

Abstract
We investigated the role of arachidonic acid metabolism and assessed the participation of mast cells and leukocytes in neurogenic inflammation in rat paw skin. We compared the effect of lipoxygenase (LOX) and cyclo-oxygenase (COX) inhibitors on oedema induced by saphenous nerve stimulation, substance P (SP), and compound 48/80.
Intravenous (i.v.) pre-treatment with a dual COX/LOX inhibitor (RWJ 63556), a dual LOX inhibitor/cysteinyl-leukotriene (CysLt) receptor antagonist (Rev 5901), a LOX inhibitor (AA 861), a five-lipoxygenase activating factor (FLAP) inhibitor (MK 886), or a glutathione S-transferase inhibitor (ethacrynic acid) significantly inhibited (40 to 60%) the development of neurogenic oedema, but did not affect cutaneous blood flow. Intradermal (i.d.) injection of LOX inhibitors reduced SP-induced oedema (up to 50% for RWJ 63556 and MK 886), whereas ethacrynic acid had a potentiating effect.
Indomethacin and rofecoxib, a highly selective COX-2 inhibitor, did not affect neurogenic and SP-induced oedema. Surprisingly, the structurally related COX-2 inhibitors, NS 398 and nimesulide, significantly reduced both neurogenic and SP-induced oedema (70% and 42% for neurogenic oedema, respectively; 49% and 46% for SP-induced oedema, respectively).
COX-2 mRNA was undetectable in saphenous nerves and paw skin biopsy samples, before and after saphenous nerve stimulation.
A mast cell stabilizer, cromolyn, and a H1 receptor antagonist, mepyramine, significantly inhibited neurogenic (51% and 43%, respectively) and SP-induced oedema (67% and 63%, respectively).
The co-injection of LOX inhibitors and compound 48/80 did not alter the effects of compound 48/80. Conversely, ethacrynic acid had a significant potentiating effect. The pharmacological profile of the effect of COX inhibitors on compound 48/80-induced oedema was similar to that of neurogenic and SP-induced oedema.
The polysaccharide, fucoidan (an inhibitor of leukocyte rolling) did not affect neurogenic or SP-induced oedema.
Thus, (i) SP-induced leukotriene synthesis is involved in the development of neurogenic oedema in rat paw skin; (ii) this leukotriene-mediated plasma extravasation might be independent of mast cell activation and/or of the adhesion of leukocytes to the endothelium; (iii) COX did not appear to play a significant role in this process.
Keywords: Neurogenic inflammation, lipoxygenase, cyclo-oxygenase, leukotriene, saphenous nerve
Introduction
Neurogenic inflammation is a well characterized mechanism, involving the activation of primary afferent C fibres and the subsequent release of vasodilating and inflammatory neuropeptides at the peripheral nerve endings. Substance P (SP) and calcitonin gene-related peptide (CGRP) are the main neuromediators responsible for the oedema and vasodilatation observed in the skin of the leg in response to antidromic electrical stimulation of the saphenous nerve in rats (Escott & Brain, 1993; Lembeck & Holzer, 1979).
The mechanism underlying the development of SP-induced oedema probably involves the direct activation of endothelial cells, the release of potent inflammatory mediators from mast cells, and the adhesion/marginalization of leukocytes. SP causes gaps to form between endothelial cells, resulting in immediate plasma and protein leakage upon the activation of the endothelial neurokinin-1 (NK1) receptors (Baluk et al., 1997). This direct NK1 receptor-mediated mechanism probably, at least partly, accounts for the development of oedema observed following intradermal injection of SP or electrical stimulation of C fibres (Regoli et al., 1996). SP also induces the release of histamine from isolated mast cells in many animal species, accounting for the mast cell-dependent H1 receptor-mediated component of neurogenic oedema (Maggi, 1997). In addition, SP-induced stimulation of cultured endothelial cells promotes the expression of adhesion molecules at the membrane surface in vitro, notably CD11, ICAM-1, and VCAM-1 (DeRose et al., 1994; Quinlan et al., 1999). It has been shown in many animal species that intradermally injected SP induces granulocyte infiltration in the inflamed skin within hours (Fröde-Saleh et al., 1999; Walsh et al., 1995). The role of neutrophil adhesion/activation in the rapid onset (within minutes) of oedema following the antidromic electrical stimulation of primary afferents remains unclear.
SP-induced stimulation of endothelial cells, mast cells and/or neutrophils may trigger the release of arachidonic acid metabolites by these cyclo-oxygenase (COX) and lipoxygenase (LOX) rich cells (Bell & Harris, 1999; Pairet & Engelhardt, 1996). Arachidonic acid derivatives are important inflammatory mediators in several models of acute and chronic inflammation. However, the role of COX and LOX pathways in the development of oedema in response to SP and the antidromic stimulation of primary afferent C fibres remains poorly understood. It has been demonstrated that the stimulation of the saphenous nerve in rats induces the concomitant accumulation of CGRP and prostaglandin (PG) E2, a well known pro-inflammatory eicosanoid from the COX pathway, in the skin (Kress et al., 1999). Nevertheless, the role of prostaglandins in purely neurogenic oedema has not been explored. It also has been shown that leukotriene (LT) B4, a potent mast cell-derived chemotactic factor, is responsible for the granulocyte marginalization observed after the intradermal injection of SP in mice (Iwamoto et al., 1993). However, it is not involved in the SP-induced skin oedema which develops within minutes of subcutaneous injection of the neuropeptide. The involvement of the LOX pathway in skin neurogenic oedema has never been described.
This study was designed to assess the role of the arachidonic acid metabolites in neurogenic inflammation. Therefore, we investigated the effects of various COX and LOX pathway inhibitors in skin oedema induced by electrical stimulation of the saphenous nerve in rats. We used a non-selective inhibitor (indomethacin), selective COX-2 inhibitors (rofecoxib, NS 398, nimesulide) a dual COX/LOX inhibitor (RWJ 63556), a dual LOX inhibitor/cysteinyl-leukotriene (CysLt) receptor antagonist (Rev 5901), a LOX inhibitor (AA 861) and a five-lipoxygenase activating factor (FLAP) inhibitor (MK 886). To determine the participation of LTC4-synthase and CysLts we used a glutathione S-transferase inhibitor, ethacrynic acid, which is a LTC4-synthase from the glutathione S-transferase family (Leung, 1986).
