

Abstract
Long-term (⩾12 h) treatment of cultured bovine adrenal chromaffin cells with A23187 (a Ca2+ ionophore) or thapsigargin (TG) [an inhibitor of sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA)] caused a time- and concentration-dependent reduction of cell surface [3H]-saxitoxin (STX) binding capacity, but did not change the KD value. In A23187- or TG-treated cells, veratridine-induced 22Na+ influx was reduced (with no change in veratridine EC50 value) while it was enhanced by α-scorpion venom, β-scorpion venom, or Ptychodiscus brevis toxin-3, like in nontreated cells.
The A23187- or TG-induced decrease of [3H]-STX binding was diminished by BAPTA-AM. EGTA also inhibited the decreasing effect of A23187. A23187 caused a rapid, monophasic and persistent increase in intracellular concentration of Ca2+ ([Ca2+]i) to a greater extent than that observed with TG. 2,5-Di-(t-butyl)-1,4-benzohydroquinone (DBHQ) (an inhibitor of SERCA) produced only a rapid monophasic increase in [Ca2+]i, without any effect on [3H]-STX binding.
Reduction in [3H]-STX binding capacity induced by A23187 or TG was attenuated by Gö6976 (an inhibitor of conventional protein kinase C) or calpastatin peptide (an inhibitor of calpain). When the internalization rate of cell surface Na+ channels was measured in the presence of brefeldin A (an inhibitor of vesicular exit from the trans-Golgi network), A23187 or TG accelerated the reduction of [3H]-STX binding capacity.
Six hours treatment with A23187 lowered Na+ channel α- and β1-subunit mRNA levels, whereas TG had no effect.
These results suggest that elevation of [Ca2+]i caused by A23187, TG or DBHQ exerted differential effects on down-regulation of cell surface functional Na+ channels and Na+ channel subunit mRNA levels.
Keywords: Sodium channel, down-regulation, calcium, A23187, thapsigargin, [3H]-saxitoxin binding, Northern blot, 22Na+ influx, protein kinase C, calpain
Introduction
Voltage-dependent Na+ channels consist of the principal α-subunit (∼260 kDa) which may be associated with a noncovalently-attached β1-subunit (∼36 kDa) and a disulfide-linked β2-subunit (∼33 kDa) (Catterall, 1992; Isom et al., 1994). The α-subunits issued from a large multigene family (Dietrich et al., 1998) contain the ion-pore and the toxin binding sites (site 1 for tetrodotoxin (TTX) and saxitoxin (STX); site 2 for veratridine; site 3 for α-scorpion toxin; site 4 for β-scorpion toxin; and site 5 for Ptychodiscus brevis toxin) (Catterall, 1992). In contrast, the β1-subunits are structurally homologous among various tissues (Makita et al., 1994; Oh & Waxman, 1994); the β2-subunit from brain is the only one which has been sequenced so far (Isom et al., 1995).
The density of cell surface Na+ channels is crucial to regulating the development and differentiation of neurons (Van Huizen et al., 1985; Mourre et al., 1987; Toledo-Aral et al., 1995), retina (Miguel-Hidalgo et al., 1995) and skeletal myocytes (Linsdell & Moody, 1994). Fluctuation of Na+ channel density has been described in pathological states such as neuropathy (Brismar, 1993), epilepsy (Sashihara et al., 1994) and ischaemia or hypoxia (Xia & Haddad, 1994; 1999; Urenjak & Obrenovitch, 1996).
Because spatiotemporal-specific heterogeneous increases of intracellular concentration of Ca2+ ([Ca2+]i) differentially modulate genetic and phenotypic responses (Ginty, 1997; Malviva & Rogue, 1998), various agents raising [Ca2+]i have been investigated for their ability to alter Na+ channel levels and their subunit mRNAs. Chronic (1 – 8 days) treatment with Ca2+ ionophore A23187 or depolarizing concentrations of K+ reduced the density of [3H]-STX binding sites (Sherman & Catterall, 1984; Sherman et al., 1985; Brodie et al., 1989) and Na+ currents (Satoh et al., 1992; Chiamvimonvat et al., 1995), as well as Na+ channel α-subunit mRNA levels (Offord & Catterall, 1989; Duff et al., 1992) in skeletal and cardiac myocytes. Treatment of neuroblastoma cells with A23187 (1 μM for 2 – 3 days) decreased Na+ currents and α-subunit mRNA levels (Hirsh & Quandt, 1996), whereas A23187 treatment had no effect on β1- and β2-subunit mRNA levels in astrocytes and neuroblastoma cells (Oh et al., 1997). Previous studies did not, however, analyse [Ca2+]i transients in relation with Na+ channel expression. In addition, little is known about the mechanisms whereby [Ca2+]i regulates cell surface expression of Na+ channels.
In adrenal chromaffin cells (embryologically derived from the neural crest), the Na+ channel α-subunit is homologous to the TTX- and STX-sensitive human neuroendocrine type Na+ channel α-subunit (hNE-Na) (Klugbauer et al., 1995). Adrenal chromaffin cells possess sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA)-2b and -3 isozymes localized in the endoplasmic reticulum (Mathiasen et al., 1993; Poulsen et al., 1995; Alonso et al., 1999) whereby cytosolic Ca2+ is taken up into Ca2+ store associated with inositol 1,4,5-trisphosphate receptor- and ryanodine receptor-coupled Ca2+ release channels (Cheek & Thastrup, 1989; Robinson & Burgoyne, 1991a,1991b; Robinson et al., 1992). Thapsigargin (TG) and 2,5-di-(t-butyl)-1,4-benzohydroquinone (DBHQ), two inhibitors of SERCA but not of the plasma membrane Ca2+-ATPase (Cheek & Thastrup, 1989; Robinson & Burgoyne, 1991a,1991b; Robinson et al., 1992), and A23187 (Heldman et al., 1996), as well as muscarinic receptor agonists that increase inositol 1,4,5-trisphosphate (Cheek & Thastrup, 1989), have previously been shown to raise [Ca2+]i in adrenal chromaffin cells. In the present study, we examined whether treatment of adrenal chromaffin cells with either A23187, TG, or DBHQ could alter [Ca2+]i, [3H]-STX binding, and Na+ channel α- and β1-subunit mRNA levels. Because either α- or β-scorpion venom, or Ptychodiscus brevis toxin-3 (PbTx-3) cooperatively enhance veratridine-induced 22Na+ influx in adrenal chromaffin cells (Wada et al., 1987; 1992), Na+ channel function was characterized by these distinct classes of venoms and toxins.
Methods
Materials
Eagle's minimum essential medium was from Nissui Seiyaku, Tokyo, Japan; calf serum from Nacalai Tesque, Kyoto, Japan; A23187 from Wako Junyaku, Tokyo, Japan; DBHQ from Biomol Research Laboratories, Plymouth Meeting, PA, U.S.A.; calpastatin peptide and Gö6976 from Calbiochem-Novabiochem Corp., San Diego, CA, U.S.A. TG, BAPTA-AM, EGTA, cytosine arabinoside, veratridine, α-scorpion venom (Leiurus quinquestriatus quinquestriatus), β-scorpion venom (Centruroides sculpturatus), ouabain, TTX, and brefeldin A (BFA) were from Sigma, St. Louis, MO, U.S.A.; PbTx-3 from Latoxan, Westbury, NY, U.S.A.; fura-2/AM from Dojindo, Kumamoto, Japan; TRIzol reagent from Life Technologies, Rockville, MD, U.S.A.; oligotex-dT30<Super> from Nippon Roche Co., Tokyo, Japan; BcaBEST labelling kit from Takara, Kyoto, Japan; [3H]-STX (20 – 40 Ci mmol−1), 22NaCl (6 – 17 Ci mmol−1), and [α-32P]-dCTP (4000 Ci mmol−1), and 1-quinuclidinyl(phenyl-4-3H)benzilate ([3H]-QNB) (30 – 60 Ci mmol−1) from Amersham, Buckinghamshire, U.K.; cDNA for human glyceraldehyde 3-phosphate dehydrogenase (GAPDH) from Clontech Laboratories, Palo Alto, CA, U.S.A. Plasmids containing hNE-Na (Klugbauer et al., 1995), and rat brain Na+ channel β1-subunit (Oh & Waxman, 1994) were generously donated by Dr F. Hofmann (Technischen Universität München) and Dr Y. Oh (University of Alabama), respectively.
Primary culture of adrenal chromaffin cells and drug treatment
Isolated bovine adrenal chromaffin cells were cultured (4×106 dish−1, Falcon; 35 mm in diameter) in Eagle's minimum essential medium supplemented with 10% calf serum under 5%O2 and 95% air in a CO2 incubator (Wada et al., 1985a,1985b). For measurement of [Ca2+]i cells were plated at the density of 4×105 cells per 35 mm culture dish on 25 mm cover glass, and cultured as described (Tanaka et al., 1998). At 60 – 62 h after plating, cells were treated with fresh medium (with or without A23187, TG, or DBHQ for up to 96 h) and test medium was changed after 48 h. A23187, TG and DBHQ were dissolved in dimethyl sulphoxide (DMSO), DMSO final concentration in the test medium being ∼0.2%. When the effects of EGTA, BAPTA-AM, Gö6976, calpastatin peptide and BFA were examined, these compounds were included in the medium. The culture medium contained 3 μM cytosine arabinoside to inhibit proliferation of nonchromaffin cells; when chromaffin cells were purified by differential plating (Yanagita et al., 2000), concentration-response curve of veratridine for 22Na+ influx was not modified, as compared to that obtained in cells plated using conventional method.
