affecting its magnitude (Figures 4 and 6C). Some mutations also affected the extent and/or reversibility of block by 10 mM SITS: those that rendered the block reversible were concentrated in the N terminus of the channel (Figure 5).
Figure 3.
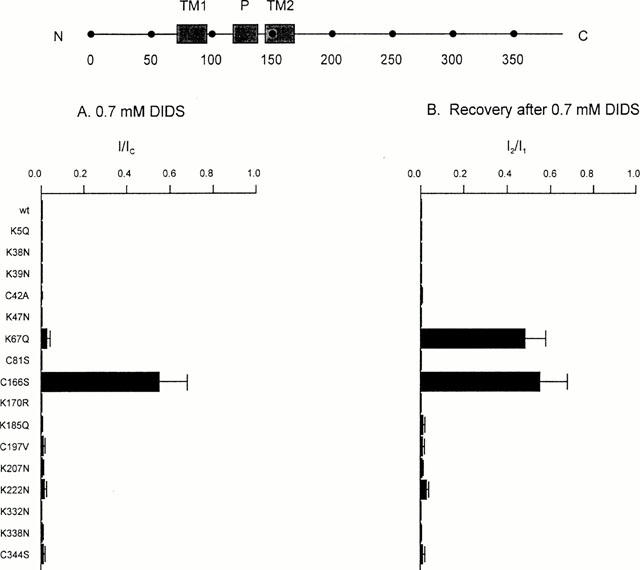
Effects of Kir6.2 mutations on DIDS block. (Top) Schematic model of Kir6.2, with the positions of the C terminus (C), N terminus (N), transmembrane domains (TM1, TM2), pore loop (P) and various residues indicated. (A) Mean KATP current remaining in the presence of 0.7 mM DIDS (I), expressed as a fraction of that in the absence of the compound (IC). Oocytes were coinjected with SUR1 and mutant Kir6.2, as indicated. The number of patches ranged between 3–6 for each mutant. (B) Mean recovery of the KATP current after removal of 0.7 mM DIDS. Steady-state currents recorded following removal of DIDS (I2) are expressed as a fraction of that before its application (I1). Oocytes were coinjected with SUR1 and mutant Kir6.2, as indicated. The number of patches ranged between 3–6 for each mutant. Currents were not corrected for rundown.
Figure 5.
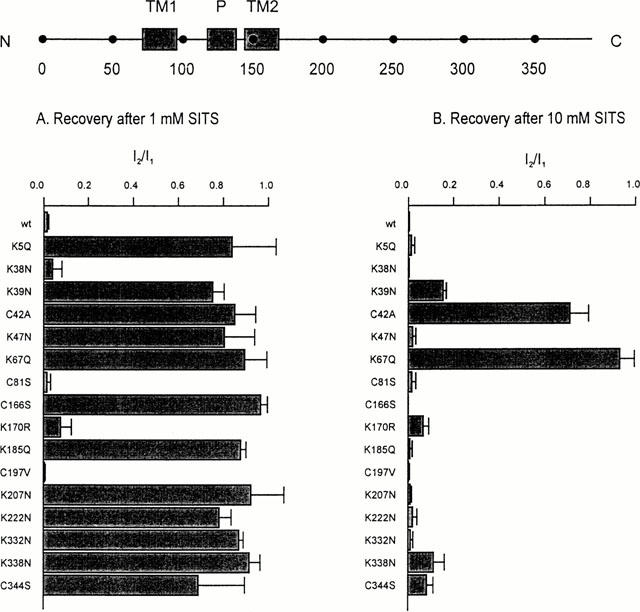
Effects of Kir6.2 mutations on the reversibility of SITS. (Top) Schematic model of Kir6.2, with the positions of the C terminus (C), N terminus (N), transmembrane domains (TM1, TM2), pore loop (P) and various residues indicated. (Bottom) Mean extent of recovery of the KATP current after inhibition by 1 mM SITS (left) or 10 mM SITS (right). Steady-state current recorded following removal of SITS (I2) is expressed as a fraction of that before its application (I1). Oocytes were coinjected with SUR1 and mutant Kir6.2, as indicated. The number of patches ranged between 3–6 for each mutant. Currents were not corrected for rundown.