The second aim of this study was to assess the role of mast cells and polymorphonuclear leukocytes (PMN) in this model and to determine whether SP-induced mast cell and/or PMN activation account for the LOX-dependent inflammatory process. We used a mast cell stabilizer, cromolyn, which is the most effective inhibitor of histamine release from mast cells (Kubes & Kanwar, 1994), an H1 receptor antagonist, mepyramine, an inhibitor of leukocyte rolling, fucoidan, and an inhibitor of leukocyte adhesion, NPC 14686 (Burch et al., 1991). The results were compared to those obtained with the same drugs on oedema provoked by intradermal injections of SP or compound 48/80, a well-known mast cell activator. Finally, COX and LOX inhibitors which significantly affected neurogenic oedema were tested on neurogenic vasodilatation to confirm their postjunctional effect. The results were compared with those obtained with a μ-opioid receptor agonist, DAMGO, and a CGRP1 receptor antagonist, CGRP8 – 37, which are prejunctional and postjunctional effect inhibitors, respectively (Barber, 1993; Escott & Brain, 1993).
Methods
Neurogenic oedema
We adapted a previously described procedure for measuring oedema induced by electrical stimulation of the saphenous nerve in the rat (Escott & Brain, 1993). Briefly, 24 h before the experiments began male Wistar rats (200 – 300 g) were injected with guanethidine (20 mg kg−1 s.c.) to prevent vasoconstriction caused by the stimulation of the sympathetic fibres of the saphenous nerve (Gamse & Saria, 1987). Animals were anaesthetized with sodium pentobarbitone (60 mg kg−1 i.p., plus maintenance dose as necessary) and the hind limbs were shaved. Body temperature was maintained at 36 to 38°C by an automatic heating pad. The saphenous nerves in the right and left paws were carefully dissected and cut centrally. The distal ends of the saphenous nerves were placed on bipolar platinum electrodes and immersed in mineral oil. Test agents or the appropriate vehicle were administered via a cannula in the jugular vein 15 min before electrical stimulation. Ten minutes before electrical stimulation a plasma marker, Evans Blue dye (20 mg kg−1; 1 ml kg−1), was injected through the dorsal vein of the penis. The right saphenous nerve was stimulated (3 V, 1 ms, 5 Hz for 5 min; Dual Impedance Research Stimulator, Harvard). The left saphenous nerve was not stimulated to determine resting plasma leakage in the skin during the experiments. Previous experiments have shown that these electrical stimulation conditions result in the induction of submaximal oedema. Immediately after the stimulation a cardiac blood sample was taken and the animal killed by administration of an overdose of anaesthetic. The blood samples were centrifuged at 3000×g for 15 min and the plasma was retained. The oedematous area of skin on the right paw (detected by Evans Blue dye extravasation) and a corresponding area of skin on the left paw were removed and weighed. We subjected these samples to an extraction procedure as previously described (Beach & Steinetz, 1961). Evans Blue dye was quantified by spectroscopy and by measuring the absorbance at 620 nm, for 100 μl plasma and skin biopsy samples.
SP- and compound 48/80-induced oedema
Evans Blue dye was used as a plasma marker to measure plasma extravasation induced by i.d. injection of SP (100 pmol site−1) or compound 48/80 (1 μg site−1). Briefly, 5 min after the injection of Evans Blue into an anaesthetized rat (as described above), an inflammatory inducer with (treated animal) or without (control animal) test agents was injected i.d. (0.1 ml site−1) into the right paw. To determine the plasma leakage generated by the injection procedure NaCl 0.9% was injected i.d. into the contralateral paw. Thirty minutes after initiation of the inflammatory cascade a cardiac blood sample was taken and the animal killed by administration of an overdose of anaesthetic. The areas of oedematous skin around the injection sites in the right and left paws were removed and weighed. Blood and skin biopsy samples were treated as above.
Neurogenic vasodilatation
Animals were prepared as for neurogenic oedema. A laser Doppler probe (Perimed, PF3) was positioned and secured in a region of hind paw skin that is innervated by the saphenous nerve. Signals were digitized (Power Lab/8s, ADInstruments) and transferred to a personal computer for off-line analysis of the neurogenic rise in blood flow (in perfusion units, PU) induced by the electrical stimulation of the saphenous nerve. A 30-min stabilization period was used to determine resting blood flow. The saphenous nerve was then stimulated (3 V, 1 ms, 5 Hz for 10 s). When blood flow returned to resting values the nerve was stimulated for a second time to check the stability of the neurogenic vasodilating response. After recovery from this second stimulation test agents or the appropriate vehicle (1 ml kg−1) were administered via a cannula in the jugular vein, and a third electrical stimulation was given 15 min later. A preliminary study showed that the three electrical consecutive stimulations each provoked the same rise in arteriolar blood flow (97±11 PU for the first SE; 96±10 PU for the second SE; 93±9 PU for the third SE, n=17), with comparable resting value (54±6 PU for the first SE; 53±5 PU for the second SE; 51±5 PU for the third SE, n=17), and needed the same time to return to the resting value (7±1 min for the first SE; 5±1 min for the second SE; 6±1 min for the third SE, n=17).
RT – PCR experiments to detect COX-1 and COX-2 mRNA
Contralateral and ipsilateral saphenous nerves and paw skin biopsy samples were removed from control animals before and after a 5-min stimulation (as described above). Tissues were immediately frozen in liquid nitrogen. Total RNA was extracted with Trizol, according to the manufacturer's instructions. Briefly, tissues were cut into small pieces and homogenized in Trizol. Total RNA was extracted with chloroform and precipitated with isopropyl alcohol. It was centrifuged and the pellet was resuspended in DEPC-treated water. The concentration was determined by measuring the absorbance at 260 nm. The quality of the RNA was checked by electrophoresis in 1% agarose gels and glyceraldehyde-3-phosphate dehydrogenase (GADPH) reverse transcription-polymerase chain reaction (RT – PCR). Rat GAPDH, COX-1, and COX-2 primers were selected on the basis of cDNA sequences in the NCBI database (accession numbers AF106860, S67721, and AF233596). Their sequence (5′ to 3′) were: GAPDH up TGA TGC TGG TGC TGA GTA TG; GAPDH down TCA TTG AGA GCA ATG CCA GC; COX-1 up ACG TGA GCT ACT ATA CTC GC; COX-1 down ACT CCT TGA TGA CAT CCT CG; COX-2 up GAC GAT CAA GAT AGT GAT CG; and COX-2 down TTA CAG CTC AGT TGA ACG CC. GAPDH, COX-1, and COX-2 primers amplified products of 650 bp, 932 bp, and 841 bp, respectively. RT was carried out for 90 min at 37°C in a volume of 30 μl. The reaction mixture contained 2 μg of total RNA, 30 U RNAsin, 250 pmol random hexamer primers, 5 mM DTT, and 400 U Moloney murine leukaemia virus (M-MLV) Reverse Transcriptase RNAseH Minus in reaction buffer (50 mM Tris-HCl, 75 mM KCl, and 3 mM MgCl2). Negative control samples, in which the RNA was replaced with sterile water or the M-MLV was omitted was used to confirm that there was no contamination. PCR was carried out with 2.5 μl (GAPDH), 5 μl (COX-1), or 10 μl (COX-2) of the cDNA mixture, in a final volume of 50 μl. The PCR reaction mixture contained 2.5 mM MgCl2, 200 μM dNTPs, 100 pmol of each primer, and 2.5 U of Taq Polymerase in reaction buffer (10 mM Tris-HCl pH 9, 50 mM KCl, and 0.1% Triton X-100). PCR was performed in a MJ Research thermocycler as follows: 94°C for 4 min, followed by 30 (GAPDH) or 40 cycles (COX-1 and COX-2) of 94°C for 50 s, 56°C for 1 min, and 72°C for 2 min; followed by a final elongation step at 72°C for 10 min. PCR products separated in a 1% agarose gel at 150 V for 60 min. Smart ladder was used as a size marker.