Treatment of adrenal chromaffin cells with A23187 (1 μM for 96 h), TG (100 nM for 96 h), DBHQ (100 μM for 96 h), EGTA (5 mM for 24 h) or BAPTA-AM (50 μM for 24 h) did not impair cell viability; more than 95% of nontreated and drug-treated cells were viable, as evidenced from the trypan blue exclusion test. In addition, carbachol (100 μM for 1 min)-induced 22Na+ influx via nicotinic receptors (Wada et al., 1985b; 1986) was comparable between nontreated and A23187- or TG-treated cells. Binding of [3H]-QNB, an antagonist of muscarinic receptors, was similar between nontreated (KD=34±2.9 pM, Bmax=52.8±3.6 fmol mg−1 protein) and A23187 (1 μM for 48 h)-treated cells (KD=30±2.2 pM, Bmax=50.9±2.6 fmol mg−1 protein) (n=3), KD and Bmax values being close to those previously reported for adrenal chromaffin cells (Ballesta et al., 1989). Briefly, nontreated and A23187-treated cells were washed, homogenized at 4°C in 50 mM Tris-HCl buffer (pH 7.4) using a Polytron (10 s×four times), and centrifuged at 900×g for 5 min. The supernatant was centrifuged at 30,000×g for 15 min; the resultant pellet was washed, suspended in buffer, and used to assay 0.01 – 1 nM [3H]-QNB binding at 37°C for 1 h, as reported previously (Ballesta et al., 1989). Bound and free ligand was separated by filtration (GF/C filter, Whatman); the filter was washed with buffer, and radioactivity was measured.
[3H]-STX binding
Cells were washed with ice-cold Krebs-Ringer phosphate (KRP) buffer (mM: NaCl 154, KCl 5.6, MgSO4 1.1, CaCl2 2.2, NaH2PO4 0.85, Na2HPO4 2.15, dextrose 5, and 0.5% bovine serum albumin (BSA), pH 7.4) and incubated with 0.5 – 25 nM [3H]-STX in 1 ml KRP buffer at 4°C for 15 min in the absence (total binding) and presence (nonspecific binding) of 1 μM TTX. The cells were then immediately washed and solubilized with 10% Triton X-100. Radioactivity was then measured. Specific binding was calculated as the total binding minus nonspecific binding.
22Na+ influx
Cells were incubated with 2 μCi 22NaCl at 37°C for 5 min in 1 ml KRP buffer with or without veratridine, α- and β-scorpion venom, PbTx-3, or ouabain. Cells were then washed, solubilized, and counted for radioactivity. Previous electrophysiological and 22Na+ influx studies have shown that venoms from Leiurus quinquestriatus quinquestriatus (Catterall, 1976) and from Centruroides sculpturatus (Meves et al., 1982) exert effects similar to those obtained with their major α- and β-scorpion toxin, respectively.
mRNA isolation and electrophoresis
Total RNA was isolated from cells by acid guanidine thiocyanate-phenol-chloroform extraction using TRIzol reagent. Poly(A)+ RNA was purified by oligotex-dT30<super>, separated by electrophoresis on 1% agarose gel containing 6.3% formaldehyde in buffer (40 mM 3-(N-morpholino)propanesulphonic acid, pH 7.2, 0.5 mM EDTA, and 5 mM sodium citrate), transferred to a nylon membrane (Hybond-N, Amersham) in 20× saline-sodium citrate (SSC; 1×SSC=0.15 M NaCl and 0.015 M sodium citrate) overnight, and cross-linked using UV cross-linker (Funakoshi, Tokyo, Japan).
Northern blot
cDNA fragments for hNE-Na [nucleotides (nt) 435 – 2666] and β1-subunit (nt 457 – 790) obtained according to Yanagita et al. (2000), as well as GAPDH cDNA (1.1 Kbp) were labelled with [α-32P]-dCTP using the BcaBEST labelling kit. The membrane was prehybridized and hybridized with hNE-Na probe for 18 h at 42°C in 6×SSC, 10×Denhardt's (2% BSA fraction V, 2% polyvinylpyrrolidone, and 2% Ficoll 400), 50% formamide, 0.5% sodium dodecyl sulphate (SDS) and 50 μg ml−1 salmon sperm DNA. Membrane was then washed in 2×, 1×, 0.2×SSC containing 0.1% SDS, each for 30 min twice, and subjected to autoradiography. The same membrane was sequentially hybridized with probes for β1-subunit and GAPDH after being thoroughly washed to remove the former probe in 0.1% SDS at 100°C. The autoradiogram was quantified by a Bioimage analyser BAS2000 (Fuji Film, Tokyo, Japan). Abundance of α- and β1-subunit mRNAs was normalized against that of GAPDH mRNA.
Measurement of [Ca2+]i
Cultured cells were preincubated at 35°C for 1 h in HEPES-buffered solution (HBS) (mM: NaCl 140, KCl 5, CaCl2 2, MgCl2 1, HEPES 10, dextrose 10, and 0.5% BSA, pH 7.4) containing 3 μM fura-2/AM, as reported previously (Tanaka et al., 1998).
Fluorescence was measured in single cells with a Ca2+-imaging system equipped with an intensified CCD camera (Quanticell/700, JEOL, Tokyo, Japan) in the perfusion chamber; cells were continuously perfused with HBS at a flow rate of 1.0 ml min−1 in the absence or presence of A23187, TG or DBHQ. Ca2+-free solution was a modified HBS containing 1 mM EGTA without CaCl2 and MgCl2. [Ca2+]i was calculated from the ratio of fluorescence intensities obtained at 510 nm with dual excitation at 340 and 380 nm, using the equation of Grynkiewicz et al. (1985).
Statistical method
All experiments were repeated at least three times (mean±s.e.mean). Significance (P<0.05) was determined by one-way or two-way analysis of variance with post hoc mean comparison using the Newman-Keuls multiple range test. Student's t-test was used when two means of group were compared.
Results
[3H]-STX binding to adrenal chromaffin cells treated with A23187, TG or DBHQ
Treatment of chromaffin cells with 1 μM A23187 decreased [3H]-STX binding in a time-dependent manner, reaching 66% reduction at 96 h (Figure 1A). Treatment with 100 nM TG caused a time-dependent reduction of [3H]-STX binding, which leveled off (35%) at 48 h. In contrast, treatment with 10 or 100 μM DBHQ for 24, 48 or 96 h did not affect [3H]-STX binding.
Figure 1.
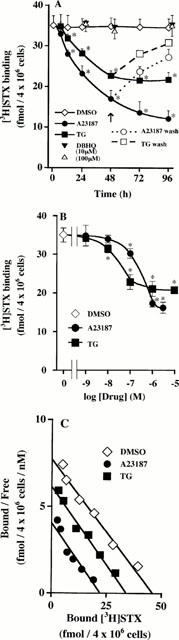
Effects of treatment with A23187, TG, and DBHQ on [3H]-STX binding capacity in adrenal chromaffin cells. (A) Cells were treated for up to 96 h with DMSO, 1 μM A23187, 100 nM TG, 10 μM or 100 μM DBHQ, then washed with KRP buffer, and tested for [3H]-STX binding assay. In a parallel experiment, cells exposed for 48 h to 1 μM A23187 or 100 nM TG were washed with culture medium (indicated by arrow), then incubated without A23187 or TG, and subjected to [3H]-STX binding assay at 72 and 96 h. Mean±s.e.mean (n=5). *P<0.05, significant decrease by A23187 or TG compared to DMSO-treated cells. (B) Cells were treated for 48 h with DMSO, A23187 or TG at indicated concentrations, and tested for [3H]-STX binding ability. Mean±s.e.mean (n=5). *P<0.05, significant decrease by A23187 or TG. (C) Scatchard plot of [3H]-STX binding to cells treated with DMSO, 1 μM A23187 or 100 nM TG for 48 h. Data are representative of one experiment from three independent experiments with similar results.
Treatment of cells with 1 μM A23187 for 12 and 24 h lowered [3H]-STX binding by 23 and 32% (Figure 1A); in contrast, when chromaffin cells were first treated with 1 μM A23187 for 6 h, then washed with culture medium and further incubated without A23187 for up to 24 h, [3H]-STX binding measured at 12 (34.5±0.9 fmol 4×106 cells−1) and 24 h (35.2±0.6 fmol 4×106 cells−1) was comparable to that measured in nontreated cells at 12 (35.0±1.1 fmol 4×106 cells−1) and 24 h (34.6±1.2 fmol 4×106 cells−1) (n=3). Treatment of cells with 100 nM TG decreased [3H]-STX binding by 8, 19 and 35% at 12, 24 and 48 h (Figure 1A). When cells were exposed to 1 μM A23187 or 100 nM TG for 48 h, subsequent washout and additional incubation in the absence of drug restored [3H]-STX binding values to 79 or 89% of control value at 96 h, respectively. As shown in Figure 1B, treatment of cells for 48 h with A23187 or TG produced a concentration-dependent decline of [3H]-STX binding capacity with EC50 values of 281.4 or 35.1 nM, respectively. Scatchard analysis (Figure 1C) revealed that A23187 (1 μM for 48 h) or TG (100 nM for 48 h) lowered Bmax values from 48.8±1.3 to 21.6±0.6 or 32.2±3.5 fmol 4×106 cells−1, without modifying KD values (5.3±0.1 nM, nontreated cells; 5.5±0.5 nM, A23187-treated cells; 5.9±0.4 nM, TG-treated cells; n=3).
Effects of A23187 and TG treatment on Na+ influx evoked by veratridine, α- and β-scorpion venom or PbTx-3
Toxins that bind to distinct sites on the Na+ channel α-subunit are useful probes to characterize pharmacological and structural properties of Na+ channel isoforms (Catterall, 1980; 1992). In adrenal chromaffin cells, we have previously shown that veratridine, a toxin acting on site 2 in segment 6 of domain I (S6I) of Na+ channel α-subunit (Trainer et al., 1996), caused a sustained influx of 22Na+ for at least 5 min. Nanomolar concentrations of TTX or STX (Wada et al., 1985a,1985b; 1987), toxins that bind to the site 1 at the extracellular loop between S5I and S6I (Noda et al., 1989; Satin et al., 1992), inhibited the effect of veratridine. Incubation of A23187- (1 μM for 48 h) or TG- (100 nM for 48 h) treated adrenal chromaffin cells with veratridine (1 – 560 μM) for 5 min caused inhibition of veratridine (⩾10 μM)-induced 22Na+ influx, as compared with nontreated cells (Figure 2A,B). The maximum influx of 22Na+ was reduced by 61 or 48% in A23187- or TG-treated cells, but EC50 values for veratridine were not significantly changed (77.5±7.2 μM, nontreated cells; 83.1±5.0 μM, A23187-treated cells; 84.2±6.4 μM, TG-treated cells; n=5). In adrenal chromaffin cells, Na+ influx increases Na+, K+-ATPase activity, so intracellular Na+ is continuously pumped out (Wada et al., 1985a; 1986). Our present study (Figure 2A,B) shows that even in the presence of ouabain (100 μM), a concentration that completely inhibits Na+,K+-ATPase activity (Wada et al., 1986), veratridine-induced 22Na+ influx was significantly reduced in A23187- or TG-treated cells, as compared to nontreated cells.
