

Abstract
2′-& 3′-O-(4-benzoylbenzoyl)-ATP (BzATP) is the prototypic agonist for P2X7 receptors. In this study we demonstrate that bovine serum albumin (BSA) can affect the potency of BzATP at P2X receptors.
BzATP potency (pEC50) to stimulate ethidium accumulation in cells expressing recombinant P2X7 receptors varied between 6.5 and 4, depending upon the species orthologue studied and ionic conditions employed. BSA (0.1 – 1 mg ml−1) and foetal bovine serum (FBS, 1 – 10% v v−1) inhibited responses to BzATP but only when the BzATP pEC50 exceeded 5.
BSA did not block ATP-stimulated ethidium accumulation, suggesting its effects were independent of P2X7 receptor blockade.
BSA did not cause breakdown of nucleotides, although FBS (10% v v−1) exhibited appreciable nucleotidase activity and caused significant breakdown of ATP.
In the presence of BSA, lipids such as 11-((5-dimethylaminonaphthalene-1-sulphonyl)amino)undecanoic acid (DAUDA) and arachidonic acid (AA) markedly increased BzATP potency. Lipids had no affect on ATP potency in the presence of BSA and had little effect on responses to BzATP in the absence of BSA.
These results suggested that the reduction in BzATP potency by BSA was due to BzATP binding to BSA and that lipids prevented this binding. Consistent with this hypothesis, BzATP inhibited binding of the fluorescent lipid, DAUDA, to BSA.
In conclusion, BSA and lipids can markedly affect BzATP potency at P2X7 receptors but this is probably a consequence of BzATP binding to BSA. This finding has important implications when using BzATP in vivo or in the presence of albumin.
Keywords: BzATP, bovine serum albumin, P2X7 receptor
Introduction
The ATP analogue 2′-& 3′-O-(4-benzoylbenzoyl)-ATP (BzATP) was originally introduced as a photoaffinity label for ATPase (Williams & Coleman, 1982) but has since become a widely used tool for studying P2 receptors. An effect of BzATP on P2 receptors was first shown by Gonzalez et al. (1989), who found that BzATP was a more potent agonist than ATP at permeabilizing transformed mouse fibroblasts. With hindsight, this effect was probably due to activation of P2X7 receptors (see below). Subsequently, BzATP was shown to be an agonist of the P2Y receptors in turkey erythrocytes and radiolabelled BzATP was used to label these sites (Boyer & Harden, 1989; Boyer et al., 1990; Erb et al., 1990). More recently, BzATP has also been shown to activate the P2Y11 receptor (Conigrave et al., 1998; Communi et al., 1999), and, in contrast to its actions of turkey erythrocyte P2Y receptors, to be an antagonist of rat and human P2Y1 receptors (Vigne et al., 1999). Finally, BzATP is an agonist of most P2X receptor types (Evans et al., 1995; Bianchi et al., 1999), exhibiting extremely high potency (pEC50=8.7) at P2X1 receptors (Bianchi et al., 1999; although see Evans et al., 1995).
The main utility of BzATP in P2 receptor research has been as an agonist of the P2X7 receptor (formerly PZ). The P2X7 receptor is an ATP-gated cation channel which is able to mediate formation of pores in cell membranes, permeable to molecules with MW of up to 800 D, and to cause cell lysis (Steinberg et al., 1987; Surprenant et al., 1996). BzATP is the most potent agonist of the P2X7 receptor, being 5 – 10 fold more potent than ATP, which possesses much lower potency at P2X7 receptors than at other members of the P2X family (Surprenant et al., 1996). Even though BzATP also possesses high potency at other P2X receptors (Bianchi et al., 1999), the very low potency of ATP (100 – 1000 μM), coupled with a higher potency of BzATP, provides a suitable method for differentiating P2X7 receptors from other P2X receptor types. It should, of course, be noted that the potencies of ATP and BzATP at the P2X7 receptor are similar to those observed at the metabotropic P2Y11 receptor (Conigrave et al., 1998; Communi et al., 1999).
In the present study we demonstrate that caution must be exercised when using BzATP as its potency at the P2X7 receptor can be markedly affected by bovine serum albumin (BSA) and foetal bovine serum (FBS). This effect, which appears to be due to BzATP binding to serum albumin, could result in a considerable underestimation of BzATP potency at P2 receptors in vivo or in studies where albumin is present. In addition, several agents, including lipids such as arachidonic acid (AA), prevent serum albumin binding of BzATP and, in the presence of BSA, can appear to exert a very marked effect on P2X7 receptor function.