Data analysis
Oedema was quantified as the difference in μl of plasma g−1 between the right and left skin biopsy samples. For example, the values for skin biopsy samples of rats treated with NaCl 0.9% were: 59±3 μl plasma g−1 of skin (left skin biopsy) vs 294±21 μl plasma g−1 of skin (right skin biopsy) for neurogenic oedema; 120±7 μl plasma g−1 of skin (left skin biopsy) vs 275±30 μl plasma g−1 of skin (right skin biopsy) for SP-induced oedema; 150±10 μl plasma g−1 of skin (left skin biopsy) vs 467±45 μl plasma g−1 of skin (right skin biopsy) for compound 48/80-induced oedema. The mean±s.e.mean oedema volume obtained for the treated group was compared with the mean±s.e.mean oedema volume in the corresponding control group. Drug effects are expressed as per cent inhibition±s.e.mean of extravasation in control animals.
For each neurogenic vasodilatation group the mean±s.e.mean amplitude of the increase in blood flow induced by the third stimulation was compared with the mean±s.e.mean amplitude of the increase in blood flow measured in response to the second stimulation. The effects of test agents or vehicles are expressed as per cent change±s.e.mean in the neurogenic response between the second and third electrical stimulation.
Statistical comparisons were made by ANOVA, followed by Bonferroni's test. P values <0.05 were considered to be significant.
Drugs
The following drugs were obtained from Sigma (Saint-Quentin Fallavier, France): CGRP8 – 37, compound 48/80, cromolyn, ethacrynic acid, Evans blue, fucoidan, guanethidine, indomethacin, mepyramine, substance P, 2-(12-hydroxydodeca-5,10-diynyl)-3,5,6-trimethyl-p-benzoquinone (AA 861), [D-Ala2, N-Me-Phe4, Gly-ol]-enkephalin (DAMGO), and N-[4-Nitro-2-phenoxyphenyl]-methanesulphonamide (nimesulide). Asp - Tyr - D - Trp - Val - D - Trp - D - Trp - Lys-NH2 (MEN 10376), 3-[1-(p-chlorobenzyl)-5-(isopropyl)-3-t-butylthioindol-2-yl]-2,2-dimethyl-propanoic acid (MK 886), N-(fluorenyl-9-methoxycarbonyl)-L-homophenylalanine (NPC 14686), and ginkgolide B (BN 52021) were purchased from Tebu (Le Perray-en-Yvelines, France); N-[2-(cyclohexyloxy)-4-nitrophenyl]-methanesulphonamide (NS 398) and α-pentyl-3-(2-quinolinylmethoxy)-benzenemethanol (Rev 5901) were obtained from Spi-Bio (Massy, France); cis-2-(diphenylmethyl)-N-[(2-iodophenyl)methyl]-1-azabicyclo[2.2.2]octan-3-amine oxalate (L-703,606) was obtained from RBI (Saint-Quentin Fallavier, France). 4-(4-methylsulfonylphenyl)-3-phenyl-2-(5H)-furanone (Rofecoxib) and N-[5-(4-fluorophenoxy)-2-thienyl] methanesulphonamide (RWJ 63556) were kindly provided by the Department of Chemistry (Laboratoire Innothéra). AA 861, BN 52021, ethacrynic acid, MEN 10376, MK 886, nimesulide, NPC 14686, NS 398, Rev 5901, rofecoxib, and RWJ 63556 were dissolved 1 : 100 v v−1 in DMSO and NaCl 0.9% (saphenous nerve-induced oedema and arteriolar dilatation experiments) and 1 : 1000 v v−1 (SP- and compound 48/80-induced oedema experiments). Indomethacin was dissolved in 0.5 M NaHCO3. All other drugs were dissolved in NaCl 0.9%. All the chemicals used in the PCR experiments were purchased from Promega (Charbonnières, France), except for Trizol reagent which was purchased from GibcoBRL (Cergy-Pontoise, France), and primers, dNTPs, and DNA ladders which were purchased from Eurogentec (Angers, France).
Results
We investigated the involvement of the LOX pathway in the development of neurogenic oedema during a 5-min stimulation period of the saphenous nerve in guanetidine-treated rats. Preliminary studies showed that our surgical protocol led to the same plasma extravasation in both paws without electrical stimulation. The difference between the oedema in right and left skin biopsy samples was that the left samples avoided intravascular dye and plasma extravasation due to the experimental manipulations. Fifteen minutes before stimulation of the saphenous nerve the rats were injected i.v. with a number of drugs that inhibit the synthesis and/or the effects of LOX-derived metabolites (Figure 1). These drugs have a direct effect on LOX (RWJ 63556, AA 861, Rev 5901), FLAP (MK 886), and glutathione S-transferase (ethacrynic acid) activity. All these LOX pathway inhibitors similarly and significantly inhibited the development of neurogenic oedema (40 – 60%). The mean volumes of extravasated plasma in skin biopsies were measured in treated and untreated animals (Table 1). We evaluated the effects of these LOX pathway inhibiting drugs on SP-induced oedema by co-injecting them i.d. with the inflammatory neuropeptide (Figure 1). RWJ 63556 and MK 886 significantly inhibited the development of SP-induced oedema (68±19 Δ μl plasma g of skin−1 (treated) vs 141±23 Δ μl plasma g of skin−1 (untreated), n=16 – 22, P<0.05 for RWJ 63556; 69±16 Δ μl plasma g of skin−1 (treated) vs 141±23 Δ μl plasma g of skin−1 (untreated), n=14 – 22, P<0.05 for MK 886). All the other inhibitors reduced the development of SP-induced oedema (20 – 40%; not significant). Finally, the non-specific glutathione S-transferase inhibitor, ethacrynic acid, caused a non-significant increase in the effects of the neuropeptide. In control experiments, i.d. injection of the same doses of LOX inhibitors without SP did not alter the basal Evans Blue leakage measured after injection of vehicle alone.
Figure 1.