Mutation of cysteine 166 to serine in Kir6.2 markedly enhances the channel open probability (Trapp et al., 1998a). This mutation also reduced the rate of DIDS block of both Kir6.2ΔC and Kir6.2/SUR1 currents (Figure 7A,B). The time course of the block of Kir6.2ΔC-C166S currents by 0.7 mM DIDS was fit by a single exponential with a time constant of 33.1±9.1 s (n=5), about eight times slower than that found for wild-type Kir6.2ΔC (∼4 s). As expected for a covalent reaction, however, the magnitude of the DIDS block was unaffected, being 97.8±1.4% (n=5) for Kir6.2ΔC-C166S currents compared with 99% for the wild-type channel. SITS had no obvious effect on Kir6.2ΔC-C166S currents at a concentration of 1 mM but at 10 mM it completely blocked the current (99.3±0.7% block, n=4).
Figure 7.
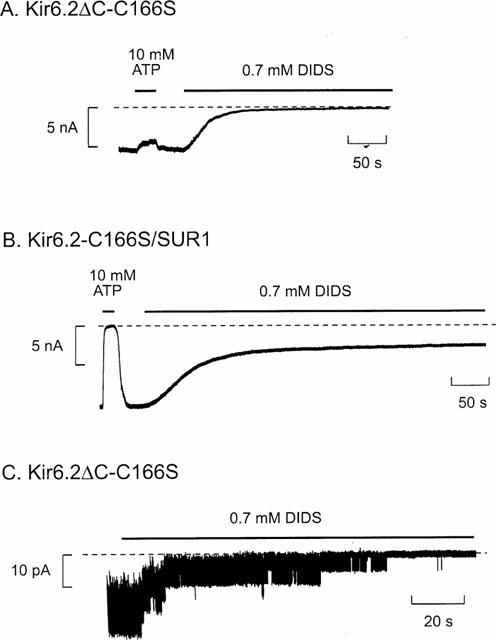
Effects of DIDS on Kir6.2ΔC-C166S and Kir6.2-C166S/SUR1 currents. Macroscopic currents (A, B) and single-channel currents (C) at −60 mV in inside-out patches from oocytes expressing Kir6.2ΔC-C166S or Kir6.2-C166S/SUR1. ATP (10 mM) or DIDS (0.7 mM) were added as indicated by the bars. The dashed line indicates the zero current level.
When Kir6.2ΔC-C166S was coexpressed with SUR1, 0.7 mM DIDS only inhibited the mutant channel by 45±13% (n=4), and a distinct component of unblocked current was observed (Figure 7B). The rate of block was best fitted by the sum of two exponentials with time constants τ1=9.8±1.0 s and τ2=53.6±7.9 s, and amplitudes A1=0.45±0.21 and A2=0.55±0.21, respectively (n=4). Kir6.2ΔC-C166S channels were fully blocked by DIDS, so that the failure of DIDS to block Kir6.2-C166S/SUR1 currents must be related to the presence of the sulphonylurea receptor. In contrast, the extent of block by 10 mM SITS was similar to that observed in the absence of SUR1, but the onset of block was slowed: τ increased from 14±3 s (n=4) to 43±15 s (n=4) when Kir6.2ΔC-C166S was coexpressed with SUR1.
At the single-channel level, Kir6.2ΔC-C166S channels are characterized by very long bursts of openings and rare long-lasting interburst closings which account for <0.5% of the total closed time (Trapp et al., 1998a). DIDS (0.7 mM) caused the channel to enter the long closed state, but several re-openings were observed before it was finally irreversibly inhibited (Figure 7C). Similar results were observed for native cardiac channels (Furukawa et al., 1993). This indicates that for a short time, at least, the channel is able to re-open after binding DIDS.
Mutation of lysine 67 to glutamine (K67Q) markedly reduced the extent of SITS block and rendered inhibition rapidly reversible (Figure 8B). A substantial recovery of current was also observed when DIDS was removed (Figure 8C), although the rate of recovery was slow. Mutation of K67 to methionine or asparagine was without effect on DIDS block of Kir6.2/SUR1 channels. As DIDS is not expected to bind covalently to these amino acids, the effects of the K67Q mutation must be mediated allosterically. The K67Q mutation did not significantly affect the ATP-sensitivity of Kir6.2/SUR1 currents (data not shown), suggesting that the ATP- and DIDS-binding sites are not identical. When Kir6.2-K67Q was coexpressed with SUR2A (rather than SUR1), the DIDS block was fast and was usually fully reversible (data not shown). This suggests that DIDS does not interact covalently with Kir6.2-K67Q/SUR2A and is consistent with studies on wild-type channels showing that SUR2A compromises DIDS block.