Methods
Tissue culture
All studies were performed using HEK293 cells expressing rat (Surprenant et al., 1996), human (Rassendren et al., 1997) or mouse (Chessell et al., 1998) recombinant P2X7 receptors. The cell lines were maintained in a 1 : 1 mixture of Dulbecco's Modified Eagle's Medium and Hams F12 (DMEM/F12) containing 10% FBS and 0.6 mg ml−1 geneticin sulphate. Cells were grown as monolayer cultures at 37°C in a humidified atmosphere containing 95% air and 5% CO2 and were harvested when confluent by incubation in Trypsin-EDTA solution and diluted 10 fold into fresh growth media.
Assay buffers
The assay buffers comprised (in mM): HEPES 10, N-methyl-D-glucamine 5, KCl 5.6, D-glucose 10, CaCl2 0.5 (pH 7.4) and were supplemented with either 280 mM sucrose (sucrose buffer) or 140 mM NaCl (NaCl buffer). Unless indicated all solutions contained 0.5 mM CaCl2 and no added MgCl2. In some studies the 0.5 mM CaCl2 was omitted and 0.1 mM EDTA included (sucrose-EDTA buffer) while in other studies the CaCl2 concentration was reduced to 0.1 mM (sucrose 0.1 CaCl2 buffer).
Ethidium accumulation
Cells expressing rat, mouse or human recombinant P2X7 receptors were harvested and added to poly-L-lysine pretreated 96-well plates (Costar, U.K.) and grown until they formed a confluent monolayer (18 – 24 h). Before use, cells were washed twice with 350 μl of assay buffer before addition of 100 μl of reaction mixture. This mixture was prepared in a separate 96-well plate and contained ethidium (100 μM final assay concentration) and other reagents in assay buffer. ATP or BzATP were added to this plate 4 min before addition to the cells. Incubations were continued until approximately 20 – 40% of maximal dye accumulation occurred (see below). The ethidium solution was removed and cellular accumulation of ethidium was determined by measuring fluorescence from below the plate with a Canberra Packard Fluorocount (excitation wavelength of 530 nm and emission wavelength of 620 nm). All incubations were at 22°C.
The rates of agonist-stimulated ethidium accumulation varied considerably between the three species and were also affected by the ionic composition of the assay buffer. Consequently, agonist exposure times were adjusted to ensure that measurements of agonist potency were determined when approximately 20 – 40% of the maximal agonist-stimulated dye accumulation had occurred. Thus, in sucrose buffer incubation times with agonist were 1, 2 and 15 min when studying rat, human and mouse receptors, respectively. In NaCl buffer, incubation times with agonist were 2 and 10 min, respectively, when studying rat and human receptors. For studies on the mouse receptor in sucrose-EDTA buffer a 5 min incubation was employed, while for studies on the human receptor in sucrose buffer containing 0.1 mM CaCl2, a 1 min incubation time was used. It should be noted that estimates of agonist potency do not vary significantly when determined after various time of agonist exposure (Michel et al., 2000; A.D. Michel, unpublished observations).
Nucleotide binding to BSA
Binding of nucleotides to BSA was assessed indirectly by studying their effect on the binding of 11-((5-dimethylaminonaphthalene-1-sulphonyl)amino)undecanoic acid (DAUDA) to BSA (Wilkinson & Wilton, 1986). The fluorescence of DAUDA increases up to 60 fold upon binding to BSA and agents which compete for DAUDA binding sites on BSA reduce this fluorescence (Wilton, 1990). Nucleotides, diluted in sucrose buffer, were added to a solution of DAUDA (3 μM) and 0.01% BSA in sucrose buffer and fluorescence was measured at an excitation wavelength of 340 nm and emission wavelength of 490 nm, using an Hitachi F2000 spectrofluorimeter.
ATP breakdown studies
The breakdown of ATP by BSA or FBS solutions was determined using the luciferin/luciferase technique. Solutions of ATP were incubated in the absence or presence of BSA or FBS in either sucrose buffer or DMEM/F12 media. Samples (10 μl) were removed at various times, added to 100 μl of luciferin/luciferase mixture (Sigma) and luminescence was measured using a Canberra Packard Topcount scintillation and luminescence counter.