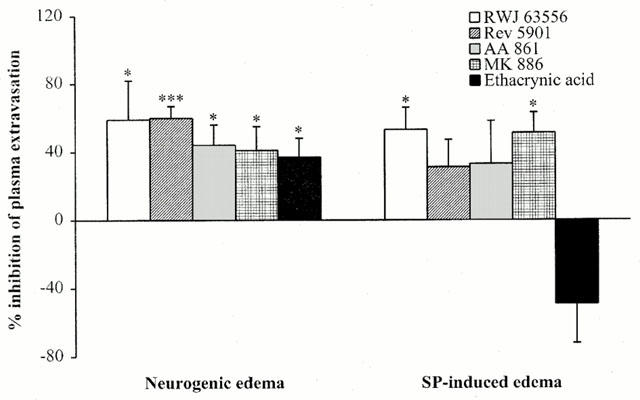
The effect of LOX pathway inhibitors on neurogenic and SP-induced oedema. RWJ 63556 (1 mg kg−1 i.v.), Rev 5901 (1 mg kg−1 i.v.), AA 861 (0.1 mg kg−1 i.v.), MK 886 (10 μg kg−1 i.v.), or ethacrynic acid (1 mg kg−1 i.v.) were injected 15 min before a 5-min stimulation (neurogenic oedema). RWJ 63556 (1 μg site−1 i.d.), Rev 5901 (1 μg site−1 i.d.), AA 861 (1 μg site−1 i.d.), MK 886 (1 μg site−1 i.d.), or ethacrynic acid (1 μg site−1 i.d.) were co-injected with SP (100 pmol site−1; SP-induced oedema). The anti-inflammatory effects are expressed as per cent inhibition of the oedema volume compared to untreated animals (see Methods). Asterisks indicate significant differences in volumes of neurogenic or SP-induced oedema between treated and untreated animals. (*P<0.05, ***P<0.001 according to ANOVA, followed by Bonferroni's test for multiple comparisons).
Table 1.
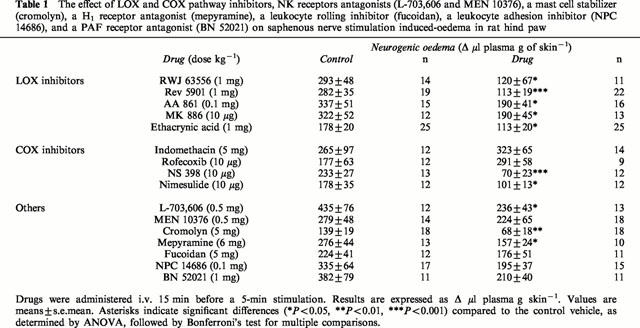
The effect of LOX and COX pathway inhibitors, NK receptors antagonists (L-703,606 and MEN 10376), a mast cell stabilizer (cromolyn), a H1 receptor antagonist (mepyramine), a leukocyte rolling inhibitor (fucoidan), a leukocyte adhesion inhibitor (NPC 14686), and a PAF receptor antagonist (BN 52021) on saphenous nerve stimulation induced-oedema in rat hind paw
We assessed the effect of the most potent LOX pathway inhibitor on the neurogenic vasodilatation induced by a 10-s electrical stimulation of the saphenous nerve. The results were compared to those obtained with the CGRP receptor antagonist, CGRP8 – 37, and the μ-opioid receptor agonist, DAMGO. These drugs were used to assess postjunctional and prejunctional inhibition of vasodilatation following electrical stimulation, respectively (Barber, 1993; Escott & Brain, 1993). MK 886 (10 μg kg−1 i.v.) did not affect basal cutaneous blood flow or the increase in blood flow induced by electrical stimulation (Figure 2a). This contrasted with the blockade observed if 0.3 mg kg−1 CGRP8 – 37 was injected i.v. (Figure 2b). MK 886 slightly, but not significantly, potentiated the neurogenic rise in microcirculatory blood flow (+38±24%, n=10), whereas CGRP8 – 37 and DAMGO significantly inhibited it (−76±6%, n=8; P<0.001 and −65±7%, n=8; P<0.001, respectively) (Figure 2c). CGRP8 – 37 and DAMGO did not affect cutaneous basal blood flow, suggesting that the C fibres do not control skin microcirculation in resting conditions.
Figure 2.
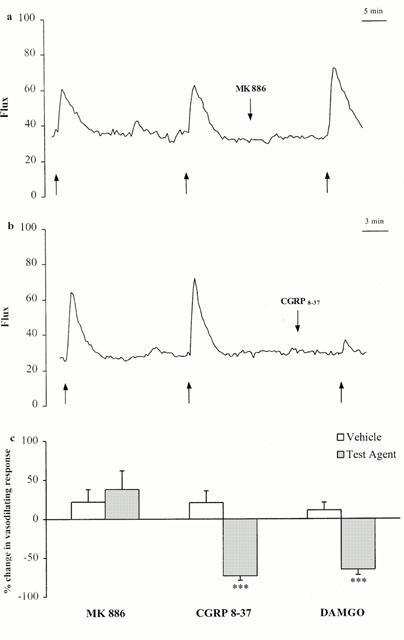
Comparison of the effects of the FLAP inhibitor, MK 886 (10 μg kg−1 i.v.), CGRP8 – 37 (0.3 mg kg−1 i.v.,) and the μ-opioid agonist, DAMGO (1 mg kg−1 i.v.), on neurogenic vasodilatation. (a, b) Representative blood flow traces obtained from individual experiments showing the effects of MK 886 and CGRP8 – 37 on neurogenic vasodilatation. Arrows (up) delineate the 10-s stimulation period. Arrows (down) indicate the time at which the test agents were injected. (c) Summarized results obtained with MK 886, CGRP8 – 37, and DAMGO. The amplitude of the increase in blood flow (in arbitrary units) induced by the third stimulation was compared with that induced by the second stimulation. The effects of the test agent and vehicle are expressed as per cent change±s.e.mean in neurogenic response between the second and third electrical stimulations. Asterisks indicate significant differences between the neurogenic increase in blood flow measured during the second and third stimulations (***P<0.001 according to ANOVA, followed by Bonferroni's test for multiple comparisons).
Alternatively, arachidonic acid may be converted by COX into PGH2, the precursor of PGE2 which is a vasodilating and proinflammatory prostaglandin. Thus, the COX pathway may be involved in neurogenic oedema, neurogenic vasodilatation, and SP-induced oedema particularly via PGE2-dependent mechanisms. We used indomethacin, a non-selective inhibitor of the constitutive (COX-1) and inducible (COX-2) isoforms of COX, to test this hypothesis (Figure 3a). Prior treatment with indomethacin (5 mg kg−1, i.v.) did not modify neurogenic oedema (Table 1). Similarly, co-injection of indomethacin and the neuropeptide did not prevent SP-induced oedema. Conflicting results were obtained with the extensively used selective COX-2 inhibitors, NS 398, nimesulide, and rofecoxib (Figure 3a). NS 398 and nimesulide, which are structurally similar, inhibited neurogenic oedema (Table 1). However, identical doses of the highly selective COX-2 inhibitor, rofecoxib, did not affect the neurogenic inflammatory response. Similar pharmacological profiles were observed for SP-induced oedema with these NSAIDs, although the inhibitory effects of nimesulide were not statistically significant. None of the COX inhibitors affected neurogenic vasodilatation (Figure 3b). We used RT – PCR to assess the expression of the COX-1 and COX-2 genes in the tissues involved in the neurogenic inflammatory response (Figure 3c). COX-1 mRNA levels were similar in ipsilateral and contralateral saphenous nerves and paw skin biopsy samples, before and after the 5-min stimulation of the saphenous nerve. COX-2 mRNA was not detected in the total RNA extracted from these samples, it was detected in total RNA samples extracted from rat brains which was used as a positive control for the constitutive expression of the COX-2 gene in neurons.
