

Abstract
We examined whether acetylcholine release in the hippocampus and striatum and noradrenaline release in the hippocampus is altered in CB1 receptor-deficient mice. The electrically evoked tritium overflow from hippocampal slices preincubated with [3H]-choline was increased by about 100% in CB1−/− compared to CB1+/+ mice whereas the electrically evoked tritium overflow from striatal slices preincubated with [3H]-choline and from hippocampal slices preincubated with [3H]-noradrenaline did not differ. The cannabinoid receptor agonist, WIN 55,212-2, inhibited, and the CB1 receptor antagonist, SR 141716, facilitated, the evoked tritium overflow from hippocampal slices (preincubated with [3H]-choline) from CB1+/+ as opposed to CB1−/− mice. Both drugs did not affect the evoked tritium overflow from striatal slices (preincubated with [3H]-choline) and from hippocampal slices (preincubated with [3H]-noradrenaline) from CB1+/+ and CB1−/− mice. The selective increase in acetylcholine release in CB1−/− mice may indicate that the presynaptic CB1 receptors on the cholinergic neurones of the mouse hippocampus are tonically activated and/or constitutively active in vivo.
Keywords: Hippocampus, striatum, acetylcholine release, noradrenaline release, cannabinoid CB1 receptors, CB1 receptor-deficient mouse, presynaptic receptors, C57BL/6J mouse, SR 141716, septohippocampal pathway
Introduction
Presynaptic cannabinoid CB1 receptors have been shown to occur on various neurones of the peripheral and central nervous system; activation of these receptors causes inhibition of the release of the respective neurotransmitter (for review, see Pertwee, 1997; Ameri, 1999). For the identification of CB1 receptors the advent of selective CB1 receptor antagonists, in particular SR 141716, was very important (for review, see Barth & Rinaldi-Carmona, 1999). When given alone, SR 141716 facilitates neurotransmitter release in a series of experimental models, e.g., acetylcholine release in the hippocampus, as shown by Gifford & Ashby (1996) (rat in vitro) and Kathmann et al. (2000) (mouse in vitro). This finding may mean that the CB1 receptors are activated by endogenously formed cannabinoids (‘endocannabinoids'; for review, see Mechoulam et al., 1998). Since SR 141716 proved to be an inverse agonist under certain circumstances (for review, see Shire et al., 1999), another explanation might be that the CB1 receptors in these experimental models are constitutively active. Both mechanisms would be in harmony with the notion that the CB1 receptors under consideration may play a physiological role.
CB1 receptor-deficient mice have been described recently (Ledent et al., 1999; Zimmer et al., 1999) and offer an additional method to check the potential physiological role of the CB1 receptor. This genetic approach offers several advantages over the pharmacological blockade of the CB1 receptor, most importantly an increased receptor specificity and completeness of receptor inactivation. Another advantage over the use of SR 141716 is that the lack of receptor and/or its associated function is not restricted to the moment of the experiment but is persistent. Using the CB1 receptor knock-out mouse generated by Zimmer et al. (1999), we examined whether CB1 receptor disruption alters acetylcholine release from the hippocampus. For the sake of comparison, acetylcholine release from the striatum and noradrenaline release from the hippocampus, both of which are not subject to modulation via CB1 receptors (Schlicker et al., 1997; Kathmann et al., 2001), were considered as well.
Methods
C57BL/6J mice with disrupted CB1 receptor gene (CB1−/− mice; obtained from A. Zimmer) and C57BL/6J mice of the wild type strain (CB1+/+ mice; purchased from Jackson, Bar Harbor, ME, U.S.A.) were bred in the animal facilities of our department. All animals were housed in a temperature- and humidity-controlled vivarium with a 12-h dark – light cycle. Food and water were available ad libitum. For the experiments, hippocampal or striatal slices (0.3 mm thick, 2 mm diameter) were prepared from 8 – 16 week old animals. In each experiment, slices from a CB1+/+ and a CB1−/− mouse differing in age by ⩽2 weeks were directly compared to each other. The slices were incubated (37°C) for 30 min with physiological salt solution (PSS) containing [3H]-choline 0.1 μM or [3H]-noradrenaline 0.025 μM. The PSS was composed as follows (mM): NaCl 118, KCl 4.8, NaHCO3 25, KH2PO4 1.2, MgCl2 1.2, CaCl2 1.3, glucose 11.1, ascorbic acid 0.06, Na2EDTA 0.03; it was aerated with 95% O2 and 5% CO2.