Figure 8.
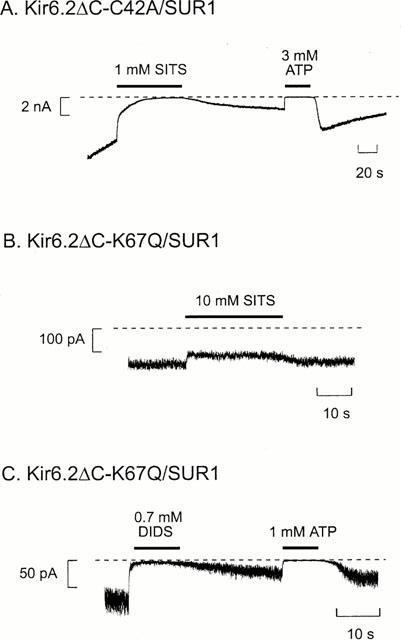
Effects of stilbene disulphonates on Kir6.2ΔC-C42A/SUR1 and Kir6.2ΔC-K67Q/SUR1 currents. Macroscopic Kir6.2ΔC-C42A/SUR1 (A) or Kir6.2ΔC-K67Q/SUR1 (B, C) current recorded at −60 mV in an inside-out patch. ATP, SITS or DIDS were added as indicated by the bars. The dashed line indicates the zero current level.
Although the extent of inhibition of Kir6.2ΔC-42A/SUR1 currents by SITS was similar to that of wild-type channels, the block was no longer irreversible (Figure 8A). This suggests that C42 may participate in a covalent reaction with SITS and is consistent with the fact that mutation of this N-terminal residue abolishes sensitivity to thiol reagents (Trapp et al., 1998b). We also observed that after removal of SITS and partial recovery of the current, subsequent application of 3 mM ATP resulted in a further increase in current when ATP was removed (e.g. Figure 8A). Because these experiments were carried out in Mg-free solution, this increase cannot be due to channel ‘reactivation', as this phenomenon requires Mg2+ (Ohno-Shosaku et al., 1987). It is possible that ATP may accelerate the unbinding of SITS from the mutant channel, consistent with the idea that the SITS- and ATP-binding sites interact. For example, ATP may induce an allosteric conformational change in Kir6.2 that influences the structure of the SITS-binding site, thereby displacing the stilbene disulphonate.
Effects of DIDS on other inward rectifiers
Finally, we tested the effect of intracellular DIDS (0.7 mM) on the related inward rectifiers Kir1.1, Kir2.1 and Kir4.1. All were fully and irreversibly blocked (data not shown). The fact that DIDS blocks several types of inward rectifiers suggests that they share a common binding site(s) for stilbene derivatives. It is possible that the cysteine residue equivalent to C42 in Kir6.2, which is shared by all Kir channels, may contribute to this binding site.
Discussion
Site of action
Our results suggest that despite their structural similarity to sulphonylureas, stilbene disulphonates do not block KATP channels by interaction with the sulphonylurea receptor subunit. Rather they interact with the Kir6.2 subunit of the channel. It is also theoretically possible that they bind to some other protein that is endogenously expressed in oocytes and that modulates the activity of Kir6.2, but this seems less likely because mutations in Kir6.2 alter both the rate and extent of block by stilbene disulphonates.
Our data suggest that stilbene disulphonates such as DIDS or SITS are common inhibitors of the inward rectifier channel family. In addition, they block other types of ion channel, such as chloride channels. This promiscuity means they are not suitable as pharmacological blockers for evaluation of the physiological roles of ion channels. However, their ability to form covalent bonds with lysine and cysteine residues can be exploited for structure-function studies.
