

Abstract
We investigated whether K+ can act as an endothelium-derived hyperpolarizing factor (EDHF) in isolated small renal arteries of Wistar-Kyoto rats.
Acetylcholine (0.001 – 3 μM) caused relaxations that were abolished by removal of the endothelium. However, acetylcholine-induced relaxations were not affected by the nitric oxide (NO) synthase inhibitor Nω-nitro-L-arginine methyl ester (L-NAME, 100 μM), by L-NAME plus the soluble guanylate cyclase inhibitor 1H-[1,2,4]oxadiazolo[4,3,-a]quinoxalin-1-one (ODQ, 1 μM) or by L-NAME plus the cyclo-oxygenase inhibitor indomethacin (10 μM). In rings precontracted with high-K+(60 mM) physiological salt solution in the presence of L-NAME, acetylcholine-induced relaxations were abolished.
L-NAME-resistant relaxations were abolished by the large-conductance Ca2+-activated K+ channel inhibitor charybdotoxin plus the small-conductance Ca2+-activated K+ channel inhibitor apamin, while the inward rectifier K+ channel inhibitor Ba2+ or the gap junction inhibitor 18α-glycyrrhetinic acid had no effect. Acetylcholine-induced relaxation was unchanged by ouabain (10 μM) but was partially inhibited by a higher concentration (100 μM).
In half of the tissues tested, K+(10 mM) itself produced L-NAME-resistant relaxations that were blocked by ouabain (10 μM) and partially reduced by charybdotoxin plus apamin, but not affected by 18α-glycyrrhetinic acid or Ba2+. However, K+ did not induce relaxations in endothelium-denuded tissues.
In conclusion, acetylcholine-induced relaxations in this tissue are largely dependent upon hyperpolarization mechanisms that are initiated in the endothelium but do not depend upon NO release. K+ release cannot account for endothelium-dependent relaxation and cannot be an EDHF in this artery. However, K+ itself can initiate endothelium-dependent relaxations via a different pathway from acetylcholine, but the mechanisms of K+-induced relaxations remain to be clarified.
Keywords: Acetylcholine, Ba2+, endothelium-dependent, hyperpolarization, EDHF, 18α-glycyrrhetinic acid, K+, nitric oxide, ouabain, renal arteries
Introduction
Acetylcholine produces arterial relaxations by releasing several endothelium-derived vasoactive factors, including nitric oxide (NO) and prostacyclin, a cyclo-oxygenase metabolite of arachidonic acid. It has also been proposed that activation of the endothelium can initiate hyperpolarization of the underlying smooth muscle leading to vasorelaxation, and that an endothelium-derived hyperpolarizing factor (EDHF) is involved (Feletou & Vanhoutte, 1999; Garland et al., 1995). While NO itself can induce hyperpolarization of many arteries (Tare et al., 1990), a number of other candidates have been proposed for this elusive chemical mediator (Feletou & Vanhoutte, 1999).
It has been proposed that EDHF initiates hyperpolarization and relaxation of smooth muscle cells by opening K+ channels (Popp et al., 1996; McCulloch et al., 1997; Nishiyama et al., 1998). This effect can be prevented by elevation of extracellular K+ concentration (Adeagbo & Triggle, 1993), or by specific K+ channel inhibitors (Popp et al., 1996; McCulloch et al., 1997; Nishiyama et al., 1998). In the rat hepatic artery, however, Edwards et al. (1998) demonstrated that, upon stimulation with acetylcholine, endothelial cells released sufficient K+ ions to activate directly the inward rectifier K+ channels and electrogenic Na+/K+ adenosine triphosphatase (Na+/K+-ATPase) in the smooth muscle cells, leading to hyperpolarization and relaxation, thus raising the possibility that K+ ion per se was acting as the EDHF in this tissue. Moreover, they demonstrated that K+ channel inhibitors acted by closing the K+ channels in the endothelial cells and preventing K+ efflux, rather than by directly blocking the K+ channels in smooth muscle cells. This novel hypothesis is supported by evidence obtained in rabbit arcuate arteries (Jimenez et al., 1999), whereas findings from other arteries conflicted with the proposed role of K+ ion as an EDHF (Quignard et al., 1999; Doughty et al., 2000; Lacy et al., 2000).
Having found that relaxations in the small renal arteries of Wistar-Kyoto rats induced by acetylcholine were completely resistant to inhibition of NO synthase (Jiang et al., 1999), we have now investigated the role of EDHF in these endothelium-dependent relaxations. The present study was carried out to clarify (1) the role of EDHF in acetylcholine-induced relaxations in isolated small renal arteries from Wistar-Kyoto rats; and (2) whether K+ ion can account for the activity of EDHF.