Subsequently, the slices were superfused at 1 ml min−1 (37°C; 110 min) and the superfusate was collected in 5-min samples. Tritium overflow was evoked by two 2-min periods of electrical field stimulation after 40 and 90 min (S1, S2). For hippocampal or striatal slices preincubated with [3H]-choline, stimulation parameters were 3 Hz, 200 mA, 2 ms and the PSS used for superfusion contained a CaCl2 concentration of 3.25 mM (if not stated otherwise) and hemicholinium-3 10 μM. For hippocampal slices preincubated with [3H]-noradrenaline, the stimulation parameters were 0.3 Hz, 50 mA, 2 ms and the PSS used for superfusion (CaCl2 concentration 1.3 mM) contained desipramine 1 μM and rauwolscine 1 μM. In hippocampal and striatal slices preincubated with [3H]-choline and in hippocampal slices preincubated with [3H]-noradrenaline, addition of tetrodotoxin 1 μM or omission of Ca2+ ions (from 62 min of superfusion onward) inhibited the electrically evoked tritium overflow by 89% or more (results not shown).
Tritium efflux was calculated as the fraction of the tritium content in the slices at the beginning of the respective collection period (fractional rate of tritium efflux). To quantify basal efflux, the fractional rates in the 5-min collection period from 85 – 90 min were determined. The electrically evoked tritium overflow was calculated by subtraction of basal from total efflux during stimulation and the subsequent 13 min and expressed as per cent of the tritium present in the slice at the onset of stimulation (basal tritium efflux was assumed to decline linearly from the 5-min collection period before to that 15 – 20 min after onset of stimulation). To quantify the stimulated tritium overflow, the tritium overflow evoked by S1 or the ratio of the overflow evoked by S2 over that evoked by S1 was determined, as explained in the Results section.
Statistics
Results are given as means±s.e.mean of n experiments (n refers to the number of animals). Student's t-test was used for comparison of mean values; the Bonferroni correction was used when two or more values were compared to the same control.
Drugs used
[Methyl-3H]-choline chloride (specific activity 75 Ci mmol−1), R-(−)-[ring-2, 5, 6-3H]-noradrenaline (spec. act. 51.8 Ci mmol−1) (NEN, Zaventem, Belgium); AF-DX 384 (5,11-dihydro-11-{[(2-{2-[(dipropylamino)methyl]-1-piperidinyl}ethyl)amino]carbonyl}-6H-pyrido(2,3-β)(1,4)benzodiazepine-6-one; Thomae, Biberach an der Riss, Germany); desipramine hydrochloride (Ciba-Geigy, Wehr, Germany); hemicholinium-3 (ChemCon, Freiburg, Germany); rauwolscine hydrochloride (Roth, Karlsruhe, Germany); SR 141716 (N-piperidino-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-3-pyrazole-carboxamide; Sanofi, Montpellier, France); tetrodotoxin (ICN, Eschwege, Germany or Roth, Karlsruhe, Germany); WIN 55,212-2 (R(+)-[2,3-dihydro-5-methyl-3-[(morpholinyl)methyl]pyrrolo[1,2,3-de]-1,4-benzoxazin-yl](1-naphthalenyl)methanone mesylate; RBI/Sigma, München, Germany). Drugs were dissolved in DMSO (SR 141716, WIN 55,212-2), citrate buffer (0.1 mM, pH 4.8; tetrodotoxin) or water (other drugs) and diluted with PSS to obtain the concentration required. Diluted DMSO and citrate buffer did not affect basal or evoked tritium overflow by themselves.