Data analysis
In studies on ethidium-accumulation, individual concentration-effect curves (CEC) were fitted to a four parameter logistic function using GraphPad Prism 3 to determine the maximum and minimum responses and to calculate the pEC50 value and the Hill slope. The data are the mean±s.e.mean of 3 – 5 experiments and statistical tests were performed using GraphPad Prism 3. For graphical purposes, CEC are presented as a percentage of the maximal response obtained in one of the experimental groups.
In studies on DAUDA binding to BSA, the inhibition data for BzATP were analysed using GraphPad Prism 3 according to a 2 site binding model. In this model the data were analysed assuming two components of DAUDA binding to BSA. The analysis provided estimates of the pIC50 for BzATP at these two components and the proportion of the total fluorescence signal that each component comprised.
Materials
All tissue culture reagents and FBS were from Gibco (Paisley, U.K.). ATP was from Promega (Southampton, U.K.). BSA, BzATP and AA were obtained from Sigma (Poole, U.K.). Unless stated, grade V, fatty acid free, BSA (Catalogue Number A0281) was used. In some studies, BSA grades IV (Sigma Catalogue Number A7511) and V (Sigma Catalogue Numbers A3059, A3294 and A7906) were also examined. DAUDA was obtained from Molecular probes (Cambridge Bioscience, Cambridge, U.K.) and was dissolved in DMSO (30 mM solution) before dilution into assay buffer.
Results
Effect of BSA on responses to BzATP and ATP in sucrose buffer
In cells expressing human or rat P2X7 receptors, BSA (0.1 – 3 mg ml−1) produced a concentration-dependent decrease in the potency of BzATP to stimulate ethidium-accumulation, with no effect on the maximal response (Figure 1). The decrease in pEC50 was significant (P<0.05, 1-way ANOVA and Dunnett's test) at BSA concentrations of 0.3 and 0.1 mg ml−1, respectively, at human and rat P2X7 receptors. In addition, at 1 and 3 mg ml−1 BSA, the slope of the BzATP CEC increased significantly. For example, at the rat P2X7 receptor, Hill slopes increased from 1.9±0.4 in the absence of BSA to 10.4±3.7 in the presence of 1 mg ml−1 BSA. These data are presented as a Schild plot in Figure 1D. At the human receptor the Schild slope was 0.99 and the estimated pA2 was 0.59±0.13 (corresponding to a BSA concentration of 0.26 mg ml−1), while at the rat receptor the Schild slope was 1.2 and the estimated pA2 was 1.28±0.14 (corresponding to a BSA concentration of 0.052 mg ml−1). At the mouse P2X7 receptor BSA was less effective (Figure 1D) and only produced a 2.1±0.4 fold shift in the BzATP DRC at 2 mg ml−1 BSA (BzATP pEC50 control=4.18±0.02; BSA=3.87±0.08; P<0.05, 1-way ANOVA and Dunnett's test).
Figure 1.
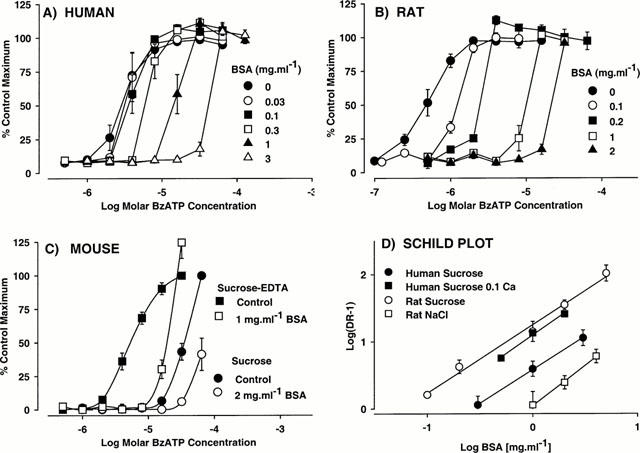
The effect of bovine serum albumin (BSA) on BzATP-stimulated ethidium accumulation in cells expressing (A) human, (B) rat or (C) mouse P2X7 receptors. Concentration-effect curves (CEC) to BzATP were obtained in the absence or presence of BSA. Data are the mean±s.e.mean of four experiments and are expressed as a percentage of the maximum response obtained in the absence of BSA. Lines through data points do not represent lines of best fit. Basal fluorescence values were not removed. In (A – C), CEC were obtained in sucrose buffer containing 0.5 mM CaCl2 (sucrose) while in (C) CEC were also obtained in calcium-free sucrose buffer containing 0.1 mM EDTA (Sucrose EDTA). In (D), the ratios of EC50 values determined in the presence of the indicated concentrations of BSA to those determined in the absence of BSA (Dose-ratio, DR) were calculated and are presented as a Schild plot. These data were obtained in either NaCl buffer (NaCl) or sucrose buffer containing either 0.5 mM CaCl2 (sucrose) or 0.1 mM CaCl2 (Sucrose 0.1 Ca).