Methods
Tissue preparation and functional studies
Male Wistar-Kyoto rats (350 – 400 g) were anaesthetized by pentobarbitone sodium (30 mg kg−1, i.p.) and killed by exsanguination. This procedure was approved by the Animal Experimentation Ethics Committee of the University of Melbourne and is in compliance with the guidelines of the Australian National Health and Medical Research Council. Small extrarenal arteries with an outer diameter of 250∼350 μm, which represent the second or third branches of the major renal artery, were dissected out in a physiological salt solution (PSS) at room temperature. Arterial segments of 1.5 mm long were taken for functional studies, with care being taken to protect the integrity of endothelium. In some experiments, the endothelium was deliberately removed by rubbing the inner surface of the segment with a stainless steel wire.
The functional study was performed in a Mulvany myograph (Model 610M, J.P. Trading I/S, Denmark). Blood vessel rings were mounted onto two parallel stainless steel wires through the lumen. One wire was fixed to a displacement micrometer and the other one was connected to a tension transducer. The tissue was placed in a chamber containing 6 ml PSS, which was maintained at 36°C±1 and gassed with 5% CO2 and 95% O2. The isometric tension was displayed and recorded with a MacLab data recording system (AD Instruments Pty. Ltd., Australia).
Experimental protocols
Four rings were obtained from each single animal. After an equilibration period of 20 min, the tissue was normalized to 90% of the inner circumference corresponding to 100 mmHg blood pressure (Mulvany & Halpern, 1977), using a non-linear curve-fitting programme developed by McPherson (1992). This setting represents a resting force of 2 – 3 mN under the present experimental condition. All relaxation responses were observed after the vessel ring was precontracted with 1 μM phenylephrine or isotonic high-K+ PSS ([K+]=60 mM) as indicated. Concentration-response curves of acetylcholine-induced relaxations were constructed in either control or drug-treated rings, and the responses were compared between the control and drug-treated tissues. Since K+-induced responses varied between different tissues, the response in each tissue was used as control for the response obtained after drug-treatment in the same tissue.
Drugs and solutions
The following drugs were used: acetylcholine perchlorate, apamin, charybdotoxin, 18α-glycyrrhetinic acid, indomethacin, L-phenylephrine hydrochloride, Nω-nitro-L-arginine methyl ester hydrochloride (L-NAME), ouabain, 1H-[1,2,4]oxadiazolo[4,3,-a]quinoxalin-l-one (ODQ), sodium nitroprusside. Acetylcholine was from BDH Chemicals Ltd. (U.K.) and charybdotoxin was from Auspep Pty. Ltd. (Melbourne, Australia). Other drugs were from Sigma (St. Louis, MO, U.S.A.).
Stock solutions of the drugs were made by dissolving them in distilled water, except for ODQ and 18α-glycyrrhetinic acid, which were dissolved in dimethylsulphoxide; and for indomethacin, which was dissolved in ethanol. Stock solutions were added directly into the PSS during experimentation. The solvents had no effect on acetylcholine and K+-induced responses. The PSS had the following composition (mM): NaCl 118.0, KCl 4.7, NaHCO3 25.0, MgSO4 1.2, KH2PO4 1.2, CaCl2 2.5 and D-glucose 5.0. The isotonic high-K+ PSS was prepared by replacing NaCl in normal PSS with equal molar KCl. The final concentration of K+ was adjusted before use by mixing high-K+ PSS with normal PSS.
Data and statistical analysis
The tension of the vessel wall was measured in mN. Relaxations were expressed as percentage reductions of phenylephrine-produced contraction. pEC50 (which is the negative logarithm molar concentration required to produce 50% of the maximal response) values were calculated using a GraphPad Prism software. Data were presented as mean±standard error of the mean (s.e.mean). The mean data were analysed with one-way analysis of variance (one-way ANOVA) followed by Student's t-test or Tukey's test as appropriate. A value of P<0.05 was regarded as statistically significant.
Results
Effects of L-NAME, ODQ and indomethacin on acetylcholine-induced relaxations
In endothelium-intact small renal artery rings, phenylephrine (1 μM) produced a mean contraction of 9.4 mN±1.0 (n=5). Acetylcholine (0.001 – 3 μM) produced concentration-dependent relaxations of phenylephrine-induced tone, which were abolished by denuding rings of endothelium. Treatment with the NO synthase inhibitor L-NAME (100 μM) significantly increased phenylephrine-induced tone to 16.9 mN±1.3 (n=9, P<0.005). However, L-NAME alone, or L-NAME plus the guanylate cyclase inhibitor ODQ (1 μM) or L-NAME plus the cyclo-oxygenase inhibitor indomethacin (10 μM), had no significant effect on acetylcholine-induced relaxations (Figure 1a). In contrast, ODQ (1 μM) nearly abolished relaxations induced by the NO donor sodium nitroprusside (1 μM) (Figure 1b).