Results
Basal tritium efflux
Basal tritium efflux was expressed as fractional rate of tritium efflux in the collection period from 85 – 90 min of superfusion (i.e., in the 5-min period preceding the second stimulation). The values (min−1) obtained for slices from CB1+/+ mice amounted to 0.0016±0.0001 (hippocampal slices preincubated with [3H]-choline; n=14), 0.0035±0.0002 (striatal slices preincubated with [3H]-choline; n=14) and 0.0023±0.0001 (hippocampal slices preincubated with [3H]-noradrenaline; n=7); virtually identical values were obtained in slices from CB1−/− mice (not shown). The following alterations of the experimental conditions did not affect the basal efflux: use of slices from female mice, alteration of Ca2+ concentration, addition of drugs (WIN 55,212-2, SR 141716, AF-DX 384) (results not shown).
Electrically evoked tritium overflow
Tritium overflow was evoked twice, after 40 and 90 min of superfusion (S1, S2). To quantify the amount of tritium release in slices from CB1−/− as opposed to CB1+/+ mice, the tritium overflow evoked by S1 (expressed as per cent of tissue tritium) was used. Both strains did not differ with respect to striatal slices preincubated with [3H]-choline and hippocampal slices preincubated with [3H]-noradrenaline (Figure 1B). On the other hand, the evoked tritium overflow in hippocampal slices from CB1−/− mice preincubated with [3H]-choline strongly exceeded that in hippocampal slices from wild type mice. This held true for the standard experimental condition (increase by 102%) but also when the Ca2+ concentration in the superfusion medium was lowered from 3.25 to 2.6 or 1.95 mM, when slices from female animals were used or when the muscarinic autoreceptors were blocked by AF-DX 384 (Figure 1A). The increase in the electrically evoked tritium overflow at the Ca2+ concentration of 1.95 mM was significantly (P<0.025) higher than that under the standard experimental condition.
Figure 1.
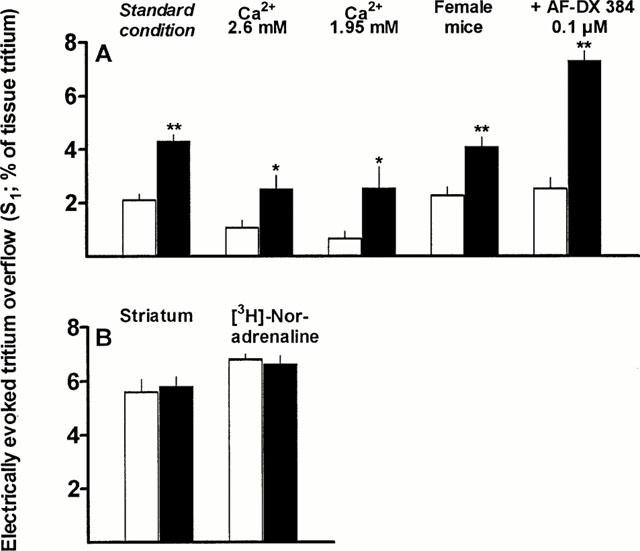
Electrically evoked tritium overflow from superfused hippocampal and striatal slices from CB1+/+ (empty columns) and CB1−/− mice (filled columns). Tritium overflow was evoked after 40 min of superfusion (S1; and again after 90 min, not shown). Hippocampal slices from male CB1+/+ and CB1−/− mice were preincubated with [3H]-choline and then superfused with medium (Ca2+ 3.25 mM) containing hemicholinium-3 10 μM; stimulation parameters were 3 Hz, 200 mA, 2 ms. The results of this series (standard condition) are depicted in A. In additional series (A and B), (i) the Ca2+ concentration was lowered, (ii) slices from female mice were used, (iii) a muscarinic receptor antagonist (AF-DX 384) was present in the medium, (iv) striatal slices were used or (v) hippocampal slices preincubated with [3H]-noradrenaline were used. In the latter series, superfusion was carried out with medium (Ca2+ 1.3 mM) containing desipramine 1 μM and rauwolscine 1 μM and the stimulation parameters were 0.3 Hz, 50 mA, 2 ms. Data are expressed as means±s.e.mean of 5 – 14 experiments. *P⩽0.05, **P<0.005.