Figure 2.
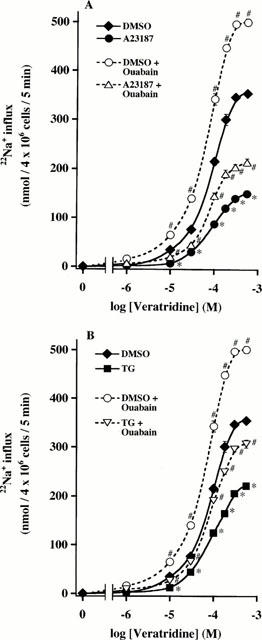
Treatment of adrenal chromaffin cells with A23187 and TG: effects on veratridine-induced 22Na+ influx in the absence and presence of ouabain. Cells were treated for 48 h with DMSO, (A) 1 μM A23187 or (B) 100 nM TG, and washed. To measure 22Na+ influx, cells were incubated with 2 μCi 22NaCl at 37°C for 5 min in the absence or presence of 1-560 μM veratridine and 100 μM ouabain. Basal 22Na+ influx values (nmol 4×106 cells−1 5 min−1; n=5) were similar among nontreated (19.6±2.0), A23187 (18.6±1.8)- and TG (18.2±2.4)-treated cells, and these values were subtracted from the data. 22Na+ influx values obtained with ouabain alone were similar among nontreated (70.8±3.2), A23187 (70.4±2.8)- and TG (70.4±2.8)-treated cells. Mean±s.e.mean (n=5). *P<0.05, significant decrease by A23187 or TG; #P<0.05, significant enhancement by ouabain of veratridine's effect within each cell group.
A previous study has shown that cooperative modulation of veratridine-induced 22Na+ influx caused by site 3-5-binding toxins occurs in a Na+ channel isoform-specific manner (Cestéle et al., 1995). As shown in Figure 3, either α-scorpion venom, which binds to site 3 between S3IV and S4IV (Rogers et al., 1996), or β-scorpion venom, which interacts with site 4 (Catterall, 1992), or PbTx-3, which binds to site 5 (Yuhi et al., 1994) between S5IVand S6I (Trainer et al., 1994), had little effect per se on 22Na+ influx in nontreated and A23187- or TG-treated cells. However, in nontreated cells, α-, β-scorpion venom or PbTx-3 enhanced veratridine (30 μM)-induced 22Na+ influx by 2.2, 2.5, or 3.9 fold, respectively, values similar to those previously reported (Wada et al., 1987; 1992). In A23187- or TG-treated cells, absolute values of 22Na+ influx were lower when compared to nontreated cells; however, α-, β-scorpion venom or PbTx-3 enhanced veratridine-induced 22Na+ influx in a synergistic mode. In addition, PbTx-3 combination with α- or β-scorpion venom further augmented veratridine-induced 22Na+ influx by 6.4 or 6.1 fold in A23187- or TG-treated cells (Wada et al., 1992).
Figure 3.
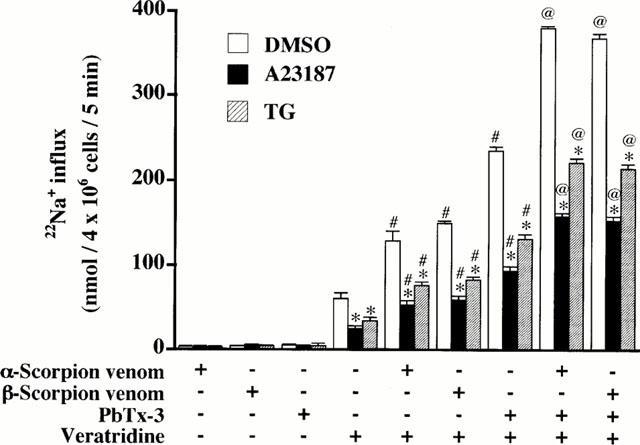
Enhancement of veratridine-induced 22Na+ influx by α-scorpion venom, β-scorpion venom and PbTx-3 in nontreated, A23187- and TG-treated adrenal chromaffin cells. Cells were treated for 48 h with DMSO, 1 μM A23187 or 100 nM TG, washed, and incubated with 2 μCi 22NaCl at 37°C for 5 min in the absence (−) or presence (+) of 0.5 μg ml−1 α-scorpion venom, 5 μg ml−1 β-scorpion venom, 1 μM PbTx-3 or 30 μM veratridine. Basal 22Na+ influx values at 37°C were subtracted for each value. Mean±s.e.mean (n=3). *P<0.05, combined with DMSO-treated cells; #P<0.05, significant enhancement by venom or toxin of veratridine's effect. @P<0.05; significant enhancement by scorpion venom compared to value obtained in cells exposed to veratridine and PbTx-3.
Effects of EGTA, or BAPTA-AM on A23187- or TG-induced reduction of [3H]-STX binding
We examined whether reduction in [3H]-STX binding capacity caused by A23187 or TG was dependent on extra- or intra-cellular Ca2+, using EGTA or BAPTA-AM, cell membrane-impermeable or -permeable Ca2+ chelators, respectively. As shown in Figure 4A, treatment of cells for 24 h with EGTA totally prevented A23187-induced reduction of [3H]-STX binding capacity. In contrast, EGTA attenuated TG-induced decrease of [3H]-STX binding capacity by 40% only. BAPTA-AM entirely blocked the decline of [3H]-STX binding capacity evoked by A23187 or TG. Because Na+ channel down-regulation caused by A23187 or TG was absolutely dependent on [Ca2+]i, we examined whether either a long-lasting or a transient increase in [Ca2+]i may be sufficient to lower [3H]-STX binding capacity. As shown in Figure 4B, when cells were exposed to A23187 or TG for the first 24 h period, then treated with or without BAPTA-AM in the presence of A23187 or TG, addition of BAPTA-AM at 24 h fully prevented A23187- or TG-induced subsequent decrease in [3H]-STX binding capacity at 48 h.
Figure 4.
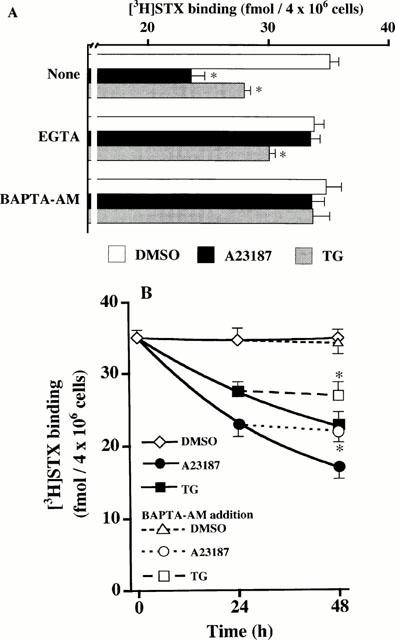
Ca2+-dependent decrease of [3H]-STX binding capacity in A23187- and TG-treated adrenal chromaffin cells. (A) Cells were treated for 24 h with DMSO, 1 μM A23187 or 100 nM TG in the absence (none) or presence of 5 mM EGTA or 50 μM BAPTA-AM, washed, and subjected to [3H]-STX binding assay. Mean±s.e.mean (n=5). *P<0.05, significant decrease by A23187 or TG within each cell group. (B) Cells were exposed to DMSO, 1 μM A23187 or 100 nM TG for the first 24 h, then treated with or without 50 μM BAPTA-AM in the continuous presence of DMSO, A23187 or TG for up to 48 h, and used for [3H]-STX binding assay. Mean±s.e.mean (n=3). *P<0.05, compared with A23187 or TG alone.
Effects of A23187, TG, or DBHQ on [Ca2+]i in the presence or absence of extracellular Ca2+
Because A23187- or TG-induced decrease in [3H]-STX binding capacity was dependent on [Ca2+]i, we measured whether A23187, TG or DBHQ could modify [Ca2+]i. In the presence of extracellular Ca2+ (Figure 5A), A23187 (1 μM) produced an immediate and significant increase in [Ca2+]i in a monophasic manner, followed by a decline to a value above the initial control [Ca2+]i. This value was sustained for up to 60 min. TG (100 nM) induced a slowly-developing monophasic rise in [Ca2+]i followed by a persistent plateau. In contrast, DBHQ (100 μM) robustly increased [Ca2+]i, an effect that gradually waned, as resting level was reattained at 60 min. As shown in Figure 5C, [Ca2+]i increases were observed in all cells studied (A23187, n=27; TG, n=21; DBHQ, n=20). The peak increase in [Ca2+]i over the basal value (Δ [Ca2+]i) was remarkably higher in A23187-treated cells than in TG- or DBHQ-treated cells, whereas peak values of [Ca2+]i were not significantly different between TG- or DBHQ-treated cells. At 30 min, the highest [Ca2+]i was in A23187-treated cells; however, [Ca2+]i went higher in TG-treated cells than in DBHQ-treated cells, as [Ca2+]i in DBHQ-treated cells gradually declined toward the basal value. In the absence of extracellular Ca2+ (Figure 5B), A23187 did not evoke any [Ca2+]i increase over the basal value in most cells examined (n=27). In some cells, however, A23187 caused a slight transient increase in [Ca2+]i, followed by a late, significant decrease below baseline at 30 min (Figure 5C). As shown in Figure 5B, omission of extracellular Ca2+ shortened the duration of TG- or DBHQ-induced monophasic increases in [Ca2+]i and returned the DBHQ-induced rise in [Ca2+]i to the basal value by 15 min. In contrast, [Ca2+]i was continuously elevated during sustained (>30 min) treatment with TG, although its peak amplitude was smaller in Ca2+-free medium, compared to that in Ca2+-containing medium. TG or DBHQ increased [Ca2+]i in all cells examined (TG, n=44; DBHQ, n=43) (Figure 5C). The peak value of [Ca2+]i was lower in TG-treated cells than in DBHQ-treated cells; in contrast, TG-induced sustained elevation of [Ca2+]i was still observed at 30 min, whereas [Ca2+]i decreased below the basal value in DBHQ-treated cells.
Figure 5.