Figure 1.
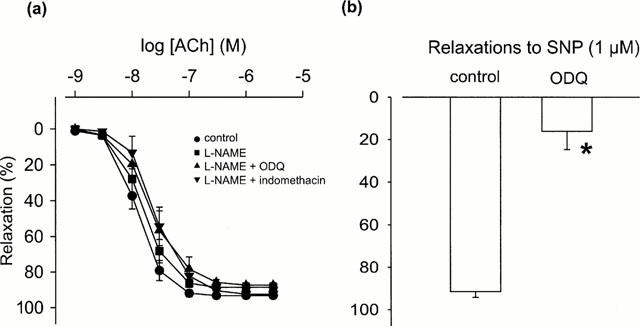
Effects of (a) the nitric oxide (NO) synthase inhibitor L-NAME (100 μM), L-NAME plus soluble guanylate cyclase inhibitor ODQ (1 μM) and L-NAME plus cyclo-oxygenase inhibitor indomethacin (10 μM) on acetylcholine (ACh)-induced relaxations (n=4 – 9); and (b) ODQ (1 μM) alone on the NO donor sodium nitroprusside (SNP, 1 μM)-induced relaxations (n=3 – 4) in endothelium-intact small renal arteries from Wistar-Kyoto rats. Relaxations are presented as percentage reductions of the precontractions produced by phenylephrine (1 μM). Data are mean±standard error of the mean (s.e.mean). *P<0.001, one-way analysis of the variance (one-way ANOVA) followed by Student's t-test.
NO independent relaxations
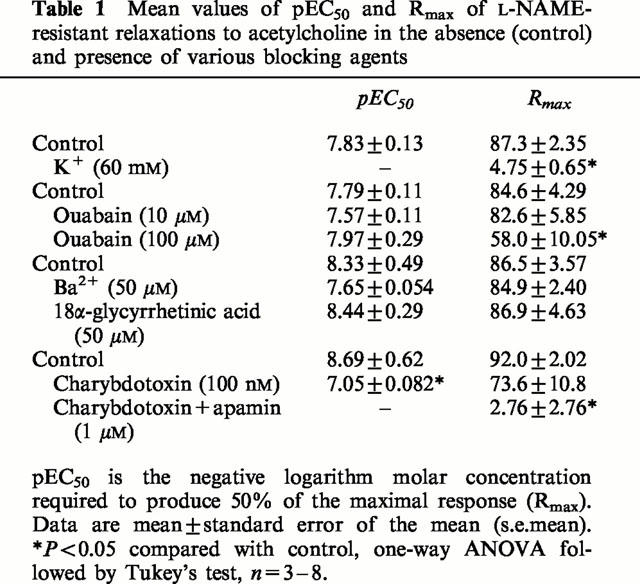
In the presence of L-NAME (100 μM), 60 mM high-K+ PSS precontracted the tissues to a level similar to that induced by phenylephrine (1 μM), the mean value being 19.4 mN±1.2 (n=4, P>0.05). In K+ precontracted tissues, acetylcholine-induced relaxations were abolished (Figure 2a). In phenylephrine-contracted tissues, L-NAME-resistant relaxations to acetylcholine were not affected by the Na+/K+-ATPase inhibitor ouabain at 10 μM (Figure 2a), but were slightly reduced by this agent at 100 μM. Comparable relaxations induced by 1 μM sodium nitroprusside were not changed by ouabain (100 μM). L-NAME-resistant relaxations were not affected by the inward rectifier K+ channel inhibitor Ba2+ (added as BaCl2, 50 μM), or the gap junction inhibitor 18α-glycyrrhetinic acid (50 μM) (Figure 2a), but were significantly reduced by the large-conductance Ca2+-activated K+ channel blocker charybdotoxin (100 nM) and were abolished by charybdotoxin plus the small-conductance Ca2+-activated K+ channel blocker apamin (1 μM) (Figure 2a). These antagonists did not significantly change phenylephrine-induced tone. The mean data of the effects of high-K+ PSS, ouabain, Ba2+, 18α-glycyrrhetinic acid, charybdotoxin and charybdotoxin plus apamin on L-NAME-resistant relaxations are shown in Figure 3. The pEC50 and Rmax values of acetylcholine-induced responses in the absence and presence of these drugs are given in Table 1.
Figure 2.