Effect of cannabinoid receptor ligands on the electrically evoked tritium overflow
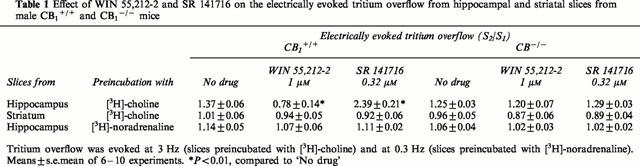
To study the effect of drugs on the evoked tritium overflow, the ratio of the overflow evoked by S2 over that evoked by S1 was determined. The cannabinoid receptor agonist WIN 55,212-2 or the CB1 receptor antagonist SR 141716 was added to the medium before and during S2. Table 1 shows that the evoked tritium overflow was inhibited by WIN 55,212-2 and facilitated by SR 141716 in hippocampal slices (preincubated with [3H]-choline) from CB1+/+ mice but was not affected in slices from CB1−/− mice. WIN 55,212-2 and SR 141716 failed to affect the evoked tritium overflow from striatal slices preincubated with [3H]-choline and from hippocampal slices preincubated with [3H]-noradrenaline, irrespective of whether the slices were obtained from CB1+/+ or CB1-/- mice.
Table 1.
Effect of WIN 55,212-2 and SR 141716 on the electrically evoked tritium overflow from hippocampal and striatal slices from male CB1+/+ and CB1−/− mice
Discussion
In the present study, we compared the electrically evoked tritium overflow from slices of CB1+/+ and CB1−/− mice preincubated with [3H]-choline (hippocampus, striatum) and with [3H]-noradrenaline (hippocampus). In slices preincubated with [3H]-choline, the high-affinity choline uptake was inhibited by hemicholinium-3 and the muscarinic autoreceptor was blocked (in part of the experiments) by AF-DX 384 to avoid interactions with these mechanisms. Similarly, desipramine and rauwolscine were used in slices preincubated with [3H]-noradrenaline to block the neuronal noradrenaline transporter and the α2-autoreceptor, respectively. It is known from the literature that, in slices preincubated with [3H]-noradrenaline and then superfused in the presence of an inhibitor of the neuronal noradrenaline transporter, about 90% of the electrically evoked tritium overflow consist of unmetabolized [3H]-noradrenaline (Baumann & Maître, 1977). In slices, preincubated with [3H]-choline and then superfused in the presence of an inhibitor of the high-affinity choline uptake (but in the absence of a cholinesterase inhibitor), the electrically evoked tritium overflow mainly consists of [3H]-choline formed from [3H]-acetylcholine immediately after release (Richardson & Szerb, 1974). Since, in addition, the electrically evoked tritium overflow was Ca2+-dependent and tetrodotoxin-sensitive in our experiments we can assume that it represents quasi-physiological acetylcholine and noradrenaline release, respectively, and we used these terms in the following text.
If one applies the same stimulation protocol in order to evoke acetylcholine and noradrenaline release (S1, expressed as per cent of tissue tritium), the amount of noradrenaline will exceed that of acetylcholine by a factor of more than 10 (unpublished results). For this reason, we used a higher stimulation frequency and a higher current strength to evoke acetylcholine release. Since, nonetheless, the amount of released acetylcholine was very low in hippocampal slices from CB1+/+ (C57BL/6J) mice (Kathmann et al., 2001), the Ca2+ concentration in the superfusion medium was stepwise increased to 1.95, 2.6 and 3.25 mM. Most of the experiments in which acetylcholine release was studied were carried out at 3.25 mM since at this Ca2+ concentration S1 values higher than 1.5% of tissue tritium were obtained. Note that, at lower S1 values, the S2/S1 ratios (used to quantify the effects of drugs on transmitter release, e.g. in Table 1) will show a very high variation.