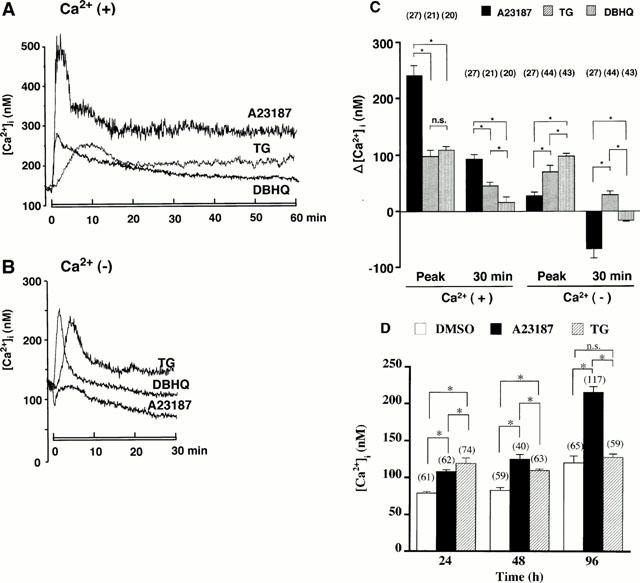
Time-course of [Ca2+]i increase in adrenal chromaffin cells: effects of A23187, TG and DBHQ treatment in the presence or absence of extracellular Ca2+. Cells preloaded with fura-2 were treated with 1 μM A23187, 100 nM TG or 100 μM DBHQ (as indicated by the horizontal bars under recordings) in (A) Ca2+-containing HEPES-buffered solution (HBS) for 60 min and (B) Ca2+-free HBS containing EGTA for 30 min. Each recording shown here was obtained in a single cell and is typical of independent multiple experiments with similar results. (C) Increment of [Ca2+]i over the basal value (Δ [Ca2+]i). Cells were treated with or without A23187, TG or DBHQ in the presence (Ca2+ (+)) or absence (Ca2+ (−)) of extracellular Ca2+. Peak, maximum increase of Δ [Ca2+]i; 30 min, increase of Δ [Ca2+]i measured at 30 min after the exposure to A23187, TG or DBHQ. Basal values of [Ca2+]i were 109.4±6.8 nM (n=68) in Ca2+-containing medium, and 100.4±5.2 nM (n=114) in Ca2+-free medium. Parenthesis (number of cells examined). (D) Culture dishes were divided into nontreated, A23187- or TG-treated cell group; cells were treated in cultured medium with DMSO, 1 μM A23187 or 100 nM TG for 24, 48 and 96 h, and then subjected to [Ca2+]i measurement. Mean±s.e.mean. *P<0.05, significant difference; n.s., no significant difference.
We then examined whether [Ca2+]i may remain continuously elevated with chronic treatment by A23187 or TG. Culture dishes were classified into nontreated (n=185), A23187 (n=219) or TG (n=196)-treated cell groups; they were incubated for 24, 48 and 96 h, and then used for [Ca2+]i measurement as indicated in Figure 5D. In the nontreated cell group, [Ca2+]i remained stable until 48 h, but unexpectedly increased at 96 h. In the A23187-treated cell group, [Ca2+]i progressively went up depending on the duration of A23187 treatment. In the TG-treated cell group, [Ca2+]i increased over the control value at 24 and 48 h, but was similar to the value of the control cell group at 96 h. The [Ca2+]i increment in the A23187-treated cell group was smaller than that in the TG-treated cell group at 24 h, but larger at 48 and 96 h.
Decrease of [3H]-STX binding evoked by A23187 or TG: prevention by Gö6976 or a calpain inhibitor
Due to the A23187- or TG-induced decrease in [3H]-STX binding capacity being entirely dependent on the long-lasting [Ca2+]i increase (Figures 4 and 5) we examined whether a Ca2+-dependent, conventional protein kinase C (cPKC) was involved in A23187- or TG-induced down-regulation of Na+ channels. Our previous [3H]-STX binding and Western blot analysis revealed that among four cPKC isozymes, adrenal chromaffin cells contain the cPKC-α isozyme only. Treatment of cells with thymeleatoxin (TMX), a selective activator of cPKC, caused the translocation of cPKC-α from cytosol to membranes, an event assumed to be the hallmark of PKC activation (Newton, 1997). This treatment caused down-regulation of Na+ channels by promoting endocytic internalization of cell surface Na+ channels (Yanagita et al., 1996; 2000). As shown in Figure 6, treatment of cells for 24 h with 1 μM Gö6976, a selective inhibitor of cPKC, had no effect per se, but reversed the inhibitory effect of A23187 or TG on [3H]-STX binding capacity by 56 or 66%. This concentration of Gö6976 completely prevents the TMX-induced decrease in [3H]-STX binding capacity (Yanagita et al., 2000).
Figure 6.
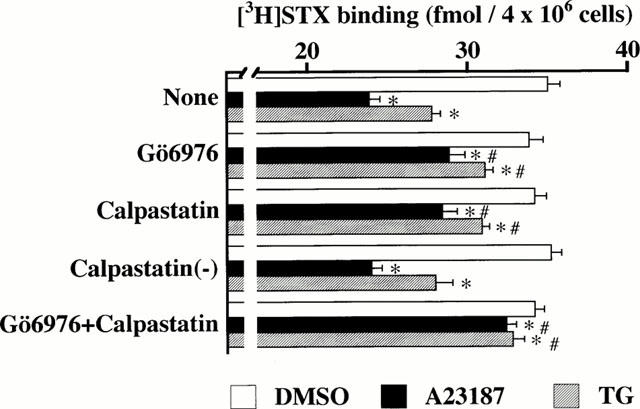
Decrease of [3H]-STX binding capacity in A23187- and TG-treated adrenal chromaffin cells: prevention by Gö6976 and calpain inhibitor. Cells were treated for 24 h with DMSO, 1 μM A23187 or 100 nM TG in the absence (None) or presence of 1 μM Gö6976, or/and 1 μM calpastatin peptide or 1 μM biologically inactive calpastatin analogue [calpastatin (−)], and used for [3H]-STX binding assay. Mean±s.e.mean (n=5). *P<0.05, significant decrease by A23187 or TG; #P<0.05, significant prevention by Gö6976 or/and calpastatin.
It has been shown that calpain, a family of Ca2+-dependent cysteine proteases (Carafoli & Molinari, 1998), catalyzes regulated proteolysis of cytoskeletal and membrane enzymes involved in signal transduction, as well as transcriptional factors, thereby modulating physiological events such as the internalization of endocytic vesicles (Nakamura et al., 1992; Sato et al., 1995; Kamal et al., 1998; Michaely et al., 1999). We then used the cell permeable calpastatin 27-amino acid peptide, an endogenous inhibitor of calpain. As shown in Figure 6, treatment with the calpastatin peptide had little effect per se, but prevented A23187- or TG-induced decrement in [3H]-STX binding by 54 or 65%. As control, the biologically inactive calpastatin peptide had no effect. The simultaneous treatment of cells with Gö6976 and calpastatin peptide prevented A23187- or TG-induced reduction in [3H]-STX binding capacity by 84 or 81%.
Effects of A23187, TG, or BFA on [3H]-STX binding capacity
The internalization rate of Na+ channels in the absence or presence of A23187 or TG was studied using BFA, an inhibitor of the guanine nucleotide exchange protein of ADP-ribosylation factor 1, a monomeric GTPase (Moss & Vaughan, 1995). BFA has been shown to block cell surface vesicular externalization from the trans-Golgi network (TGN) of newly-synthesized renal epithelial Na+ channels (Shimkets et al., 1997; Staub et al., 1997), receptors (Schonhorn & Wessling-Resnick, 1994; Hirasawa et al., 1998), and transporters (Chakrabarti et al., 1994), whereas it has no effect on ADP-ribosylation factor 6-catalyzed endocytosis (Schonhorn & Wessling-Resnick, 1994; Cavenagh et al., 1996; Hirasawa et al., 1998). As shown in Figure 7, treatment for 6 h with A23187 or TG did not lower [3H]-STX binding capacity. Treatment for 6 h with BFA alone decreased [3H]-STX binding capacity by 16%, whereas 6 h treatment with A23187 plus BFA or TG plus BFA lowered [3H]-STX binding by 31 or 25%, respectively. After 12 h treatment, A23187, TG, or BFA reduced [3H]-STX binding by 22, 9 or 31%, respectively. Treatment of cells with A23187 plus BFA or TG plus BFA remarkably reduced [3H]-STX binding capacity by 59 or 50%, respectively.
Figure 7.
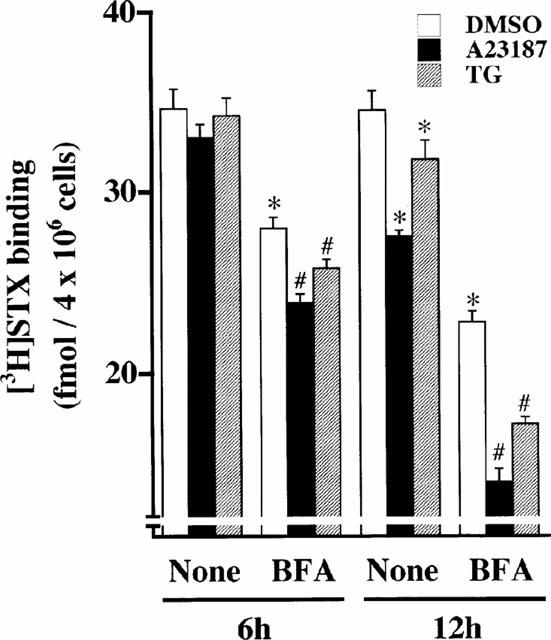
Enhancement by BFA of A23187-, and TG-induced decrease of [3H]-STX binding capacity in adrenal chromaffin cells. Cells were treated with DMSO, 1 μM A23187 or 100 nM TG for 6 or 12 h in the absence (None) or presence of 10 μg ml−1 BFA, and subjected to [3H]-STX binding assay. Mean±s.e.mean (n=5). *P<0.05, significant decrease by BFA, A23187 or TG; #P<0.05, compared with BFA alone.