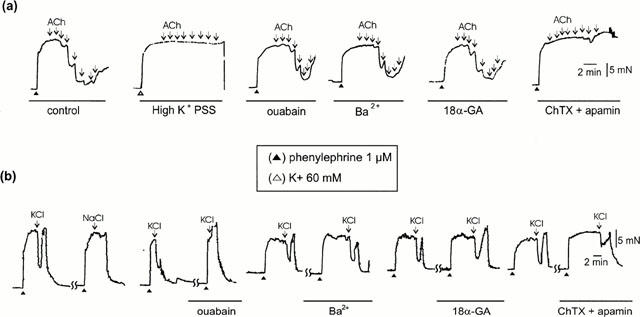
Original records showing the relaxations induced by (a) acetylcholine (ACh, 1 nM – 3 μM, as shown by downward arrows) and (b) 10 mM K+ (KCl) in tissues treated with high-K+ (60 mM) physiological salt solution (PSS), the Na+/K+-ATPase inhibitor ouabain (10 μM), the inward rectifier K+ channel inhibitor Ba2+ (50 μM), the gap junction inhibitor 18α-glycyrrhetinic acid (18α-GA, 50 μM), or the large-conductance Ca2+-activated K+ channel inhibitor charybdotoxin (100 nM) plus the small-conductance Ca2+-activated K+ channel inhibitor apamin (1 μM) (ChTX+apamin). All experiments were in the presence of L-NAME (100 μM). The first trace in the lower panel shows that 10 mM K+-induced relaxations could not be mimicked by equal molar NaCl. (▴) precontractions produced by 1 μM phenylephrine; (Δ) precontraction produced by high-K+ PSS.
Figure 3.
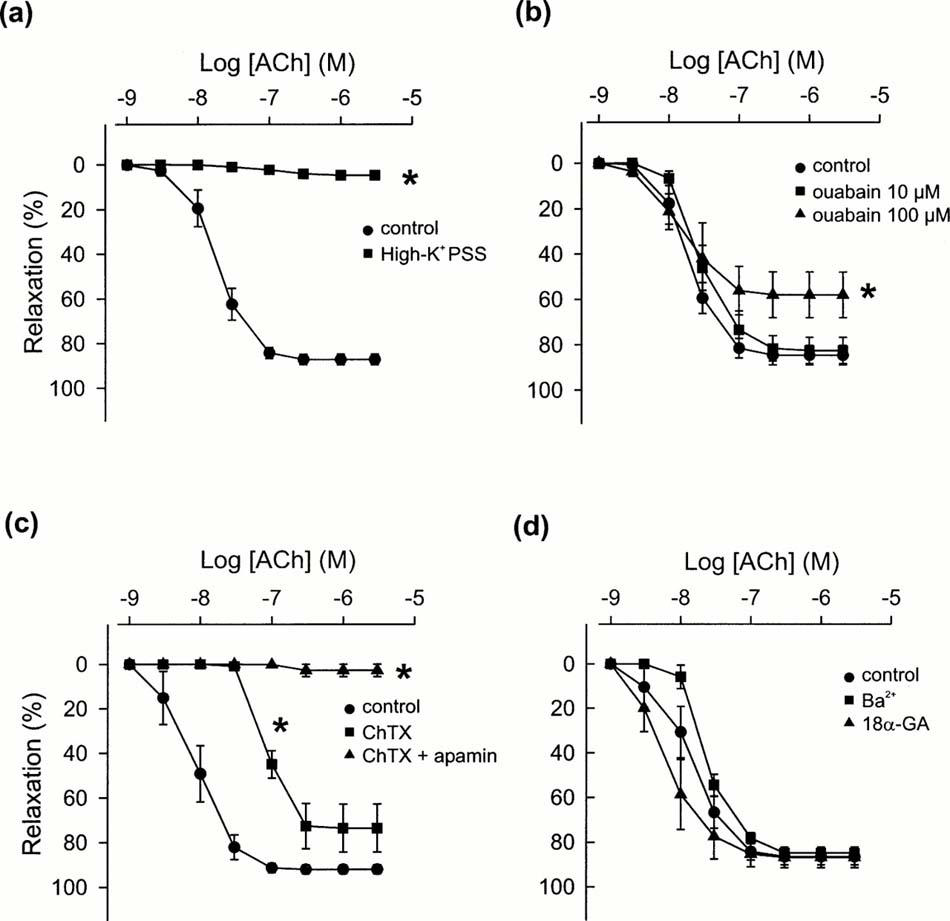
Effects of (a) high-K+ (60 mM), PSS, (b) ouabain (10 and 100 μM), (c) charybdotoxin (ChTX, 100 nM) and charybdotoxin plus apamin (1 μM), and (d) Ba2+ (50 μM) or 18α-glycyrrhetinic acid (18α-GA, 50 μM) on acetylcholine (ACh)-induced relaxations in the presence of 100 μM L-NAME. Tissues were precontracted with phenylephrine (1 μM) except for those in high-K+ PSS experiments in which tissues were contracted with high-K+ PSS. Relaxations are presented as percentage reductions of the precontractions. Data are mean±s.e.mean. (n=3 – 8). *P<0.05 vs control, one-way ANOVA followed by Tukey's test.