The major finding of the present investigation is that acetylcholine release was about twice as high in the hippocampus from CB1 receptor-deficient mice when compared to wild type animals. This increase was found under three different Ca2+ concentrations in the superfusion medium and in slices from male and female mice. One might think that the increase in transmitter release in the knock-out animals is a more general phenomenon. This, however, does not appear to hold true since the release of the same transmitter in another brain region (striatum) and the release of another transmitter (noradrenaline) in the same brain region did not show a difference between CB1+/+ and CB1−/− mice. Moreover, one might assume that the increase in acetylcholine release is related to a compromised function of the muscarinic autoreceptor. This possibility can be excluded since the increase in acetylcholine release also occurred when the muscarinic autoreceptor was blocked in slices from CB1 receptor-deficient and wild type mice. In addition, the muscarinic receptor agonist oxotremorine caused a very marked inhibition of acetylcholine release in hippocampal slices from CB1−/− mice (maximum inhibition by 80%; unpublished results), which was comparable to that found in the hippocampus from NMRI mice (Kathmann et al., 2001).
A plausible explanation for the selective increase in hippocampal acetylcholine release in CB1−/− mice would be that the release of this transmitter is dis-inhibited due to the disruption of tonically activated or constitutively active presynaptic CB1 receptors on cholinergic neurones (see Introduction). The following findings are in line with this notion. (i) The CB1 receptor antagonist SR 141716 facilitated acetylcholine release in hippocampal slices from CB1+/+ mice (Kathmann et al., 2001; present study). (ii) SR 141716 did not facilitate (and the cannabinoid receptor agonist WIN 55,212-2 did not inhibit) acetylcholine release in hippocampal slices from CB1−/− mice. (iii) SR 141716 (and WIN 55,212-2) did not affect acetylcholine release in striatal slices and noradrenaline release in hippocampal slices from CB1+/+ mice. An auxiliary experimental series on hippocampal slices from CB1+/+ mice (not shown) revealed that WIN 55,212-2 inhibited acetylcholine release in a concentration-dependent manner and that its concentration-response curve was shifted to the right by SR 141716, yielding an apparent pA2 value of 8.1, which is similar to that obtained in other CB1 receptor models (e.g., Schlicker et al., 1997; Kathmann et al., 2001).
The possibility cannot be excluded that other mechanisms may account for the selective increase in hippocampal acetylcholine release in CB1−/− mice, which, e.g., might represent a compensatory change in response to the alteration of the release of other transmitters. Irrespective of the mechanism, our finding is interesting inasmuch as several lines of evidence suggest that the effects of cannabinoids on learning and memory may be causally related to an alteration of the septohippocampal cholinergic system. Thus, learning and memory processes are negatively influenced by cannabinoid receptor agonists (for reviews, see Hampson & Deadwyler, 1999; Sullivan, 2000) but facilitated by SR 141716 (Terranova et al., 1996; Lichtman, 2000) or CB1 receptor disruption (Reibaud et al., 1999) and hippocampal acetylcholine release is inhibited by cannabinoid receptor agonists but facilitated by SR 141716 (Gifford & Ashby, 1996; Gessa et al., 1997; Kathmann et al., 2001; present study) or CB1 receptor disruption (present study). Moreover, the impairment of working memory in the rat by a cannabinoid receptor agonist is reversed by the cholinesterase inhibitor eptastigmine (Braida & Sala, 2000) and the facilitatory effect of SR 141716 on memory processes in the rat is partially antagonized by the muscarinic receptor antagonist scopolamine (Terranova et al., 1996). It has to be stressed, however, that the evidence is not unequivocal. Thus, in the study by Acquas et al. (2000) cannabinoid receptor agonists did not inhibit but rather increased hippocampal acetylcholine release in the rat in vivo. Furthermore, the Δ9-tetrahydrocannabinol-induced impairment of a memory task in the rat was not counteracted by the cholinesterase inhibitor physostigmine and the impairment of the same task by scopolamine was not counteracted by SR 141716 (Lichtman & Martin, 1996). Even if the role of our finding with respect to learning and memory is not yet clear, this finding represents a new piece of evidence in favour of tonically activated or constitutively active CB1 receptors.