Figure 3.
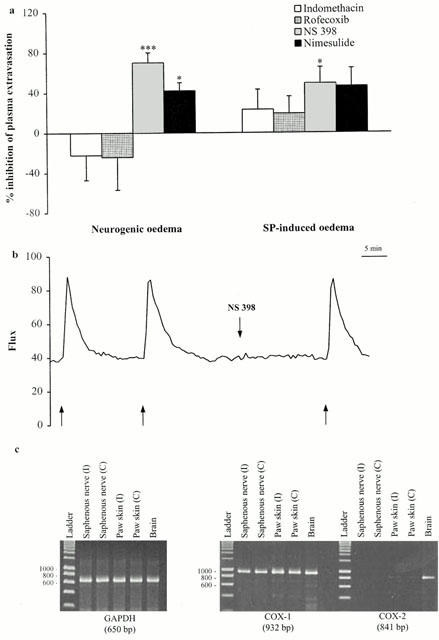
(a) Effect of COX pathway inhibitors on neurogenic oedema and SP-induced oedema. Indomethacin (5 mg kg−1 i.v.), rofecoxib (10 μg kg−1 i.v.), NS 398 (10 μkg−1 i.v.), or nimesulide (10 μg kg−1 i.v.) were injected 15 min before a 5-min stimulation (neurogenic oedema). Indomethacin (1 μg site−1 i.d.), rofecoxib (1 μg site−1 i.d.), NS 398 (1 μg site−1 i.d.), or nimesulide (3 μg site−1 i.d.) were co-injected with SP (100 pmol site−1; SP-induced oedema). The anti-inflammatory effects are expressed as per cent inhibition of the oedema volume measured in untreated animals (see Methods). Asterisks indicate significant differences in the volume of neurogenic or SP-induced oedema between treated and untreated animals. (*P<0.05, ***P<0.001 according to ANOVA, followed by Bonferroni's test for multiple comparisons). (b) A representative trace from an individual experiment showing the lack of effect of NS 398 (10 μg kg−1 i.v.) on neurogenic vasodilatation. Arrows (up) delineate the 10-s stimulation periods. Arrows (down) indicate the time at which the test agent was injected. (c) Analysis of COX-1 and COX-2 mRNA levels in rat paw skin and saphenous nerve by RT – PCR. Total RNA was extracted from the saphenous nerve and skin biopsy samples of ipsilateral (I) and contralateral (C) paws before and after a 5-min stimulation. COX-1 and COX-2 mRNA levels were studied by RT – PCR with specific rat COX-1 and COX-2 mRNA primers. Rat brain RNA was used as a positive control for COX-1 and COX-2.
We used a NK1 receptor antagonist, L-703,606, and a mast cell stabilizer, cromolyn, to assess the involvement of NK1 receptors and mast cells in neurogenic inflammation (Figure 4a). As expected, L-703,606 inhibited neurogenic and SP-induced oedema (Table 1). Conversely, the NK2 receptor antagonist, MEN 10376, had no effect on neurogenic oedema (Table 1). Prior treatment with cromolyn reduced neurogenic oedema (Table 1), but did not affect neurogenic vasodilatation (+8±16%, n=6, not significant; data not shown). The H1 receptor antagonist, mepyramine, which inhibits neurogenic and SP-induced oedema was used to confirm mast cell-dependent extravasation (Figure 4a). We assessed the effects of LOX and COX pathway inhibitors on compound 48/80-induced oedema, a mast cell activator, to determine whether leukotriene-mediated extravasation in neurogenic oedema resulted from SP-induced mast cell activation (Figure 4b). As expected, mepyramine inhibited compound 48/80-induced oedema (128±21 Δ μl plasma g of skin−1 (treated) vs 317±42 Δ μl plasma g of skin−1 (untreated), n=9 – 18, P<0.01). None of the LOX pathway inhibitors inhibited compound 48/80-induced extravasation, but ethacrynic acid significantly potentiated the inflammatory response (503±52 Δ μl plasma g of skin−1 (treated) vs 356±32 Δ μl plasma g of skin−1 (untreated), n=8 – 21, P<0.05). The pharmacological profile of COX inhibitors on compound 48/80-induced oedema was similar to those for neurogenic and SP-induced oedema. NS 398 and nimesulide inhibited this purely mast cell-dependent extravasation, whereas indomethacin and rofecoxib were had no effect. Intravenous injection of the polysaccharide, fucoidan at a dose known to inhibit PMN rolling (Yamaki et al., 1998), did not affect neurogenic oedema (Figure 4a). Co-injection of fucoidan with the inflammatory mediator did not affect SP-induced oedema, but significantly inhibited the effects of compound 48/80. Finally, pre-treatment with a combination of mepyramine (to block the mast cell-dependent component) and MK 886 (to block the leukotriene-dependent component) did not inhibit of neurogenic inflammation more than pre-treatment with mepyramine or MK 886 alone (46±12% inhibition, n=10 for pre-treatment with mepyramine and MK 886 together, 43±9% inhibition, n=10 for pre-treatment with mepyramine alone and 41±14% inhibition, n=13 for pre-treatment with MK 886 alone).
Figure 4.