Effects of A23187 or TG treatment on Na+ channel α- and β1-subunit mRNA levels
We measured the steady-state levels of Na+ channel α- and β1-subunit mRNAs in cells treated with or without A23187 or TG for up to 48 h. As shown in Figure 8A, hNE-Na probe hybridized to one major transcript (∼9.4 kb) corresponding to the α-subunit as reported previously (Klugbauer et al., 1995; Yanagita et al., 2000). When α-subunit mRNA level was normalized against that of GAPDH mRNA, A23187 treatment progressively decreased α-subunit mRNA level by ∼48% between 3 and 48 h (Figure 8B). In contrast, TG treatment had no measurable effect on α-subunit mRNA level between 3 and 48 h. Figure 8A shows that β1-subunit probe hybridized to a single transcript (∼1.5 kb), as reported previously (Makita et al., 1994; Oh & Waxman, 1994; Yanagita et al., 2000). A23187 decreased the relative level of the β1-subunit mRNA by ∼50% between 6 and 48 h, whereas TG had no effect (Figure 8B).
Figure 8.
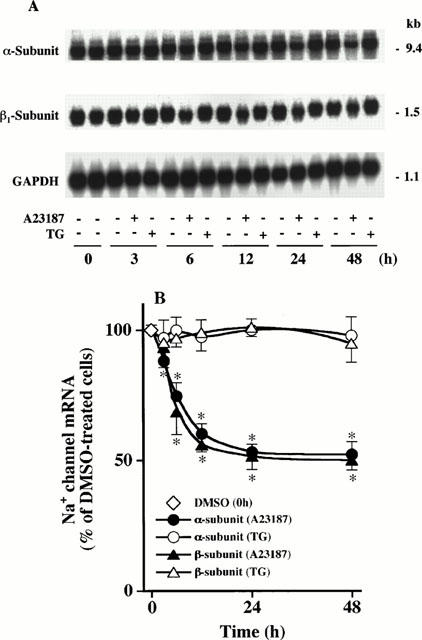
Northern blot analysis: distinct effects of A23187 and TG treatment on Na+ channel α- and β1-subunit mRNA levels in adrenal chromaffin cells. (A) Cells were treated with DMSO (−), 1 μM A23187 (+) or 100 nM TG (+) for up to 48 h; poly(A)+ RNA was extracted, separated by electrophoresis on agarose gel, and transferred to membrane. The membrane was sequentially hybridized to each 32P-labelled cDNA probe for hNE-Na (top), rat brain β1-subunit (middle), and GAPDH (bottom) after removal of the former probe. Data shown are typical one from three independent analyses with similar results. (B) Levels of α- and β1-subunit mRNAs and GAPDH mRNA in (A) were quantified by a Bioimage analyser, and the relative amount of α- or β1-subunit mRNA GAPDH mRNA−1 in A23187- or TG-treated cells is shown. A value of 100% represents the relative level obtained in DMSO-treated cells at the left lane of each incubation time. Mean±s.e.mean (n=3). *P<0.05, compared with DMSO-treated cells.
Discussion
In the present study, treatment of adrenal chromaffin cells with A23187 or TG caused a time- and concentration-dependent decrease in [3H]-STX binding capacity. Scatchard analysis revealed that A23187 or TG lowered the Bmax value of [3H]-STX binding, but did not alter the KD value. A23187 or TG also attenuated veratridine-induced 22Na+ influx, but did not change the EC50 value of veratridine. It has been shown that veratridine causes persistent activation of Na+ channels at resting membrane potential by blocking Na+ channel inactivation and shifting the voltage dependence of Na+ channel activation to a more negative membrane potential (Catterall, 1992). In our present study, the maximum values of Na+ influx caused by veratridine in the presence of ouabain (Figure 2) were 500.2, 214.3 and 306.6 nmol per 5 min per 4×106 nontreated, A23187- or TG-treated cells, respectively. Because STX binds to Na+ channels with the molecular ratio of 1 : 1 (Catterall, 1980), the Bmax values of [3H]-STX binding (Figure 1C) show that there exist approximately 292.8×108, 129.6×108 and 193.2×108 of Na+ channels per 4×106 nontreated, A23187- or TG-treated cells, respectively. Thus, the calculated Na+ influx rates (10−18 mol single Na+ channel−1 min−1) are comparable among nontreated (3.41), A23187 (3.32)- or TG (3.26)-treated cells. Also, either α- or β-scorpion venom, or PbTx-3 alone, and a combination of either venom with PbTx-3 remarkably potentiated veratridine-induced 22Na+ influx in A23187- or TG-treated cells in the same manner as in nontreated cells (Wada et al., 1987; 1992). These results suggest that long-term treatment with A23187 or TG down-regulates cell surface Na+ channels without altering the activity and the pharmacological property of the Na+ channel molecule.
Either EGTA or BAPTA-AM completely prevented the A23187-induced decline in [3H]-STX binding capacity. A23187 elevated [Ca2+]i in the presence (but not in the absence) of extracellular Ca2+ for at least 96 h, consistent with A23187-induced long-lasting (>96 h) down-regulation of cell surface [3H]-STX binding. These results suggest that A23187-induced influx of extracellular Ca2+ and the subsequent rise in [Ca2+]i were responsible for the down-regulation of Na+ channels. TG-induced reduction in [3H]-STX binding capacity was fully abolished by BAPTA-AM, thereby showing that this reduction was dependent on [Ca2+]i. In contrast, EGTA prevented the TG-induced reduction of [3H]-STX binding by 40% only, suggesting that part of the TG-induced decrease of [3H]-STX binding capacity could be attributed to the TG-induced influx of extracellular Ca2+ presumably via a mechanism termed capacitative Ca2+ entry (Parekh & Penner, 1997), previously described in adrenal chromaffin cells (Cheek & Thastrup, 1989; Robinson et al., 1992). In the presence of EGTA, the TG-induced monophasic increase in [Ca2+]i and subsequent sustained plateau were reduced, compared to those in Ca2+-containing medium. [Ca2+]i remained elevated for up to 30 min. Thus, the remaining 60% reduction in [3H]-STX binding capacity induced by TG in the presence of EGTA might be attributed to the TG-induced sustained increase in [Ca2+]i due to mobilization from intracellular Ca2+ pools.
In Ca2+-containing medium, DBHQ robustly increased [Ca2+]i with an amplitude comparable to that induced with TG, while this compound did not alter [3H]-STX binding capacity. The DBHQ-induced monophasic rise of [Ca2+]i, however, decayed rapidly, and was followed by the gradual decline of [Ca2+]i toward basal levels. At 30 min the DBHQ-induced increase in [Ca2+]i was significantly smaller, compared to the TG-induced rise of [Ca2+]i. Moreover, the observation that TG-induced [Ca2+]i rise lasted for at least 48 h is in agreement with the fact that TG-induced down-regulation of cell surface [3H]-STX binding capacity levelled off between 48 and 96 h. Continuous chronic elevation of [Ca2+]i may be necessary to induce down-regulation of Na+ channels, because (1) BAPTA-AM, when added 24 h after addition of A23187 or TG, completely blocked the subsequent decreasing effect of A23187 or TG on [3H]-STX binding capacity; (2) the decreasing effect of A23187 or TG was readily reversed when A23187 or TG was washed out, even after 48 h treatment; and (3) pulse treatment for 6 h with A23187 was insufficient to diminish [3H]-STX binding, when assayed at 12 and 24 h. In contrast, treatment of adrenal chromaffin cells with DBHQ (100 μM for 24 h) decreased the number of cell surface insulin receptors to a larger extent than TG treatment (100 nM for 24 h), as shown by 125I-insulin binding assay (authors' unpublished observation). Similar results have been obtained in lymphocytes, in which duration and amplitude of [Ca2+]i increase were found to control the activation of a distinct subset of transcriptional factors independently (Timmerman et al., 1996; Dolmetsch et al., 1997).
In the present study, A23187 treatment lowered Na+ channel α- and β1-subunit mRNA levels as early as 3 and 6 h, respectively, when [3H]-STX binding capacity was not yet decreased. The reduction of both transcript levels lasted for at least 48 h in these conditions. In contrast, TG treatment failed to decrease Na+ channel α- and β1-subunit mRNA levels between 3 and 48 h. On the basis that A23187 evoked a rapid monophasic increase and sustained (>60 min) plateau of [Ca2+]i with approximately a 2 fold greater amplitude than did TG, it may be argued that A23187-induced higher level of [Ca2+]i increase was required for the down-regulation of Na+ channel subunit transcripts. A previous expression study of Xenopus oocytes showed that injection of decreasing amounts of cardiac Na+ channel β1-subunit mRNA, in conjunction with the constant amount of the α-subunit mRNA, produces a graded reduction in cell surface expression of Na+ channels (Qu et al., 1995). Our present study showed that compared to TG-induced decrease in [3H]-STX binding capacity, A23187-induced reduction of [3H]-STX binding occurred to a greater extent at each incubation time, and lasted for a longer period. These results suggest that A23187-induced decrease in Na+ channel subunit mRNA levels contributes to the down-regulation of cell surface Na+ channels.
In adrenal chromaffin cells, our previous study shows that the TMX-induced, rapid (<15 min) and sustained (>15 h) translocation of cPKC-α from cytosol to membranes promotes endocytic internalization of cell surface Na+ channels, and causes down-regulation of Na+ channels, as evidenced by using Gö6976 and BFA (Yanagita et al., 2000). In the present study, reduction of [3H]-STX binding capacity caused by A23187 or TG was attenuated by concurrent 24 h treatment with Gö6976. Although the mechanisms whereby [Ca2+]i lowered [3H]-STX binding may vary during chronic (∼96 h) treatment with A23187 or TG, we measured whether A23187 or TG treatment could stimulate internalization of Na+ channels at 6 and 12 h. BFA treatment (10 μg ml−1 for ∼12 h) decreased [3H]-STX binding per se, and enhanced A23187- or TG-induced reduction in [3H]-STX binding. BFA treatment (2.5 – 10 μg ml−1 for 2 – 36 h) has been shown to block cell surface vesicular trafficking from the TGN of glucose transporter-4 (Chakrabarti et al., 1994), transferrin receptors (Schonhorn & Wessling-Resnick et al., 1994), renal epithelial Na+ channels (Shimkets et al., 1997; Staub et al., 1997), and α1B-adrenoceptors (Hirasawa et al., 1998), whereas a similar BFA treatment does not alter the internalization rate of these ion channels and receptors (Schonhorn & Wessling-Resnick, 1994; Cavenagh et al., 1996; Hirasawa et al., 1998). Thus, the attenuation evoked by Gö6976 and the enhancement by BFA of A23187- or TG-induced decline in [3H]-STX binding capacity suggest that A23187- or TG-induced rise of [Ca2+]i, and the subsequent activation of cPKC-α accelerated internalization of cell surface Na+ channels, thereby contributing to the down-regulation of Na+ channels during 6 and 12 h treatment with A23187 or TG.