Regulation by SUR
It seems probable that the time course of DIDS block of the KATP current represents the rate of the covalent reaction of the stilbene disulphonate with the channel, because there was no recovery when DIDS was removed and because ATP was unable to reactivate the channel if it was applied after DIDS. Both of these effects would be expected if DIDS bound reversibly to the channel. The slower rate of DIDS block observed for Kir6.2/SUR2A currents suggests that the covalent reaction occurs much more slowly when SUR2A is present. This might indicate that SUR2A (but not SUR1) reduces access of DIDS to its binding site, or accelerates the off-rate of DIDS from its binding site so that the covalent reaction occurs with lower frequency. The fact that two exponentials are required to fit the time course of block of Kir6.2/SUR2A currents further suggests that more than one covalent reaction takes place. It is not clear whether this is also the case for Kir6.2ΔC/SUR1, as the data could be fitted with a single exponential time course.
There are at least two explanations for the pedestal observed for Kir6.2/SUR2A currents. First, DIDS may not bind to all channels. Secondly, DIDS may bind to all channels but not block them completely; a more elaborate version of this idea would be that the channels remain open when DIDS binds but are closed when the covalent reaction takes place, and that not all channels undergo the covalent reaction. Our results favour the idea that DIDS is covalently bound to all channels because both the rate and extent of ATP inhibition is impaired, even after DIDS is removed (Figure 1). This implies that DIDS remains linked to the channel even after washing with control solution. We therefore conclude that even though DIDS is covalently linked to Kir6.2/SUR2A the channel does not completely close. Because no residual pedestal of current is observed for Kir6.2ΔC, this implies that SUR2A must impair the transduction of DIDS binding into closure of the Kir6.2 pore.
Although SUR1 did not modify DIDS block of wild-type Kir6.2, it markedly affected the DIDS block of the mutant channel Kir6.2-C166S, as discussed below. It is perhaps worth noting that this is not the only example of such modulation – the ATP block of Kir6.2 is also influenced by SUR1, although in this case inhibition is enhanced (Tucker et al., 1997).
Effects of gating
There is evidence that the DIDS binding site is accessible when the channel is closed, because tolbutamide does not protect against DIDS inhibition. There is also evidence that DIDS can bind to the open state since it reduces the burst duration in single-channel recordings of both wild-type (Furukawa et al., 1993) and mutant channels (Figure 7). Thus DIDS binding appears to be independent of whether the channel is open or closed.
Kir6.2ΔC-C166S currents are characterized by prolonged bursts of opening with very few long closed states (Trapp et al., 1998a). The fact that the rate of DIDS block was slower for Kir6.2ΔC-C166S currents is consistent with the idea that DIDS block is influenced by the rate of transition between the open and long closed states, as is the case for ATP (Trapp et al., 1998a). However, the fact that Kir6.2ΔC-C166S/SUR1 currents also show a decreased steady-state block cannot be explained merely by effects on gating. It appears that, as is the case of Kir6.2/SUR2A, the transduction between the DIDS-binding site and the channel gate is impaired, enabling the channel to reopen with DIDS bound to it.
Effects of Kir6.2 mutations
The block by DIDS and SITS is voltage-independent, and is therefore unlikely to lie within the pore of Kir6.2. Furthermore, mutation of C81, which lies within the first transmembrane domain of the channel, had no apparent effect on disulphonate inhibition. Both SITS and DIDS are membrane impermeant and are effective when added to the intracellular solution. Thus, they must react with the cytosolic domains of the channel.
Because covalent reaction of stilbene disulphonates is thought to involve lysine or cysteine residues, we mutated every lysine and cysteine lying within the intracellular N and C terminus of Kir6.2. Although only two mutations affected DIDS block, a large number of mutations had effects on SITS block (especially at mM concentration). There are a number of possible explanations for this finding. First, multiple residues may contribute to the stilbene disulphonate-binding site. Secondly, some mutations may influence the single-channel kinetics and affect stilbene disulphonate block by an indirect mechanism (as in the case for C166). Thirdly, since stilbene disulphonates are negatively charged, neutralization of lysine residues might influence electrostatic interactions between these compounds and the channel. This may explain why some mutations slowed the time-course of channel inhibition (e.g. mutation K185E, Figure 2C). Fourthly, SITS may interact directly with only a few residues, but these may be located in a different part of the protein and brought into proximity by folding of the N and C terminal cytosolic domains. Consequently, mutations in many residues might perturb protein folding and thereby allosterically decrease binding.