Acknowledgments
This study was supported by grants from the Deutsche Forschungsgemeinschaft (GRK 246, Schl 266/5-3) and from the Land Nordrhein-Westfalen (BONFOR programme). We wish to thank Mrs Doris Petri and Mrs Petra Zeidler for their skilled technical assistance and the companies Ciba-Geigy, Sanofi and Thomae for gifts of drugs.
Abbreviations
- ACh
acetylcholine
- AF-DX 384
5,11-dihydro-11-{[(2-{2-[(dipropylamino)methyl]-1-piperidinyl}ethyl)amino]carbonyl}-6H-pyrido(2,3-β)(1,4)benzodiazepine-6-one
- C57BL/6J
mouse strain
- CB1−/−
CB1+/+ mouse, CB1 receptor-deficient and wild type mouse
- NMRI
mouse strain
- PSS
physiological salt solution
- S1
S2, first and second stimulation, respectively
- SR 141716
N-piperidino-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-3-pyrazole-carboxamide
- WIN 55,212-2
R(+)-[2,3-dihydro-5-methyl-3-[(morpholinyl)methyl]pyrrolo[1,2,3-de]-1,4-benzoxazin-yl](1-naphthalenyl)methanone
References
- ACQUAS E., PISANU A., MARROCU P., DI CHIARA G. Cannabinoid CB1 receptor agonists increase rat cortical and hippocampal acetylcholine release in vivo. Eur. J. Pharmacol. 2000;401:179–185. doi: 10.1016/s0014-2999(00)00403-9. [DOI] [PubMed] [Google Scholar]
- AMERI A. The effects of cannabinoids on the brain. Prog. Neurobiol. 1999;58:315–348. doi: 10.1016/s0301-0082(98)00087-2. [DOI] [PubMed] [Google Scholar]
- BARTH F., RINALDI-CARMONA M. The development of cannabinoid antagonists. Curr. Med. Chem. 1999;6:745–755. [PubMed] [Google Scholar]
- BAUMANN P.A., MAÎTRE L. Blockade of presynaptic α-receptors and of amine uptake in the rat brain by the antidepressant mianserine. Naunyn-Schmiedeberg's Arch. Pharmacol. 1977;300:31–37. doi: 10.1007/BF00505077. [DOI] [PubMed] [Google Scholar]
- BRAIDA D., SALA M. Cannabinoid-induced working memory impairment is reversed by a second generation cholinesterase inhibitor in rats. Neuroreport. 2000;11:2025–2029. doi: 10.1097/00001756-200006260-00044. [DOI] [PubMed] [Google Scholar]
- GESSA G.L., MASCIA M.S., CASU M.A., CARTA G. Inhibition of hippocampal acetylcholine release by cannabinoids: reversal by SR 141716A. Eur. J. Pharmacol. 1997;327:R1–R2. doi: 10.1016/s0014-2999(97)89683-5. [DOI] [PubMed] [Google Scholar]
- GIFFORD A.N., ASHBY C.R. Electrically evoked acetylcholine release from hippocampal slices is inhibited by the cannabinoid receptor agonist, WIN 55,212-2, and is potentiated by the cannabinoid antagonist, SR 141716 A. J. Pharmacol. Exp. Ther. 1996;277:1431–1436. [PubMed] [Google Scholar]
- HAMPSON R.E., DEADWYLER S.A. Cannabinoids, hippocampal function and memory. Life Sci. 1999;65:715–723. doi: 10.1016/s0024-3205(99)00294-5. [DOI] [PubMed] [Google Scholar]
- KATHMANN M., WEBER B., SCHLICKER E. Cannabinoid CB1 receptor-mediated inhibition of acetylcholine release in the brain of NMRI, CD-1 and C57BL/6J mice. Naunyn-Schmiedeberg's Arch. Pharmacol. 2001;363:50–56. doi: 10.1007/s002100000304. [DOI] [PubMed] [Google Scholar]