Our present study also showed that decrease in [3H]-STX binding capacity caused by A23187 or TG was diminished by simultaneous 24 h treatment with calpastatin peptide. One possible mechanism of calpain-induced down-regulation of Na+ channels is internalization of Na+ channels, because calpain stimulates budding of clathrin-coated vesicles, an event triggering the internalization of ion channels (e.g. renal epithelial Na+ channels) (Lamaze & Schmid, 1995; Liu & Robinson, 1995; Shimkets et al., 1997; Staub et al., 1997). In the brain, calpain binds to clathrin-coated vesicles in a Ca2+-dependent manner, and catalyzes the degradation of membrane proteins, thus promoting generation of clathrin-coated vesicles (Nakamura et al., 1992; Sato et al., 1995). In vivo and in vitro studies in fibroblasts have shown that calpain-catalyzed proteolytic removal of spectrin, a cytoskeletal protein, is the prerequisite for budding of clathrin-coated vesicles, and the effect of calpain is stimulated by annexin VI, a Ca2+-dependent phospholipid-binding protein (Kamal et al., 1998). In brain and fibroblasts, clathrin is associated with ankyrin, a protein that accelerates calpain-induced degradation of spectrin and endocytosis of clathrin-coated vesicles that harboured low-density lipoprotein (Michaely et al., 1999). In brain and skeletal muscle, cell surface Na+ channels are associated with ankyrin and spectrin, which is thought to maintain high density of Na+ channels at the axon initial segment and node of Ranvier, as well as in the postsynaptic folds of the neuromuscular junction (Srinivasan et al., 1988; Wood & Slater, 1998). However, cerebellum-specific knockout of ankyrin in mouse has documented that ankyrin also directs appropriate targeting of Na+ channels to the axon initial segment (Zhou et al., 1998). Although little is known about the molecular machinery regulating internalization of Na+ channels, these findings may raise the question of whether calpain and ankyrin are involved in the internalization of Na+ channels in adrenal chromaffin cells, as previously reported for the endocytosis of clathrin-coated vesicles in brain and fibroblasts (Nakamura et al., 1992; Sato et al., 1995; Kamal et al., 1998; Michaely et al., 1999).
Finally, the biological relevance of our present study should be addressed. Aberrant down-regulation of Na+ channels may impair normal development and differentiation of neurons (Van Huizen et al., 1985; Mourre et al., 1987; Miguel-Hidalgo et al., 1995; Toledo-Aral et al., 1995) and skeletal muscle cells (Lindsell & Moody, 1994). However, down-regulation of brain Na+ channels may be a compensatory defensive mechanism against hypoxia- or ischaemia-induced neuronal injury (Xia & Haddad, 1994; 1999; Urenjak & Obrenovitch, 1996). Hypoxia or ischaemia increases noninactivating Na+ currents via TTX- and STX-sensitive Na+ channels, thus causing Ca2+ overload via reversed operation of Na+-Ca2+ exchanger (Urenjak & Obrenovitch, 1996). Hypoxia- or ischaemia-induced depletion of ATP perturbs Ca2+ sequestration into the endoplasmic reticulum catalyzed by SERCA, and also compromises extracellular Ca2+ extrusion by plasma membrane Ca2+-ATPase, thus aggravating Ca2+ overload (Urenjak & Obrenovitch, 1996). Our present study suggests that the increases in [Ca2+]i regulate the density of functional Na+ channels via distinct mechanisms.
Acknowledgments
We thank Drs Franz Hofmann (Institüt für Pharmakologie und Toxikologie der Technischen Universität München) and Youngsuk Oh (Department of Medicine, University of Alabama) for donating the hNE-Na and the β1-subunit plasmids, respectively. We also thank Dr Dominique Aunis (INSERUM, Strasbourg) for linguistic improvement of this manuscript. Technical and secretarial assistance by Ms Keiko Kawabata and Mr Keizo Masumoto is greatly appreciated. This study was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science and Culture, Japan.
Abbreviations
- BAPTA-AM
1,2-bis(2-aminophenoxy)-ethane-N,N,N′,N′-tetra-acetic acid tetrakis-acetoxymethyl ester
- BFA
brefeldin A
- BSA
bovine serum albumin
- [Ca2+]i
intracellular concentration of Ca2+
- cPKC
conventional protein kinase C
- DBHQ
2,5-di-(t-butyl)-1,4-benzohydroquinone
- DMSO
dimethyl sulphoxide
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- HBS
HEPES-buffered solution
- hNE-Na
tetrodotoxin- and saxitoxin-sensitive human neuroendocrine type sodium channel α-subunit
- KRP
Krebs-Ringer phosphate
- nt
nucleotides
- PbTx-3
Ptychodiscus brevis toxin-3
- QNB
quinuclidiny(phenyl-4-3H)benzilate
- SDS
sodium dodecyl sulphate
- SERCA
sarco(endo)plasmic reticulum Ca2+-ATPase
- SSC
saline-sodium citrate
- STX
saxitoxin
- TGN
trans-Golgi network
- TMX
thymeleatoxin
- TTX
tetrodotoxin
References
- ALONSO M.T., BARRERO M.J., MICHELENA P., CARNICERO E., CUCHILLO I., GARCÍA A.G., GARCÍA-SANCHO J., MONTERO M., ALVAREZ J. Ca2+-induced Ca2+ release in chromaffin cells seen from inside the ER with targeted aequorin. J. Cell Biol. 1999;144:241–254. doi: 10.1083/jcb.144.2.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BALLESTA J.J., BORGES R., GARCÍA A.G., HIDALGO M.J. Secretory and radioligand binding studies on muscarinic receptors in bovine and feline chromaffin cells. J. Physiol. (Lond.) 1989;418:411–426. doi: 10.1113/jphysiol.1989.sp017849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRISMAR T. Abnormal Na-currents in diabetic rat nerve nodal membrane. Diabetic Med. 1993;10 Suppl. 2:110S–112S. doi: 10.1111/j.1464-5491.1993.tb00216.x. [DOI] [PubMed] [Google Scholar]
- BRODIE C., BRODY M., SAMPSON S.R. Characterization of the relation between sodium channels and electrical activity in cultured rat skeletal myotubes: regulatory aspects. Brain Res. 1989;488:186–194. doi: 10.1016/0006-8993(89)90708-7. [DOI] [PubMed] [Google Scholar]
- CARAFOLI E., MOLINARI M. Calpain: a protease in search of a function. Biochem. Biophys. Res. Commun. 1998;247:193–203. doi: 10.1006/bbrc.1998.8378. [DOI] [PubMed] [Google Scholar]
- CATTERALL W.A. Purification of a toxic protein from scorpion venom which activates the action potential Na+ ionophore. J. Biol. Chem. 1976;251:5528–5536. [PubMed] [Google Scholar]
- CATTERALL W.A. Neurotoxins that act on voltage-sensitive sodium channels in excitable membranes. Annu. Rev. Pharmacol. Toxicol. 1980;20:15–43. doi: 10.1146/annurev.pa.20.040180.000311. [DOI] [PubMed] [Google Scholar]
- CATTERALL W.A. Cellular and molecular biology of voltage-gated sodium channels. Physiol. Rev. 1992;72:S15–S48. doi: 10.1152/physrev.1992.72.suppl_4.S15. [DOI] [PubMed] [Google Scholar]
- CAVENAGH M.M., WHITNEY J.A., CARROLL K., ZHANG C.-J., BOMAN A.L., ROSENWALD A.G., MELLMAN I., KAHN R.A. Intracellular distribution of Arf proteins in mammalian cells. Arf6 is uniquely localized to the plasma membrane. J. Biol. Chem. 1996;271:21767–21774. doi: 10.1074/jbc.271.36.21767. [DOI] [PubMed] [Google Scholar]
- CESTÉLE S., KHALIFA R.B., PELHATE M., ROCHAT H., GORDON D. α-Scorpion toxins binding on rat brain and insect sodium channels reveal divergent allosteric modulations by brevetoxin and veratridine. J. Biol. Chem. 1995;270:15153–15161. doi: 10.1074/jbc.270.25.15153. [DOI] [PubMed] [Google Scholar]
- CHAKRABARTI R., BUXTON J., JOLY M., CORVERA S. Insulin-sensitive association of GLUT-4 with endocytic clathrin-coated vesicles revealed with the use of brefeldin A. J. Biol. Chem. 1994;269:7926–7933. [PubMed] [Google Scholar]
- CHEEK T.R., THASTRUP O. Internal Ca2+ mobilization and secretion in bovine adrenal chromaffin cells. Cell Calcium. 1989;10:213–221. doi: 10.1016/0143-4160(89)90004-3. [DOI] [PubMed] [Google Scholar]
- CHIAMVIMONVAT N., KARGACIN M.E., CLARK R.B., DUFF H.J. Effects of intracellular calcium on sodium current density in cultured neonatal rat cardiac myocytes. J. Physiol. (Lond.) 1995;483:307–318. doi: 10.1113/jphysiol.1995.sp020587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DIETRICH P.S., MCGIVERN J.G., DELGADO S.G., KOCH B.D., EGLEN R.M., HUNTER J.C., SANGAMESWARAN L. Functional analysis of a voltage-gated sodium channel and its splice variant from rat dorsal root ganglia. J. Neurochem. 1998;70:2262–2272. doi: 10.1046/j.1471-4159.1998.70062262.x. [DOI] [PubMed] [Google Scholar]