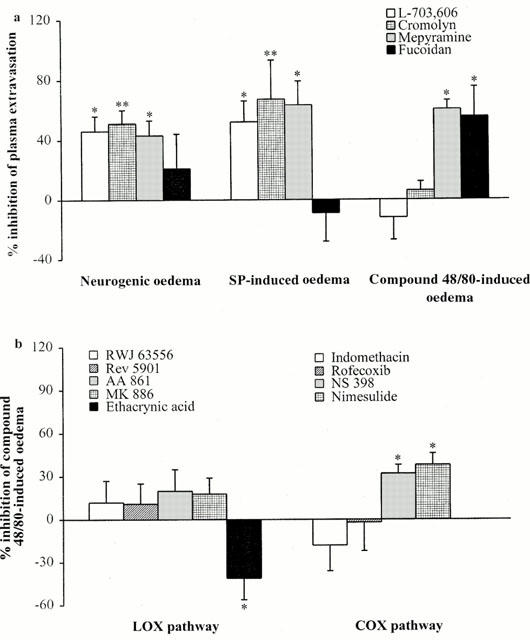
(a) Involvement of NK1 receptors, mast cells and PMN adhesion in neurogenic and SP-induced oedema. The NK1 receptor antagonist, L-703,606 (0.5 mg kg−1 i.v.), the mast cell stabilizer, cromolyn (5 mg kg−1 i.v.), the H1 receptor antagonist, mepyramine (6 mg kg−1 i.v.), or the polysaccharide fucoidan (5 mg kg−1 i.v.) were injected 15 min before a 5-min stimulation (neurogenic oedema). L-703,606 (1 μg site−1 i.d.), cromolyn (10 μg site−1 i.d.), mepyramine (1 μg site−1 i.d.), or fucoidan (10 μg site−1 i.d.) were co-injected with SP (100 pmol site−1 i.d., SP-induced oedema) or compound 48/80 (1 μg site−1 i.d.; compound 48/80-induced oedema). The anti-inflammatory effects are expressed as per cent inhibition of the oedema volume in untreated animals (see Methods). Asterisks indicate significant differences in volume of neurogenic or SP-induced oedema between treated and untreated animals. (*P<0.05, **P<0.01 according to ANOVA, followed by Bonferroni's test for multiple comparisons). (b) Effect of LOX and COX pathway inhibitors on compound 48/80-induced oedema. Drugs were co-injected with compound 48/80 (1 μg site−1 i.d.), at the same doses used in Figures 1 and 3 for SP-induced oedema. The anti-inflammatory effects are expressed as per cent inhibition of the oedema volume in untreated animals (see Methods). Asterisks indicate significant differences in the volume of compound 48/80-induced oedema between treated and untreated animals. (*P<0.05 according to ANOVA, followed by Bonferroni's test for multiple comparisons).
Discussion
The analysis of the differential effects of LOX and COX pathway inhibitors on oedema induced by electrical stimulation of the saphenous nerve and by the intradermal injection of SP suggests the involvement of LOX pathway in the development of a purely neurogenic oedema induced by short-term (5 min) antidromic electrical stimulation of afferent C fibres and excludes the participation of the COX pathway in this process.
Pre-treatment by intravenous injection of a direct LOX inhibitor (AA 861), a FLAP inhibitor (MK 886), a dual LOX/CysLt receptor antagonist (Rev 5901), or a dual COX/LOX inhibitor (RWJ 63556) significantly reduced the formation of neurogenic oedema. The CGRP-mediated neurogenic vasodilatation induced by stimulation of the saphenous nerve was not affected by LOX pathway inhibition thus, these effects cannot be due to the inhibition of neuropeptide release. In addition, the glutathione S-transferase inhibitor, ethacrynic acid, and Rev 5901 similarly inhibited the formation of neurogenic oedema, suggesting that CysLt mediates this LOX-dependent leakage. Consistent with this, LTC4 and LTD4 stimulate plasma leakage directly through the endothelium in hamster skin microcirculation, within minutes. These direct permeabilizing effects are antagonized by Rev 5901 (Gimeno et al., 1999). Our results suggest that the synthesis of SP-induced leukotriene may be involved in neurogenic oedema. RWJ 63556 and MK 886 significantly inhibited SP-induced oedema, however, all the other inhibitors of the LOX pathway either inhibited this inflammatory process, but not significantly, (20 – 40%) (AA 861 and Rev 5901) or potentiated it (ethacrynic acid). The potentiating effects of ethacrynic acid on SP- and compound 48/80-induced oedema may be due to the overall effects of this non-specific compound on glutathione metabolism. Our results suggest that leukotriene-dependent neurogenic extravasation is due to the release of SP from nerve endings, which results in the activation of LOX-rich inflammatory cells, such as mast cells and granulocytes.
Several points must be made concerning the control experiments used to assess the origin of the leukotriene-mediated component of neurogenic inflammation. As expected, the NK1 receptor antagonist, L-703,606, but not the NK2 receptor antagonist, MEN 10376, inhibited neurogenic and SP-induced oedema. L-703,606 did not affect the compound 48/80-induced oedema. Thus, neurogenic inflammation resulted from the release of SP from sensitive neurons; and primary afferent neurons and SP were not involved in the oedema produced by the direct activation of cutaneous mast cells. Conversely, mast cell-dependent neurogenic oedema (Maggi, 1997) was observed after exposure to the mast cell stabilizer, cromolyn. This agent significantly inhibited neurogenic and SP-induced oedema. Cromolyn may not have inhibited compound 48/80-induced oedema due to the high dose of compound 48/80 used; the dose of cromolyn being too low to prevent compound 48/80 from inducing extensive mast cell activation. It has been suggested that cromolyn stabilizes sensitive nerve endings and inhibits neuropeptide release (Dixon et al., 1980). In control experiments, the same dose of cromolyn that inhibited neurogenic oedema did not affect the CGRP-dependent neurogenic vasodilatation induced by electrical stimulation of the saphenous nerve. We conclude that, in our study, the effects of cromolyn on neurogenic oedema were mediated by preventing SP from activating mast cells rather than by a prejunctional stabilizing effect on sensitive neurons. The involvement of SP-mediated mast cell-dependent extravasation was further supported by the inhibitory effects of the histamine H1 receptor antagonist, mepyramine, on neurogenic and SP-induced oedema. This mast cell-dependent component of neurogenic inflammation is probably not dependent on the release of LOX products. Indeed, pre- treatment with LOX pathway inhibitors did not affect the inflammatory response that follows intradermal injection of compound 48/80. The effects of LOX inhibitors on neurogenic, SP- and compound 48/80-induced oedema suggest that the release of SP by sensitive neurons activates two different inflammatory cascades: a histamine-mediated mast cell-dependent extravasation and a leukotriene-mediated leakage. However, pre-treatment with both mepyramine and MK 886 did not inhibit the effects any more than the H1 receptor antagonist or the FLAP inhibitor alone. We cannot exclude the possibility of crosstalk between the mast cell-dependent and LOX-dependent components of neurogenic oedema.