Table 1.
Mean values of pEC50 and Rmax of L-NAME-resistant relaxations to acetylcholine in the absence (control) and presence of various blocking agents
K+-induced relaxations
In phenylephrine contracted tissues, K+ (10 mM), added as KCl to normal PSS, produced relaxations in the presence of L-NAME. In 15 out of 32 tissues from eight animals, K+ produced relaxations with a mean value of 56.1±5.6% (Figure 2b). This relaxation could not be reproduced by equal molar NaCl, suggesting that this response is specific for K+ ions and is not due to the change of ionic concentrations in PSS. K+-induced relaxations were totally blocked by ouabain at 10 μM, but were not changed by Ba2+ (50 μM) or 18α-glycyrrhetinic acid (50 μM). This response was partially reduced by charybdotoxin (100 nM) plus apamin (1 μM) (Figure 2b). The mean data are shown in Figure 4. However, K+-induced relaxations were never observed in endothelium-denuded tissues.
Figure 4.
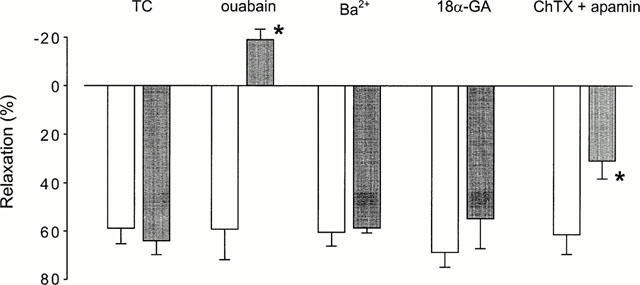
Effects of ouabain (10 μM), Ba2+ (50 μM), 18α-glycyrrhetinic acid (18α-GA, 50 μM) and charybdotoxin (ChTX, 100 nM) plus apamin (1 μM) on relaxations induced by 10 mM K+ in L-NAME-pretreated tissues. Open bars are control responses and closed bars are time control (TC) or drug-treated responses. Relaxations are expressed as percentages of 1 μM phenylephrine-induced precontractions, while the negative value means a contraction response. Data are mean±s.e.mean. *P<0.05, one-way ANOVA followed by Student's t-test, n=4 – 6.
Discussion
In the present study with isolated small renal arteries of Wistar-Kyoto rats, we have demonstrated that acetylcholine-induced relaxation is largely mediated by endothelium-dependent hyperpolarizing mechanisms, while endothelium-derived NO or prostacyclin makes little (or a redundant) contribution to the vasorelaxation. This was indicated by the following observations: (1) acetylcholine-induced endothelium-dependent relaxations were not affected by either the NO synthase inhibitor L-NAME alone or L-NAME in combination with the soluble guanylate cyclase inhibitor ODQ, which markedly blocked the relaxations induced by the NO donor sodium nitroprusside; and (2) this L-NAME-resistant response was not changed by indomethacin, but was abolished in tissues depolarized with 60 mM K+, or by the combination of the large-conductance Ca2+-activated K+ channel blocker charybdotoxin plus the small-conductance Ca2+-activated K+ channel blocker apamin (Petersson et al., 1997). These results provide further evidence supporting the view that in small resistance arteries, hyperpolarizing mechanisms have a predominant role in vasodilatation, and are more important than NO (Garland et al., 1995). However, it should be noted that these findings do not exclude a role for NO in basal tone, because L-NAME nearly doubled the contraction force induced by phenylephrine.
We also investigated the relaxations induced by elevation of the extracellular K+ concentration in a normal physiological ionic environment. However, it is clear that in this tissue release of K+ is unlikely to account for EDHF activity as suggested by Edwards et al. (1998), because firstly, K+-induced relaxations, unlike those induced by acetylcholine, could not be observed consistently in all preparations, and appeared to be somewhat variable in a single tissue during the time course of experimentation. Secondly, K+-induced relaxations were abolished by low concentrations of ouabain whereas the responses to acetylcholine were not changed by ouabain at the same concentration and only partially reduced by ouabain at higher concentrations. Thirdly, K+-induced relaxations appeared to be dependent on the integrity of endothelial cells. These findings are similar to those obtained in rat mesenteric arteries (Lacy et al., 2000), suggesting that K+ is not an EDHF in these tissues, but may cause vasorelaxation by initiating hyperpolarization of endothelial cells.