- DOLMETSCH R.E., LEWIS R.S., GOODNOW C.C., HEALY J.I. Differential activation of transcriptional factors induced by Ca2+ response amplitude and duration. Nature. 1997;386:855–858. doi: 10.1038/386855a0. [DOI] [PubMed] [Google Scholar]
- DUFF H.J., OFFORD J., WEST J., CATTERALL W.A. Class I and IV antiarrhythmic drugs and cytosolic calcium regulate mRNA encoding the sodium channel α subunit in rat cardiac muscle. Mol. Pharmacol. 1992;42:570–574. [PubMed] [Google Scholar]
- GINTY D.D. Calcium regulation of gene expression: Isn't that spatial. Neuron. 1997;18:183–186. doi: 10.1016/s0896-6273(00)80258-5. [DOI] [PubMed] [Google Scholar]
- GRYNKIEWICZ G., POENIE M., TSIEN R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- HELDMAN E., BARG J., VOGEL Z., POLLARD H.B., ZIMLICHMAN R. Correlation between secretagogue-induced Ca2+ influx, intracellular Ca2+ levels and secretion of catecholamines in cultured adrenal chromaffin cells. Neurochem. Int. 1996;28:325–334. doi: 10.1016/0197-0186(96)83614-x. [DOI] [PubMed] [Google Scholar]
- HIRASAWA A., AWAJI T., SUGAWARA T., TSUJIMOTO A., TSUJIMOTO G. Differential mechanism for the cell surface sorting and agonist-promoted internalization of the α1B-adrenoceptor. Br. J. Pharmacol. 1998;124:55–62. doi: 10.1038/sj.bjp.0701795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HIRSH J.K., QUANDT F.N. Down-regulation of Na channel expression by A23187 in N1E-115 neuroblastoma cells. Brain Res. 1996;706:343–346. doi: 10.1016/0006-8993(95)01340-7. [DOI] [PubMed] [Google Scholar]
- ISOM L.L., DE JONGH K.S., CATTERALL W.A. Auxiliary subunits of voltage-gated ion channels. Neuron. 1994;12:1183–1194. doi: 10.1016/0896-6273(94)90436-7. [DOI] [PubMed] [Google Scholar]
- ISOM L.L., RAGSDALE D.S., DE JONGH K.S., WESTENBROEK R.E., REBER B.F.X., SCHEUER T., CATTERALL W.A. Structure and function of the β2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM motif. Cell. 1995;83:433–442. doi: 10.1016/0092-8674(95)90121-3. [DOI] [PubMed] [Google Scholar]
- KAMAL A., YING Y., ANDERSON R.G. Annexin VI-mediated loss of spectrin during coated pit budding is coupled to delivery of LDL to lysosomes. J. Cell Biol. 1998;142:937–947. doi: 10.1083/jcb.142.4.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KLUGBAUER N., LACINOVA L., FLOCKERZI V., HOFMANN F. Structure and functional expression of a new member of the tetrodotoxin-sensitive voltage-activated sodium channel family from human neuroendocrine cell. EMBO J. 1995;14:1084–1090. doi: 10.1002/j.1460-2075.1995.tb07091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAMAZE C., SCHMID S.L. The emergence of clathrin-independent pinocytic pathways. Curr. Opin. Cell Biol. 1995;7:573–580. doi: 10.1016/0955-0674(95)80015-8. [DOI] [PubMed] [Google Scholar]
- LINSDELL P., MOODY W.J. Na+ channel mis-expression accelerates K+ channel development in embryonic Xenopus laevis skeletal muscle. J. Physiol. (Lond.) 1994;480:405–410. doi: 10.1113/jphysiol.1994.sp020370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU J.P., ROBINSON P.J. Dynamin and endocytosis. Endocr. Rev. 1995;16:590–607. doi: 10.1210/edrv-16-5-590. [DOI] [PubMed] [Google Scholar]
- MAKITA N., BENNETT P.B., JR, GEORGE A.L., JR Voltage-gated Na+ channel β1 subunit mRNA expressed in adult human skeletal muscle, heart, and brain is encoded by a single gene. J. Biol. Chem. 1994;269:7571–7578. [PubMed] [Google Scholar]
- MALVIYA A.N., ROGUE P.J. ‘Tell me where is calcium bred': clarifying the roles of nuclear calcium. Cell. 1998;92:17–23. doi: 10.1016/s0092-8674(00)80895-8. [DOI] [PubMed] [Google Scholar]
- MATHIASEN D., RØSSUM L.M., ROBINSON I.M., BURGOYNE R.D., EAST J.M., MØLLER M., RASMUSSEN H.N., TREIMAN M. Isolation of chromaffin cell thapsigargin-sensitive Ca2+ store in light microsomes from bovine adrenal medulla. Int. J. Biochem. 1993;25:641–652. doi: 10.1016/0020-711x(93)90348-i. [DOI] [PubMed] [Google Scholar]
- MEVES H., RUBLY N., WATT D.D. Effect of toxins isolated from the venom of the scorpion Centruroides sculpturatus on the Na currents of the node of Ranvier. Pflügers Arch. 1982;393:56–62. doi: 10.1007/BF00582392. [DOI] [PubMed] [Google Scholar]
- MICHAELY P., KAMAL A., ANDERSON R.G., BENNETT V. A requirement for ankyrin binding to clathrin during coated pit budding. J. Biol. Chem. 1999;274:35908–35913. doi: 10.1074/jbc.274.50.35908. [DOI] [PubMed] [Google Scholar]
- MIGUEL-HIDALGO J.J., ANGELIDES K.J., CHALUPA L.M. Distinct temporal patterns of expression of sodium channel-like immunoreactivity during the prenatal development of the monkey and cat retina. Eur. J. Neurosci. 1995;7:535–546. doi: 10.1111/j.1460-9568.1995.tb00658.x. [DOI] [PubMed] [Google Scholar]
- MOSS J., VAUGHAN M. Structure and function of ARF proteins: activators of cholera toxin and critical components of intracellular vesicular transport processes. J. Biol. Chem. 1995;270:12327–12330. doi: 10.1074/jbc.270.21.12327. [DOI] [PubMed] [Google Scholar]
- MOURRE C., CERVERA P., LAZDUNSKI M. Autoradiographic analysis in rat brain of the postnatal ontogeny of voltage-dependent Na+ channels, Ca2+-dependent K+ channels and slow Ca2+ channels identified as receptors for tetrodotoxin, apamin, and (−)-desmethoxyverapamil. Brain Res. 1987;417:21–32. doi: 10.1016/0006-8993(87)90175-2. [DOI] [PubMed] [Google Scholar]
- NAKAMURA M., MORI M., MORISHITA Y., MORI S., KAWASHIMA S. Specific increase in calcium-activated neutral protease with low calcium sensitivity (m-calpain) in proerythroblastic K562 cell line cells induced to differentiation by phorbol 12-myristate 13-acetate. Exp. Cell Res. 1992;200:513–522. doi: 10.1016/0014-4827(92)90203-k. [DOI] [PubMed] [Google Scholar]
- NEWTON A.C. Regulation of protein kinase C. Curr. Opin. Cell Biol. 1997;9:161–167. doi: 10.1016/s0955-0674(97)80058-0. [DOI] [PubMed] [Google Scholar]
- NODA M., SUZUKI H., NUMA S., STÜHMER W. A single point mutation confers tetrodotoxin and saxitoxin insensitivity on the sodium channel II. FEBS Lett. 1989;259:213–216. doi: 10.1016/0014-5793(89)81531-5. [DOI] [PubMed] [Google Scholar]
- OFFORD J., CATTERALL W.A. Electrical activity, cAMP, and cytosolic calcium regulate mRNA encoding sodium channel α subunits in rat muscle cells. Neuron. 1989;2:1447–1452. doi: 10.1016/0896-6273(89)90190-6. [DOI] [PubMed] [Google Scholar]
- OH Y., WAXMAN S.G. The β1 subunit mRNA of the rat brain Na+ channel is expressed in glial cells. Proc. Natl. Acad. Sci. U.S.A. 1994;91:9985–9989. doi: 10.1073/pnas.91.21.9985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OH Y., LEE Y.-J., WAXMAN S.G. Regulation of Na+ channel β1 and β2 subunit mRNA levels in cultured rat astrocytes. Neurosci. Lett. 1997;234:107–110. doi: 10.1016/s0304-3940(97)00694-0. [DOI] [PubMed] [Google Scholar]
- PAREKH A.B., PENNER R. Store depletion and calcium influx. Physiol. Rev. 1997;77:901–930. doi: 10.1152/physrev.1997.77.4.901. [DOI] [PubMed] [Google Scholar]
- POULSEN J.-C.J., CASPERSEN C., MATHIASEN D., EAST J.M., TUNWELL R.E.A., LAI F.A., MAEDA N., MIKOSHIBA K., TREIMAN M. Thapsigargin-sensitive Ca2+-ATPases account for Ca2+ uptake to inositol 1,4,5-trisphosphate-sensitive and caffeine-sensitive Ca2+ stores in adrenal chromaffin cells. Biochem. J. 1995;307:749–758. doi: 10.1042/bj3070749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QU Y., ISOM L.L., WESTENBROEK R.E., ROGERS J.C., TANDA T.N., MCCORMICK K.A., SCHEUER T., CATTERALL W.A. Modulation of cardiac Na+ channel expression in Xenopus oocytes by β1 subunits. J. Biol. Chem. 1995;270:25696–25701. doi: 10.1074/jbc.270.43.25696. [DOI] [PubMed] [Google Scholar]
- ROBINSON I.M., BURGOYNE R.D. Characterization of distinct inositol 1,4,5-trisphosphate-sensitive and caffeine-sensitive calcium stores in digitonin-permeabilized adrenal chromaffin cells. J. Neurochem. 1991a;56:1587–1593. doi: 10.1111/j.1471-4159.1991.tb02055.x. [DOI] [PubMed] [Google Scholar]
- ROBINSON I.M., BURGOYNE R.D. A distinct 2,5-di-(tert-butyl)-1,4-benzohydroquinone-sensitive calcium store in bovine adrenal chromaffin cells. FEBS Lett. 1991b;289:151–154. doi: 10.1016/0014-5793(91)81057-f. [DOI] [PubMed] [Google Scholar]
- ROBINSON I.M., CHEEK T.R., BURGOYNE R.D. Ca2+ influx induced by the Ca2+-ATPase inhibitors 2,5-di-(t-butyl)-1,4-benzohydroquinone and thapsigargin in bovine adrenal chromaffin cells. Biochem. J. 1992;288:457–463. doi: 10.1042/bj2880457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROGERS J.C., QU Y., TANADA T.N., SCHEUER T., CATTERALL W.A. Molecular determinants of high affinity binding of α-scorpion toxin and sea anemone toxin in the S3-S4 extracellular loop in domain IV of the Na+ channel α subunit. J. Biol. Chem. 1996;271:15950–15962. doi: 10.1074/jbc.271.27.15950. [DOI] [PubMed] [Google Scholar]