The fact that no single point mutation abolished the irreversible block by DIDS argues that more than one cysteine or lysine residue forms a covalent link with this compound. This is also supported by the fact that the block of Kir6.2/SUR2A currents follows a double exponential time course. Because many mutations disrupt SITS block, we suggest that the N and C terminus may interact to produce a highly defined structure and some mutations may influence SITS binding allosterically. The fact that many mutations affect SITS but not DIDS block may be explained by the much lower ability of SITS to form covalent bonds (Cabantchik & Rothstein, 1972). Previous studies with SITS in a variety of biological systems have shown that this compound must first bind tightly to its binding site before a second, slower, covalent reaction can occur. Compared to DIDS, the covalent reaction is slower, less effective and can frequently be reversed by prolonged washing. Point mutations in the N and C terminus that disrupt the binding site for SITS will therefore also prevent the covalent reaction from occurring.
A number of mutations affected the reversibility of SITS block. The most interesting of these is C42A which did not alter the extent of block, yet rendered the block almost fully reversible. This suggests that C42 may be the residue with which SITS forms a covalent reaction, an idea that is in agreement with previous studies which have implicated C42A in block by sulfhydryl reagents (Trapp et al., 1998b). However, SITS clearly reacts with additional residues to block the channel because the C42A mutation does not influence the extent of block. Furthermore, although DIDS and SITS might be expected to form a covalent reaction with the same residue(s), the C42A mutation only affected the reversibility of SITS block and not that of DIDS. This suggests that an additional residue(s) is involved in the covalent reaction with DIDS.
The K67Q mutation substantially reduced SITS block. In contrast, the amplitude of DIDS inhibition was unaffected, although the block was rendered slowly reversible. This effect appears to be allosteric, because mutation of K67 to methionine or asparagine had no apparent effect on DIDS inhibition. The K67Q mutation also reduced the functional expression of the channel which might indicate that it caused a change in the channel conformation.
It is of interest that mutations which reduce the ability of SITS to inhibit the channel irreversibly cluster in the N terminus, in a region which has been shown to interact with the C terminus of the channel (Tucker & Ashcroft, 1999).
Interactions between the ATP and DIDS binding sites
Our results demonstrate that ATP protects against irreversible block of the channel by DIDS. Mutation of K185 in Kir6.2 to glutamine, which reduces ATP-inhibition of the channel (Tucker et al., 1997) and ATP-binding to Kir6.2 (Tanabe et al., 1999), abolishes the protective effect of ATP. This suggests that access of DIDS to its binding site is decreased when ATP is bound to Kir6.2. Similarly, the rate and extent of block of Kir6.2/SUR2A currents by ATP is dramatically reduced in the presence of DIDS. A number of mutations that alter SITS block also affected the channel ATP-sensitivity (Tucker et al., 1998; Drain et al., 1998). Taken together, these results provide strong support for the idea that the ATP- and stilbene disulphonate-binding sites interact. They are therefore likely to lie close together, but they are not identical because mutations such as K67Q, which affect DIDS block, do not influence ATP inhibition.
We also observed that when ATP is added soon after DIDS application it causes a fast and extensive inhibition of the channel, whereas when ATP is added later and greater DIDS inhibition has occurred, the inhibition by ATP is slower and less complete (data not shown). This is consistent with the idea that the time course of DIDS block represents the rate of binding of DIDS to the channel: the more drug molecules that have bound to the channel, the lower the affinity for ATP (likewise the more channels that have bound DIDS, the fewer available to bind ATP).
In conclusion, our results argue that the binding site for DIDS is likely to lie close to that for ATP. Because DIDS binds irreversibly to the protein, and because it can be readily obtained in radiolabeled form, DIDS may be a useful tool for identifying the ATP-binding site.
Acknowledgments
We thank the Wellcome Trust, the BEIT Trust and the British Diabetic Association for support. Peter Proks holds a BEIT Memorial Fellowship.