Several mechanisms have been identified to account for K+-induced relaxations in arterial smooth muscle cells, including the activation of smooth muscle Na+/K+-ATPase (Webb & Bohr, 1978) and the opening of Ba2+-sensitive inward rectifier K+ channels (Knot et al., 1996). Our results have shown that K+-induced relaxations were completely blocked by ouabain, indicating that activation of Na+/K+-ATPase was involved in this response.
However, it is unlikely that the relaxation produced by K+ is due to a direct stimulation of the Na+/K+-ATPase in the smooth muscle cells since this relaxation appeared to be endothelium-dependent. Similar endothelium-dependent relaxations induced by K+ ions were also observed in rat mesenteric small arteries (Lacy et al., 2000). The mechanisms of this phenomenon are not clear. Given the evidence that endothelial cells also express Na+/K+-ATPase (Mayol et al., 1998), one possible explanation of the present results is that K+ stimulates endothelial Na+/K+-ATPase leading to endothelial hyperpolarization, which subsequently produces smooth muscle hyperpolarization and relaxation via myo-endothelial gap junctions (Edwards et al., 2000). However, other mechanisms could not be excluded in this study because firstly, K+-induced relaxations were also partially inhibited by charybdotoxin plus apamin, of which the mechanisms were unknown; secondly, the gap junction inhibitor 18α-glycyrrhetinic acid (Guo et al., 1999) had no effect on K+-induced relaxations. However, the specificity of this drug in blocking vascular myo-endothelial gap junctions remains to be clearly established.
In contrast to the results in rat mesenteric arteries (Lacy et al., 2000), in the present study Ba2+ did not change either acetylcholine- or K+-induced relaxations, suggesting that inward rectifier K+ channels are not involved (Prior et al., 1998). Our findings however did not provide direct evidence for the identity of the putative EDHF or the mechanisms by which EDHF produces biological effects. EDHF-mediated relaxations were not reduced by the cytochrome P450 inhibitor miconazole (10 μM) (data not shown), suggesting that in this tissue the EDHF is unlikely to be a cytochrome P450 metabolite as suggested by other authors (Popp et al., 1996). On the other hand, EDHF-mediated relaxations were reduced by charybdotoxin and abolished by charybdotoxin plus apamin, suggesting that EDHF-induced relaxation is associated with the opening of particular K+ channels (Andersson et al., 2000). Given that K+ cannot account for EDHF in this tissue, it is suggested that the effect of charybdotoxin plus apamin is due to the blockade of K+ channels in smooth muscle cells rather than those on endothelial cells (Quignard et al., 2000). The weak inhibitory effect of ouabain suggests that Na+/K+-ATPase does not have a significant role in the EDHF-elicited relaxations, although some studies using other arteries and other species demonstrated that activation of Na+/K+-ATPase might be involved in EDHF-mediated relaxations (Jiang et al., 2000; Kitagawa et al., 1994; Van de Voorde & Vanheel, 2000). The partial inhibition of acetylcholine-induced relaxations by ouabain at a high concentration may be due to the partial depolarization of the endothelial cells which attenuates the release of endothelium-derived factors (Luckhoff & Busse, 1990). This is also consistent with the observation that addition of K+ could not directly activate the smooth muscle Na+/K+-ATPase to induce hyperpolarization and relaxation, suggesting a minor functional role of Na+/K+-ATPase in the smooth muscle cells in this vessel. On the other hand, NO and prostacyclin-independent relaxations to acetylcholine were not affected by 18α-glycyrrhetinic acid. This is similar to the results reported in the rat mesenteric arteries (Tanaka et al., 1999), raising doubt about whether gap junctions are involved in endothelium-dependent relaxations in these arteries.
In summary, in the small renal arteries of Wistar-Kyoto rats, hyperpolarizing mechanisms have a predominant role in mediating acetylcholine-induced relaxations through the activation of particular K+ channels in smooth muscle cells. In this tissue, K+ ion is unlikely to act as an EDHF. It appears that K+ itself may produce relaxations via mechanisms dependent on the endothelium that remain to be clarified.
Acknowledgments
This work was supported by an institute block grant from the National Health and Medical Research Council of Australia (No. 983001).