- LEDENT C., VALVERDE O., COSSU C., PETITET F., AUBERT L.F., BESLOT F., BÖHME G.A., IMPERATO A., PEDRAZZINI T., ROQUES B.P., VASSART G., FRATTA W., PARMENTIER M. Unresponsiveness to cannabinoids and reduced addictive effects of opiates in CB1 receptor knockout mice. Science. 1999;283:401–404. doi: 10.1126/science.283.5400.401. [DOI] [PubMed] [Google Scholar]
- LICHTMAN A.H. SR 141716A enhances spatial memory as assessed in a radial-arm maze task in rats. Eur. J. Pharmacol. 2000;404:175–179. doi: 10.1016/s0014-2999(00)00615-4. [DOI] [PubMed] [Google Scholar]
- LICHTMAN A.H., MARTIN B.R. Δ9-Tetrahydrocannabinol impairs spatial memory through a cannabinoid receptor mechanism. Psychopharmacology. 1996;126:125–131. doi: 10.1007/BF02246347. [DOI] [PubMed] [Google Scholar]
- MECHOULAM R., FRIDE E., DI MARZO V. Endocannabinoids. Eur. J. Pharmacol. 1998;359:1–18. doi: 10.1016/s0014-2999(98)00649-9. [DOI] [PubMed] [Google Scholar]
- PERTWEE R.G. Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol. Ther. 1997;74:129–180. doi: 10.1016/s0163-7258(97)82001-3. [DOI] [PubMed] [Google Scholar]
- REIBAUD M., OBINU M.C., LEDENT C., PARMENTIER M., BÖHME G.A., IMPERATO A. Enhancement of memory in cannabinoid CB1 receptor knock-out mice. Eur. J. Pharmacol. 1999;379:R1–R2. doi: 10.1016/s0014-2999(99)00496-3. [DOI] [PubMed] [Google Scholar]
- RICHARDSON I.W., SZERB J.C. The release of labelled acetylcholine and choline from cerebral cortical slices stimulated electrically. Br. J. Pharmacol. 1974;52:499–507. doi: 10.1111/j.1476-5381.1974.tb09717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHLICKER E., TIMM J., ZENTNER J., GÖTHERT M. Cannabinoid CB1 receptor-mediated inhibition of noradrenaline release in the human and guinea-pig hippocampus. Naunyn-Schmiedeberg's Arch. Pharmacol. 1997;356:583–589. doi: 10.1007/pl00005093. [DOI] [PubMed] [Google Scholar]
- SHIRE D., CALANDRA B., BOUABOULA M., BARTH F., RINALDI-CARMONA M., CASELLAS P., FERRARA P. Cannabinoid receptor interactions with the antagonists SR 141716A and SR 144528. Life Sci. 1999;65:627–635. doi: 10.1016/s0024-3205(99)00285-4. [DOI] [PubMed] [Google Scholar]
- SULLIVAN J.M. Cellular and molecular mechanisms underlying learning and memory impairments produced by cannabinoids. Learning Memory. 2000;7:132–139. doi: 10.1101/lm.7.3.132. [DOI] [PubMed] [Google Scholar]
- TERRANOVA J.P., STORME J.J., LAFON N., PÉRIO A., RINALDI-CARMONA M., LE FUR G., SOUBRIÉ P. Improvement of memory in rodents by the selective CB1 cannabinoid receptor antagonist, SR 141716. Psychopharmacology. 1996;126:165–172. doi: 10.1007/BF02246352. [DOI] [PubMed] [Google Scholar]
- ZIMMER A., ZIMMER A.M., HOHMANN A.G., HERKENHAM M., BONNER T.I. Increased mortality, hypoactivity, and hypoalgesia in cannabinoid CB1 receptor knockout mice. Proc. Natl. Acad. Sci. 1999;96:5780–5785. doi: 10.1073/pnas.96.10.5780. [DOI] [PMC free article] [PubMed] [Google Scholar]