SP promotes the production of adhesion molecules on cultured endothelial cells and subsequent granulocyte adhesion/activation in vitro (DeRose et al., 1994; Quinlan et al., 1999). In vivo, SP induces PMN infiltration and associated extravasation in the skin of guinea-pigs, several hours after intradermal injection (Walsh et al., 1995). This effect is probably mediated by the potent chemotactic lipoxygenase product, LTB4 (Iwamoto et al., 1993). This PMN adhesion/activation may account for the leukotriene-dependent extravasation that follows a 5-min electrical stimulation of the saphenous nerve, and possibly after the intradermal injection of SP. To determine whether this was the case we used a dose of fucoidan that is known to inhibit PMN rolling (Yamaki et al., 1998). Fucoidan did not significantly reduce neurogenic or SP-induced oedema. Similarly, NPC 14686, an which inhibits the adhesion of neutrophils to the endothelium, did not significantly inhibit neurogenic oedema (Table 1). Therefore, SP-induced neutrophil adhesion does not seem to play a major role in the leukotriene-mediated extravasation observed after a 5-min stimulation of the saphenous nerve. The absence of a significant effect of fucoidan on neurogenic and SP-induced oedema appear to conflict with its significant effects on compound 48/80-induced inflammation. It is possible that, in contrast to the extensive mast cell activation induced by compound 48/80, the mast cell activation induced by a 5-min electrical stimulation of the saphenous nerve or intradermal injection of a low dose of SP was not sufficient to promote PMN adhesion to the endothelium. Walsh et al. (1995) demonstrated that the intradermal injection of less than 1×10−10 mole site−1 SP induces oedema without granulocyte marginalization, whereas higher doses induce concomitant oedema formation and PMN accumulation in guinea-pig skin. PMN adhesion is not required for leukotriene-mediated extravasation in neurogenic oedema, however, direct priming of circulating neutrophils by SP may be involved in this LOX-dependent extravasation. The direct activation of endothelial cells by SP may also induce the rapid release of the potent neutrophil primer, platelet activating factor (PAF), from intracellular stores. This is supported by the inhibitory effects of the PAF receptor antagonist, BN 52021, on neurogenic oedema (Table 1). Finally, SP has been shown to activate the LOX pathway and to inhibit the COX pathway in isolated human platelets (Gecse et al., 1999).
The electrical stimulation of primary afferents induces the concomitant accumulation of CGRP and PGE2 in rat skin (Kress et al., 1999). Thus, the pro-inflammatory and vasodilating effects of PGE2 may be involved in the development of neurogenic oedema and vasodilatation. Pre-treatment with the non-specific inhibitor of the constitutive and inducible forms of COX, indomethacin, did not significantly affect neurogenic, SP-, or compound 48/80-induced oedema, thus the COX pathway is not involved in these extravasation mechanisms. Similarly, indomethacin did not affect the vasodilatation response to electrical stimulation of the saphenous nerve. The widely used, structurally similar, specific inhibitors of the inducible cyclooxygenase, NS 398 and nimesulide, significantly inhibited these inflammatory processes. These results were surprising because the short-term nature of the experiments should have precluded de novo synthesis of COX-2 in the skin. The RT – PCR experiments demonstrated that COX-1 mRNA levels were identical in the skin of the stimulated and contralateral hind paws and remained constant before and after electrical stimulation of the saphenous nerve. COX-2 mRNA was not detected in the skin biopsy tissue before or after electrical stimulation. One explanation for the inhibitory effects of NS 398 and nimesulide on neurogenic oedema is that constitutive neuronal COX-2 activity may stimulate neuropeptide release at nerve endings. However, this is unlikely to be the case for the following reasons: (1) NS 398 and nimesulide did not affect the neurogenic vasodilatation response, suggesting that CGRP release was not inhibited; (2) RT – PCR experiments detected lower levels of COX-2 mRNA than of COX-1 mRNA in nervous tissues, except for the brain; and (3) the highly specific COX-2 inhibitor, rofecoxib, did not display similar inhibitory effects to the same doses of NS 398 and nimesulide. Thus, the anti-inflammatory effects of NS 398 or nimesulide, which may be attributable to effects other than specific COX-2 inhibition, should be interpreted with caution.
These results clearly demonstrate that arachidonic acid metabolism is involved in an acute peripheral neurogenic inflammation model and that a SP-dependent LOX pathway activation is implicated in this inflammatory process. Furthermore, this study underlines the importance of histamine release from mast cells in neurogenic inflammation in rat paw skin. The mast cell- and LOX-dependent components appear to be equally implicated in neurogenic inflammation. However, the possibility of crosstalk between both of them needs to be investigated. This may be of crucial importance for determining the pathophysiological role of primary afferent neurons in acute and chronic inflammatory disorders that sensitive neurons may be involved in (for review, Brain & Moore, 1999).
Acknowledgments
Rofecoxib and RWJ 63556 were from the Department of Chemistry, Laboratoire Innothéra.
Abbreviations
- AA 861
2- (12 -hydroxydodeca -5,10 -diynyl) -3,5,6 -trimethyl -p -benzoquinone
- BN 52021
ginkgolide B
- CGRP
calcitonin gene-related peptide
- COX
cyclo-oxygenase
- CysLt
cysteinyl-leukotriene
- DAMGO
[D-Ala2, N-Me-Phe4, Gly-ol]-enkephalin
- DMSO
dimethyl sulphoxide
- FLAP
five-lipoxygenase activating protein
- GADPH
glyceraldehyde-3-phosphate dehydrogenase
- i.d.
intradermal
- i.v.