Abbreviations
- DIDS
4,4′-diisothiocyanato-stilbene-2,2′-disulphonic acid
- SITS
4-acetamido-4′-isothiocyanato-stilbene-2,2′ disulphonic acid
- KATP channel
ATP-sensitive potassium channel
- SUR
sulphonylurea receptor
References
- AGUILAR-BRYAN L., BRYAN J. Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocrine Rev. 1999;20:101–135. doi: 10.1210/edrv.20.2.0361. [DOI] [PubMed] [Google Scholar]
- AGUILAR-BRYAN L., NICHOLS C.G., WECHSLER S.W., CLEMENT J.P., BOYD A.E., GONZÁLEZ G., HERRERA-SOSA H., NGUY K., BRYAN J., NELSON D.A. Cloning of the β-cell high-affinity sulphonylurea receptor: a regulator of insulin secretion. Science. 1995;268:423–425. doi: 10.1126/science.7716547. [DOI] [PubMed] [Google Scholar]
- ASHCROFT F.M., GRIBBLE F.M. Correlating structure and function in ATP-sensitive K+ channels. Trends Neurosci. 1998;21:288–294. doi: 10.1016/s0166-2236(98)01225-9. [DOI] [PubMed] [Google Scholar]
- ASHCROFT F.M., GRIBBLE F.M. ATP-sensitive K+ channels and insulin secretion. Diabetologia. 1999;42:903–919. doi: 10.1007/s001250051247. [DOI] [PubMed] [Google Scholar]
- CABANTCHIK Z.I., ROTHSTEIN A. The nature of the membrane sites controlling anion permeability of human red blood cells as determined by studies with disulfonic stilbene derivatives. J. Membrane Biol. 1972;10:311–330. doi: 10.1007/BF01867863. [DOI] [PubMed] [Google Scholar]
- CLEMENT IV J.P., KUNJILWAR K., GONZALEZ G., SCHWANSTECHER M., PANTEN U., AGUILAR-BRYAN L., BRYAN J. Association and stoichiometry of KATP channel subunits. Neuron. 1997;18:827–838. doi: 10.1016/s0896-6273(00)80321-9. [DOI] [PubMed] [Google Scholar]
- DRAIN P., LI L., WANG J. KATP channel inhibition by ATP requires distinct functional domains of the cytoplasmic C terminus of the pore-forming subunit. Proc. Natl. Acad. Sci. U.S.A. 1998;95:13953–13958. doi: 10.1073/pnas.95.23.13953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FURUKAWA T., VIRAG L., SAWANOBORI T., HIRAOKA M. Stilbene disulphonates block ATP-sensitive K+ channels in guinea-pig ventricular myocytes. J. Membrane Biol. 1993;136:289–302. doi: 10.1007/BF00233668. [DOI] [PubMed] [Google Scholar]
- GATTO C., LUTSENKO S., KAPLAN J.H. Chemical modification with dihydro-4,4′-diisothiocyanostilbene-2,2′-disulfonate reveals the distance between K480 and K501 in the ATP-binding domain of the Na,K-ATP-ase. Arch. Biochem. Biophys. 1997;340:90–100. doi: 10.1006/abbi.1997.9879. [DOI] [PubMed] [Google Scholar]
- GRIBBLE F.M., ASHFIELD R., ÄMMÄLÄ C., ASHCROFT F.M. Properties of cloned ATP-sensitive K-currents expressed in Xenopus oocytes. J. Physiol. 1997a;498:87–98. doi: 10.1113/jphysiol.1997.sp021843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRIBBLE F.M., TUCKER S.J., ASHCROFT F.M. The interaction of nucleotides with the tolbutamide block of K-ATP currents: a reinterpretation. J. Physiol. 1997b;504:35–45. doi: 10.1111/j.1469-7793.1997.00035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- INAGAKI N., GONOI T., CLEMENT IV J.P., NAMBA N., INAZAWA J., GONZALEZ G., AGUILAR-BRYAN L., SEINO S., BRYAN J. Reconstitution of IKATP: an inward rectifier subunit plus the sulphonylurea receptor. Science. 1995;270:1166–1169. doi: 10.1126/science.270.5239.1166. [DOI] [PubMed] [Google Scholar]
- INAGAKI N., GONOI T., CLEMENT IV J.P., WANG C.Z., AGUILAR-BRYAN L., BRYAN J., SEINO S. A family of sulfonylurea receptors determines the properties of ATP-sensitive K+ channels. Neuron. 1996;16:1011–1017. doi: 10.1016/s0896-6273(00)80124-5. [DOI] [PubMed] [Google Scholar]