Abbreviations
- EC50
concentration producing 50% of maximal response
- EDHF
endothelium-derived hyperpolarizing factor
- Na+/K+-ATPase
Na+/K+ adenosine triphosphatase
- NO
nitric oxide
- L-NAME
Nω-nitro-L-arginine methyl ester
- ODQ
1H-[1,2,4]oxadiazolo[4,3,-a]quinoxalin-1-one
- PSS
physiological salt solution
- Rmax
maximal response
References
- ADEAGBO A.S., TRIGGLE C.R. Varying extracellular [K+]: a functional approach to separating EDHF- and EDNO-related mechanisms in perfused rat mesenteric arterial bed. J. Cardiovasc. Pharmacol. 1993;21:423–429. [PubMed] [Google Scholar]
- ANDERSSON D.A., ZYGMUNT P.M., MOVAHED P., ANDERSSON T.L., HOGESTATT E.D. Effects of inhibitors of small- and intermediate-conductance calcium-activated potassium channels, inwardly-rectifying potassium channels and Na+/K+ ATPase on EDHF relaxations in the rat hepatic artery. Br. J. Pharmacol. 2000;129:1490–1496. doi: 10.1038/sj.bjp.0703226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOUGHTY J.M., BOYLE J.P., LANGTON P.D. Potassium does not mimic EDHF in rat mesenteric arteries. Br. J. Pharmacol. 2000;130:1174–1182. doi: 10.1038/sj.bjp.0703412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDWARDS G., DORA K.A., GARDENER M.J., GARLAND C.J., WESTON A.H. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- EDWARDS G., THOLLON C., GARDENER M.J., FELETOU M., VILAINE J., VANHOUTTE P.M., WESTON A.H. Role of gap junctions and EETs in endothelium-dependent hyperpolarization of porcine coronary artery. Br. J. Pharmacol. 2000;129:1145–1154. doi: 10.1038/sj.bjp.0703188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FELETOU M., VANHOUTTE P.M. The third pathway: endothelium-dependent hyperpolarization. J. Physiol. Pharmacol. 1999;50:525–534. [PubMed] [Google Scholar]
- GARLAND C.J., PLANE F., KEMP B.K., COCKS T.M. Endothelium-dependent hyperpolarization: a role in the control of vascular tone. Trends Pharmacol. Sci. 1995;16:23–30. doi: 10.1016/s0165-6147(00)88969-5. [DOI] [PubMed] [Google Scholar]
- GUO Y., MARTINEZ-WILLIAMS C., GILBERT K.A., RANNELS D.E. Inhibition of gap junction communication in alveolar epithelial cells by 18α-glycyrrhetinic acid. Am. J. Physiol. 1999;276:L1018–L1026. doi: 10.1152/ajplung.1999.276.6.L1018. [DOI] [PubMed] [Google Scholar]
- JIANG F., BILSZTA J.L.C., DUSTING G.J. Induction of heat shock proteins and vascular tone. Proc. Australasian Soc. Clin. Exp. Pharmacologists Toxicologists. 1999;6:76. [Google Scholar]
- JIANG F., LI C.G., RAND M.J. Mechanisms of nitric oxide-independent relaxations induced by carbachol and acetylcholine in rat isolated renal arteries. Br. J. Pharmacol. 2000;130:1191–1200. doi: 10.1038/sj.bjp.0703408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JIMENEZ R., DORA K.A., GARLAND C.J. A role for K+ in relaxation responses to acetylcholine in rabbit isolated arcuate artery. Br. J. Pharmacol. 1999;127 Suppl.:107P. [Google Scholar]
- KITAGAWA S., YAMAGUCHI Y., KUNITOMO M., SAMESHIMA E., FUJIWARA M. NG-nitro-L-arginine-resistant endothelium-dependent relaxation induced by acetylcholine in the rabbit renal artery. Life Sci. 1994;55:491–498. doi: 10.1016/0024-3205(94)00741-1. [DOI] [PubMed] [Google Scholar]
- KNOT H.J., ZIMMERMANN P.A., NELSON M.T. Extracellular K+-induced hyperpolarizations and dilatations of rat coronary and cerebral arteries involve inward rectifier K+ channels. J. Physiol. (Lond) 1996;492:419–430. doi: 10.1113/jphysiol.1996.sp021318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LACY P.S., PILKINGTON G., HANVESAKUL R., FISH H.J., BOYLE J.P., THURSTON H. Evidence against potassium as an endothelium-derived hyperpolarizing factor in rat mesenteric small arteries. Br. J. Pharmacol. 2000;129:605–611. doi: 10.1038/sj.bjp.0703076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUCKHOFF A., BUSSE R. Calcium influx into endothelial cells and formation of endothelium-derived relaxing factor is controlled by the membrane potential. Pflügers Arch. 1990;416:305–311. doi: 10.1007/BF00392067. [DOI] [PubMed] [Google Scholar]