- SASHIHARA S., YANAGIHARA N., IZUMI F., MURAI Y., MITA T. Differential up-regulation of voltage-dependent Na+ channels induced by phenytoin in brains of genetically seizure-susceptible (E1) and control (ddY) mice. Neuroscience. 1994;62:803–811. doi: 10.1016/0306-4522(94)90478-2. [DOI] [PubMed] [Google Scholar]
- SATIN J., KYLE J.W., SCHEN M., BELL P., CRIBBS L.L., FOZZARD H.A., ROGART R.B. A mutant of TTX-resistant cardiac sodium channels with TTX-sensitive properties. Science. 1992;256:1202–1205. doi: 10.1126/science.256.5060.1202. [DOI] [PubMed] [Google Scholar]
- SATO K., SAITO Y., KAWASHIMA S. Identification and characterization of membrane-bound calpains in clathrin-coated vesicles from bovine brain. Eur. J. Biochem. 1995;230:25–31. doi: 10.1111/j.1432-1033.1995.tb20529.x. [DOI] [PubMed] [Google Scholar]
- SATOH R., NAKABAYASHI Y., KANO M. Chronic treatment with D600 enhances development of sodium channels in cultured chick skeletal muscle cells. Neurosci. Lett. 1992;138:249–252. doi: 10.1016/0304-3940(92)90926-x. [DOI] [PubMed] [Google Scholar]
- SCHONHORN J.E., WESSLING-RESNICK M. Brefeldin A down-regulates the transferrin receptor in K562 cells. Mol. Cell. Biochem. 1994;135:159–169. doi: 10.1007/BF00926519. [DOI] [PubMed] [Google Scholar]
- SHERMAN S.J., CATTERALL W.A. Electrical activity and cytosolic calcium regulate levels of tetrodotoxin-sensitive sodium channels in cultured rat muscle cells. Proc. Natl. Acad. Sci. U.S.A. 1984;81:262–266. doi: 10.1073/pnas.81.1.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHERMAN S.J., CHRIVIA J., CATTERALL W.A. Cyclic adenosine 3′:5′-monophosphate and cytosolic calcium exert opposing effects on biosynthesis of tetrodotoxin-sensitive sodium channels in rat muscle cells. J. Neurosci. 1985;5:1570–1576. doi: 10.1523/JNEUROSCI.05-06-01570.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIMKETS R.A., LIFTON R.P., CANESSA C.M. The activity of the epithelial sodium channels is regulated by clathrin-mediated endocytosis. J. Biol. Chem. 1997;272:25537–25541. doi: 10.1074/jbc.272.41.25537. [DOI] [PubMed] [Google Scholar]
- SRINIVASAN Y., ELMER L., DAVIS J., BENNETT V., ANGELIDES K. Ankyrin and spectrin associate with voltage-dependent sodium channels in brain. Nature. 1998;333:177–180. doi: 10.1038/333177a0. [DOI] [PubMed] [Google Scholar]
- STAUB O., GAUTSCHI I., ISHIKAWA T., BREITSCHOPH K., CIECHANOVER A., SCHILD L., ROTIN D. Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. EMBO J. 1997;16:6325–6336. doi: 10.1093/emboj/16.21.6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TANAKA K., SHIBUYA I., UEZONO Y., UETA Y., TOYOHIRA Y., YANAGIHARA N., IZUMI F., KANNO T., YAMASHITA H. Pituitary adenylate cyclase-activating polypeptide causes Ca2+ release from ryanodine/caffeine stores through a novel pathway independent of both inositol trisphosphates and cyclic AMP in bovine adrenal medullary cells. J. Neurochem. 1998;70:1652–1661. doi: 10.1046/j.1471-4159.1998.70041652.x. [DOI] [PubMed] [Google Scholar]
- TIMMERMAN L.A., CLIPSTONE N.A., HO S.N., NORTHROP J.P., CRABTREE G.R. Rapid shuttling of NF-AT in discrimination of Ca2+ signals and immunosuppression. Nature. 1996;383:837–840. doi: 10.1038/383837a0. [DOI] [PubMed] [Google Scholar]
- TOLEDO-ARAL J.J., BREHM P., HALEGOUA S., MANDEL G. A single pulse of nerve growth factor triggers long-term neuronal excitability through sodium channel gene induction. Neuron. 1995;14:607–611. doi: 10.1016/0896-6273(95)90317-8. [DOI] [PubMed] [Google Scholar]
- TRAINER V.L., BADEN D.G., CATTERALL W.A. Identification of peptide components of the brevetoxin receptor site of rat brain sodium channels. J. Biol. Chem. 1994;269:19904–19909. [PubMed] [Google Scholar]
- TRAINER V.L., BROWN G.B., CATTERALL W.A. Site of covalent labeling by a photoreactive batrachotoxin derivative near transmembrane segment IS6 of the sodium channel α subunit. J. Biol. Chem. 1996;271:11261–11267. doi: 10.1074/jbc.271.19.11261. [DOI] [PubMed] [Google Scholar]
- URENJAK J., OBRENOVITCH T.P. Pharmacological modulation of voltage-gated Na+ channels: a rational and effective strategy against ischemic brain damage. Pharmacol. Rev. 1996;48:21–67. [PubMed] [Google Scholar]
- VAN HUIZEN F., ROMIJN H.J., HABETS A.M.M.C. Synaptogenesis in rat cerebral cortex cultures is affected during chronic blockade of spontaneous bioelectric activity by tetrodotoxin. Dev. Brain Res. 1985;19:67–80. doi: 10.1016/0165-3806(85)90232-9. [DOI] [PubMed] [Google Scholar]
- WADA A., ARITA M., KOBAYASHI H., IZUMI F. Binding of [3H]saxitoxin to the voltage-dependent Na channels and inhibition of 22Na influx in bovine adrenal medullary cells. Neuroscience. 1987;23:327–331. doi: 10.1016/0306-4522(87)90293-4. [DOI] [PubMed] [Google Scholar]
- WADA A., IZUMI F., YANAGIHARA N., KOBAYASHI H. Modulation by ouabain and diphenylhydantoin of veratridine-induced 22Na influx and its relation to 45Ca influx and the secretion of catecholamines in cultured bovine adrenal medullary cells. Naunyn-Schmiedeberg's Arch. Pharmacol. 1985a;328:273–278. doi: 10.1007/BF00515553. [DOI] [PubMed] [Google Scholar]
- WADA A., TAKARA H., IZUMI F., KOBAYASHI H., YANAGIHARA N. Influx of 22Na through acetylcholine receptor-associated Na channels: relationship between 22Na influx, 45Ca influx and secretion of catecholamines in cultured bovine adrenal medulla cells. Neuroscience. 1985b;15:283–292. doi: 10.1016/0306-4522(85)90135-6. [DOI] [PubMed] [Google Scholar]
- WADA A., TAKARA H., YANAGIHARA N., KOBAYASHI H., IZUMI F. Inhibition of Na+-pump enhances carbachol-induced influx of 45Ca2+ and secretion of catecholamines by elevation of cellular accumulation of 22Na+ in cultured bovine adrenal medullary cells. Naunyn-Schmiedeberg's Arch. Pharmacol. 1986;332:351–356. doi: 10.1007/BF00500086. [DOI] [PubMed] [Google Scholar]
- WADA A., UEZONO Y., ARITA M., YUHI T., KOBAYASHI H., YANAGIHARA N., IZUMI F. Cooperative modulation of voltage-dependent sodium channels by brevetoxin and classical neurotoxins in cultured bovine adrenal medullary cells. J. Pharmacol. Exp. Ther. 1992;263:1347–1351. [PubMed] [Google Scholar]
- WOOD S.J., SLATER C.R. β-Spectrin is colocalized with both voltage-gated sodium channels and ankyrinG at the adult rat neuromuscular junction. J. Cell Biol. 1998;140:675–684. doi: 10.1083/jcb.140.3.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XIA Y., HADDAD G.G. Voltage-sensitive Na+ channels increase in number in newborn rat brain after in utero hypoxia. Brain Res. 1994;635:339–344. doi: 10.1016/0006-8993(94)91459-1. [DOI] [PubMed] [Google Scholar]
- XIA Y., HADDAD G.G. Effect of prolonged O2 deprivation on Na+ channels: differential regulation in adult versus fetal rat brain. Neuroscience. 1999;94:1231–1243. doi: 10.1016/s0306-4522(99)00375-9. [DOI] [PubMed] [Google Scholar]
- YANAGITA T., KOBAYASHI H., YAMAMOTO R., KATAOKA H., YOKOO H., SHIRAISHI S., MINAMI S., KOONO M., WADA A. Protein kinase C-α and -ε down-regulate cell surface sodium channels via differential mechanisms in adrenal chromaffin cells. J. Neurochem. 2000;74:1674–1684. doi: 10.1046/j.1471-4159.2000.0741674.x. [DOI] [PubMed] [Google Scholar]
- YANAGITA T., WADA A., YAMAMOTO R., KOBAYASHI H., YUHI T., URABE M., NIINA H. Protein kinase C-mediated down-regulation of voltage-dependent sodium channels in adrenal chromaffin cells. J. Neurochem. 1996;66:1249–1253. doi: 10.1046/j.1471-4159.1996.66031249.x. [DOI] [PubMed] [Google Scholar]
- YUHI T., WADA A., YAMAMOTO R., URABE M., NIINA H., IZUMI F., YANAGITA T. Characterization of [3H]brevetoxin binding to voltage-dependent sodium channels in adrenal medullary cells. Naunyn-Schmiedeberg's Arch. Pharmacol. 1994;350:209–212. doi: 10.1007/BF00241098. [DOI] [PubMed] [Google Scholar]
- ZHOU D., LAMBERT S., MALEN P.L., CARPENTER S., BOLAND L.M., BENNETT V. AnkyrinG is required for clustering of voltage-gated Na channels at axon initial segments and for normal action potential firing. J. Cell Biol. 1998;143:1295–1304. doi: 10.1083/jcb.143.5.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]