- INAGAKI N., GONOI T., SEINO S. Subunit stoichiometry of the pancreatic β-cell ATP-sensitive K+ channel. FEBS Lett. 1997;409:232–236. doi: 10.1016/s0014-5793(97)00488-2. [DOI] [PubMed] [Google Scholar]
- KOZLOWSKI R.Z., ASHFORD M.L. ATP-sensitive K+ channel rundown is Mg2+ dependent. Proc. Royal Soc. B. 1990;240:397–410. doi: 10.1098/rspb.1990.0044. [DOI] [PubMed] [Google Scholar]
- NICHOLS C.G., SHYNG S.L., NESTOROWICZ A., GLASSER B., CLEMENT IV J.P., GONZALEZ G., AGUILAR-BRYAN L., PERMUTT M.A., BRYAN J. Adenosine diphosphate as an intracellular regulator of insulin secretion. Science. 1996;272:1785–1787. doi: 10.1126/science.272.5269.1785. [DOI] [PubMed] [Google Scholar]
- OHNO-SHOSAKU T., ZÜNKLER B., TRUBE J.G. Dual effects of ATP on K+ currents of mouse pancreatic β-cells. Pflügers Arch. 1987;408:133–138. doi: 10.1007/BF00581342. [DOI] [PubMed] [Google Scholar]
- REIMANN F., RYDER T.J., TUCKER S.J., ASHCROFT F.M. The role of lysine 185 in the Kir6.2 subunit of the ATP-sensitive K-channel in channel inhibition by ATP. J. Physiol. 1999;520:661–669. doi: 10.1111/j.1469-7793.1999.00661.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAKURA H., ÄMMÄLÄ C., SMITH P.A., GRIBBLE F.M., ASHCROFT F.M. Cloning and functional expression of the cDNA encoding a novel ATP-sensitive potassium channel expressed in pancreatic β-cells, brain heart and skeletal muscle. FEBS Lett. 1995;377:338–344. doi: 10.1016/0014-5793(95)01369-5. [DOI] [PubMed] [Google Scholar]
- SHYNG S.L., FERRIGNI T., NICHOLS C.G. Regulation of KATP channel activity by diazoxide and MgADP. Distinct functions of the two nucleotide binding folds of the sulfonylurea receptor. J. Gen. Physiol. 1997;110:643–654. doi: 10.1085/jgp.110.6.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHYNG S.L., NICHOLS C.G. Octameric stoichiometry of the KATP channel complex. J. Gen. Physiol. 1997;110:655–664. doi: 10.1085/jgp.110.6.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TANABE K., TUCKER S.J., MATSUO M., PROKS P., ASHCROFT F.M., SEINO S., AMACHI T., UEDA K. Direct photoaffinity labelling of the Kir6.2 subunit of the ATP-sensitive K+ channel by 8-azido-ATP. J. Biol. Chem. 1999;274:3931–3933. doi: 10.1074/jbc.274.7.3931. [DOI] [PubMed] [Google Scholar]
- TRAPP S., PROKS P., TUCKER S.J., ASHCROFT F.M. Molecular analysis of KATP channel gating and implications for channel inhibition by ATP. J. Gen. Physiol. 1998a;112:333–349. doi: 10.1085/jgp.112.3.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TRAPP S., TUCKER S.J., ASHCROFT F.M. Mechanism of ATP-sensitive K channel inhibition by sulphydryl modification. J. Gen. Physiol. 1998b;112:325–332. doi: 10.1085/jgp.112.3.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TUCKER S.J., ASHCROFT F.M. Mapping of the physical interaction between the intracellular domains of an inwardly rectifying potassium channel, Kir6.2. J. Biol. Chem. 1999;274:33393–33394. doi: 10.1074/jbc.274.47.33393. [DOI] [PubMed] [Google Scholar]
- TUCKER S.J., GRIBBLE F.M., PROKS P., TRAPP S., RYDER T.J., HAUG T., RIEMANN F., ASHCROFT F.M. Molecular determinants of KATP channel inhibition. EMBO J. 1998;17:3290–3296. doi: 10.1093/emboj/17.12.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TUCKER S.J., GRIBBLE F.M., ZHAO C., TRAPP S., ASHCROFT F.M. Truncation of Kir6.2 produces ATP-sensitive K-channels in the absence of the sulphonylurea receptor. Nature. 1997;387:179–181. doi: 10.1038/387179a0. [DOI] [PubMed] [Google Scholar]
- ZERANGUE N., SCHWAPPACH B., JAN Y.N., JAN L.Y. A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane KATP channels. Neuron. 1999;22:537–548. doi: 10.1016/s0896-6273(00)80708-4. [DOI] [PubMed] [Google Scholar]