- MAYOL V., DIGNAT-GEORGE F., GERBI A., MARTIN-VASALLO P., LESAULE G., SAMPOL J., MAIXENT J.M. Evidence that human endothelial cells express different isoforms of Na,K-ATPase. J. Hypertens. 1998;16:145–150. doi: 10.1097/00004872-199816020-00003. [DOI] [PubMed] [Google Scholar]
- MCCULLOCH A.I., BOTTRILL F.E., RANDALL M.D., HILEY C.R. Characterization and modulation of EDHF-mediated relaxations in the rat isolated superior mesenteric arterial bed. Br. J. Pharmacol. 1997;120:1431–1438. doi: 10.1038/sj.bjp.0701066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCPHERSON G.A. Optimal conditions for assessing vascular reactivity in small resistance arteries in the small vessel myograph. Clin. Exp. Pharmacol. Physiol. 1992;19:815–825. doi: 10.1111/j.1440-1681.1992.tb00420.x. [DOI] [PubMed] [Google Scholar]
- MULVANY M.J., HALPERN W. Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circ. Res. 1977;41:19–26. doi: 10.1161/01.res.41.1.19. [DOI] [PubMed] [Google Scholar]
- NISHIYAMA M., HASHITANI H., FUKUTA H., YAMAMOTO Y., SUZUKI H. Potassium channels activated in the endothelium-dependent hyperpolarization in guinea-pig coronary artery. J. Physiol. (Lond) 1998;510:455–465. doi: 10.1111/j.1469-7793.1998.455bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PETERSSON J., ZYGMUNT P.M., HOGESTATT E.D. Characterization of the potassium channels involved in EDHF-mediated relaxation in cerebral arteries. Br. J. Pharmacol. 1997;120:1344–1350. doi: 10.1038/sj.bjp.0701032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POPP R., BAUERSACHS J., HECKER M., FLEMING I., BUSSE R. A transferable, β-naphthoflavone-inducible, hyperpolarizing factor is synthesized by native and cultured porcine coronary endothelial cells. J. Physiol. (Lond) 1996;497:699–709. doi: 10.1113/jphysiol.1996.sp021801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRIOR H.M., WEBSTER N., QUINN K., BEECH D.J., YATES M.S. K+-induced dilation of a small renal artery: no role for inward rectifier K+ channels. Cardiovasc. Res. 1998;37:780–790. doi: 10.1016/s0008-6363(97)00237-x. [DOI] [PubMed] [Google Scholar]
- QUIGNARD J.F., FELETOU M., EDWARDS G., DUHAULT J., WESTON A.H., VANHOUTTE P.M. Role of endothelial cell hyperpolarization in EDHF-mediated responses in the guinea-pig carotid artery. Br. J. Pharmacol. 2000;129:1103–1112. doi: 10.1038/sj.bjp.0703175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QUIGNARD J.F., FELETOU M., THOLLON C., VILAINE J.P., DUHAULT J., VANHOUTTE P.M. Potassium ions and endothelium-derived hyperpolarizing factor in guinea-pig carotid and porcine coronary arteries. Br. J. Pharmacol. 1999;127:27–34. doi: 10.1038/sj.bjp.0702493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TANAKA Y., OTSUKA A., TANAKA H., SHIGENOBU K. Glycyrrhetinic acid-sensitive mechanism does not make a major contribution to non-prostanoid, non-nitric oxide mediated endothelium-dependent relaxation of rat mesenteric artery in response to acetylcholine. Res. Commun. Mol. Pathol. Pharmacol. 1999;103:227–239. [PubMed] [Google Scholar]
- TARE M., PARKINGTON H.C., COLEMAN H.A., NEILD T.O., DUSTING G.J. Hyperpolarization and relaxation of arterial smooth muscle caused by nitric oxide derived from the endothelium. Nature. 1990;346:69–71. doi: 10.1038/346069a0. [DOI] [PubMed] [Google Scholar]
- VAN DE VOORDE J., VANHEEL B. EDHF-mediated relaxation in rat gastric small arteries: influence of ouabain/Ba2+ and relation to potassium ions. J. Cardiovasc. Pharmacol. 2000;35:543–548. doi: 10.1097/00005344-200004000-00005. [DOI] [PubMed] [Google Scholar]
- WEBB R.C., BOHR D.F. Potassium-induced relaxation as an indicator of Na+-K+ ATPase activity in vascular smooth muscle. Blood Vessels. 1978;15:198–207. doi: 10.1159/000158166. [DOI] [PubMed] [Google Scholar]