intravenous
- L-703,606
cis-2-(diphenylmethyl)-N-[(2-iodophenyl)methyl]-1-azabicyclo[2.2.2]octan-3-amine oxalate
- LT
leukotriene
- LOX
lipoxygenase
- MEN 10376
Asp-Tyr-D-Trp-Val-D-Trp-D-Trp-Lys-NH2
- MK 886
3-[1-(p-chlorobenzyl)-5-(isopropyl)-3-t-butylthioindol-2-yl]-2,2-dimethyl-propanoic acid
- NK
neurokinin
- Nimesulide
N-[4-Nitro-2-phenoxyphenyl]-methanesulphonamide
- NPC 14686
N-(fluorenyl-9-methoxycarbonyl)-L-homophenylalanine
- NS 398
N-[2-(cyclohexyloxy)-4-nitrophenyl]-methanesulphonamide
- PAF
platelet activating factor
- PCR
polymerase chain reaction
- PG
prostaglandin
- PMN
polymorphonuclear leukocyte
- Rev 5901
α-pentyl-3-(2-quinolinylmethoxy)-benzenemethanol
- Rofecoxib
4-(4-methylsulphonylphenyl)-3-phenyl-2-(5H)-furanone
- RT – PCR
reverse transcription-polymerase chain reaction
- RWJ 63556
N-[5-(4-fluorophenoxy)-2-thienyl] methanesulphonamide
- SP
substance P
References
- BALUK P., HIRATA A., THURSTON G., FUJIWARA T., NEAL C.R., MICHEL C.C., MCDONALD D.M. Endothelial gaps: time course of formation and closure in inflamed venules of rats. Am. J. Physiol. 1997;272:L155–L170. doi: 10.1152/ajplung.1997.272.1.L155. [DOI] [PubMed] [Google Scholar]
- BARBER A. μ- and κ-opioid receptor agonists produce peripheral inhibition of neurogenic plasma extravasation in rat skin. Eur. J. Pharmacol. 1993;236:113–120. doi: 10.1016/0014-2999(93)90233-8. [DOI] [PubMed] [Google Scholar]
- BEACH V.L., STEINETZ B.G. Quantitative measurement of Evans Blue space in the tissues of the rat: influence of 5-hydroxytryptamine antagonists and phenelzine on experimental inflammation. J. Pharmacol. Exp. Ther. 1961;131:400–406. [PubMed] [Google Scholar]
- BELL R.L., HARRIS R.R. The enzymology and pharmacology of 5-lipoxygenase and 5-lipoxygenase activating protein. Clin. Rev. Allergy Immunol. 1999;17:91–109. doi: 10.1007/BF02737599. [DOI] [PubMed] [Google Scholar]
- BRAIN S.D., MOORE P.K. Pain and Neurogenic Inflammation. Basel: Birkhäuser Verlag; 1999. [Google Scholar]
- BURCH R.M., WEITZBERG M., BLOK N., MUHLHAUSER R., MARTIN D., FARMER S.G., BATOR J.M., CONNOR J.R., GREEN M., KO C. N-(fluorenyl-9-methoxycarbonyl) amino acids, a class of antiinflammatory agents with a different mechanism of action. Proc. Natl. Acad. Sci. U.S.A. 1991;88:355–359. doi: 10.1073/pnas.88.2.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEROSE V., ROBBINS R.A., SNIDER R.M., SPURZEM J.R., THIELE G.M., RENNARD S.I., RUBINSTEIN I. Substance P increases neutrophil adhesion to bronchial epithelial cells. J. Immunol. 1994;152:1339–1346. [PubMed] [Google Scholar]
- DIXON M., JACKSON D.M., RICHARDS I.M. The action of sodium cromoglycate on ‘C' fibre endings in the dog lung. Br. J. Pharmacol. 1980;70:11–13. doi: 10.1111/j.1476-5381.1980.tb10898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ESCOTT K.J., BRAIN S.D. Effect of a calcitonin gene-related peptide antagonist (CGRP 8–37) on skin vasodilatation and oedema induced by stimulation of the rat saphenous nerve. Br. J. Pharmacol. 1993;110:772–776. doi: 10.1111/j.1476-5381.1993.tb13878.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FRODE-SALEH T.S., CALIXTO J.B., MEDEIROS Y.S. Analysis of the inflammatory response induced by substance P in the mouse pleural cavity. Peptides. 1999;20:259–265. doi: 10.1016/s0196-9781(98)00170-3. [DOI] [PubMed] [Google Scholar]
- GAMSE R., SARIA A. Antidromic vasodilatation in the rat hindpaw measured by laser Doppler flowmetry: pharmacological modulation. J. Auton. Nerv. Syst. 1987;19:105–111. doi: 10.1016/0165-1838(87)90003-8. [DOI] [PubMed] [Google Scholar]
- GECSE A., KIS B., MEZEI Z., TELEGDY G. Effects of inflammatory neuropeptides on the arachidonate cascade of platelets. Int. Arch. Allergy Immunol. 1999;118:166–170. doi: 10.1159/000024057. [DOI] [PubMed] [Google Scholar]
- GIMENO G., CARPENTIER P.H., DESQUAND-BILLIALD S., FINET M., HANF R. Respective role of lipoxygenase and nitric oxide-synthase pathways in plasma histamine-induced macromolecular leakage in conscious hamsters. Br. J. Pharmacol. 1999;126:1801–1809. doi: 10.1038/sj.bjp.0702380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IWAMOTO I., TOMOE S., TOMIOKA H., YOSHIDA S. Leukotriene B4 mediates substance P-induced granulocyte infiltration into mouse skin. Comparison with antigen-induced granulocyte infiltration. J. Immunol. 1993;151:2116–2123. [PubMed] [Google Scholar]
- KRESS M., GUTHMANN C., AVERBECK B., REEH P.W. Calcitonin gene-related peptide and prostaglandin E2 but not substance P release induced by antidromic nerve stimulation from rat skin in vitro. Neuroscience. 1999;89:303–310. doi: 10.1016/s0306-4522(98)00280-2. [DOI] [PubMed] [Google Scholar]
- KUBES P., KANWAR S. Histamine induces leukocyte rolling in post-capillary venules: a P-selectin-mediated event. J. Immunol. 1994;152:3570–3577. [PubMed] [Google Scholar]
- LEMBECK F., HOLZER P. Substance P as neurogenic mediator of antidromic vasodilatation and neurogenic plasma extravasation. Naunyn-Schmiedeberg's Arch. Pharmacol. 1979;310:175–183. doi: 10.1007/BF00500282. [DOI] [PubMed] [Google Scholar]
- LEUNG K.H. Selective inhibition of leukotriene C4 synthesis in human neutrophils by ethacrynic acid. Biochem. Biophys. Res. Commun. 1986;137:195–200. doi: 10.1016/0006-291x(86)91195-2. [DOI] [PubMed] [Google Scholar]
- MAGGI C.A. The effects of tachykinins on inflammatory and immune cells. Regul. Pept. 1997;70:75–90. doi: 10.1016/s0167-0115(97)00029-3. [DOI] [PubMed] [Google Scholar]
- PAIRET M., ENGELHARDT G. Distinct isoforms (COX-1 and COX-2) of cyclooxygenase: possible physiological and therapeutic implications. Fundam. Clin. Pharmacol. 1996;10:1–15. doi: 10.1111/j.1472-8206.1996.tb00144.x. [DOI] [PubMed] [Google Scholar]
- QUINLAN K.L., SONG I.S., NAIK S.M., LETRAN E.L., OLERUD J.E., BUNNETT N.W., ARMSTRONG C.A., CAUGHMAN S.W., ANSEL J.C. VCAM-1 expression on human dermal microvascular endothelial cells is directly and specifically up-regulated by substance P. J. Immunol. 1999;162:1656–1661. [PubMed] [Google Scholar]
- REGOLI D., NGUYEN Q.T., CALO G.Pharmacology of Tachykinins Neurogenic Inflammation 1996Boca Raton: CRC Press; 91–99.ed. Geppetti, P. & Holzer, P. pp [Google Scholar]
- WALSH D.T., WEG V.B., WILLIAMS T.J., NOURSHARGH S. Substance P-induced inflammatory responses in guinea-pig skin: the effect of specific NK1 receptor antagonists and the role of endogenous mediators. Br. J. Pharmacol. 1995;114:1343–1350. doi: 10.1111/j.1476-5381.1995.tb13354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAMAKI K., LINDBOM L., THORLACIUS H., HEDQVIST P., RAUD J. An approach for studies of mediator-induced leukocyte rolling in the undisturbed microcirculation of the rat mesentery. Br. J. Pharmacol. 1998;123:381–389. doi: 10.1038/sj.bjp.0701617. [DOI] [PMC free article] [PubMed] [Google Scholar]