In contrast to its effect on responses to BzATP, BSA (1 mg ml−1) had no effect on responses to ATP at rat, human or mouse P2X7 receptors. Thus, in the absence of BSA the pEC50 values for ATP at rat, human or mouse receptors were 5.13±0.16, 4.68±0.08 and 3.72±0.08, respectively, and these values were not significantly different to the respective pEC50 values of 5.04±0.09, 4.74±0.07 and 3.83±0.17 determined in the presence of BSA (1 mg ml−1).
Relationship between BzATP potency and the ability of BSA to block BzATP responses
The potency of BSA to block responses to BzATP at each species orthologue could be modified by selecting extracellular ionic conditions (see Michel et al., 1999) which change the potency of BzATP. Thus, the potency of BzATP at human receptors (Figure 2) was increased (P<0.05, 1-way ANOVA and Dunnett's test) by decreasing the concentration of CaCl2 in the sucrose buffer from 0.5 to 0.1 mM (pEC50 5.51±0.08 in sucrose; pEC50 6.10±0.03 in sucrose 0.1 mM CaCl2). In sucrose 0.1 mM CaCl2 buffer (Figures 1D and 2A) the potency of BSA to block responses to BzATP (pA2 1.02±0.07, corresponding to 0.095 mg ml−1) was approximately 3 fold greater than in regular sucrose buffer containing 0.5 mM CaCl2 (pA2 0.59±0.13, corresponding to 0.26 mg ml−1). Conversely, the potency of BzATP at the human P2X7 receptor was significantly lower (P<0.05, 1-way ANOVA and Dunnett's test) in sucrose-free buffer containing 140 mM NaCl (pEC50 4.48±0.12) than in sucrose buffer (pEC50 5.51±0.0.08). In the sucrose-free NaCl buffer, BSA had no effect on BzATP potency at concentrations up to 1 mg ml−1 (data not shown).
Figure 2.
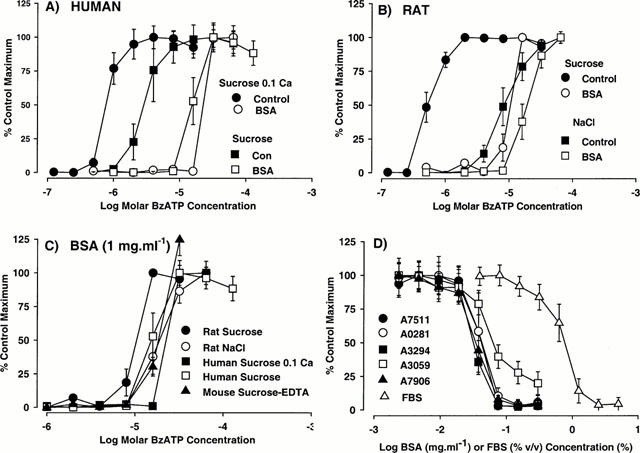
The effect of bovine serum albumin (BSA) on BzATP-stimulated ethidium accumulation in cells expressing recombinant P2X7 receptors. (A) Concentration-effect curves (CEC) to BzATP at the human P2X7 receptor were obtained in the absence (control) or presence of 1 mg ml−1 BSA (BSA) in sucrose buffer containing either 0.5 mM CaCl2 (sucrose) or 0.1 mM CaCl2. (Sucrose 0.1 Ca). (B) CEC to BzATP at the rat P2X7 receptor were obtained in the absence (control) or presence of 1 mg ml−1 BSA (BSA) in either sucrose (sucrose) or NaCl (NaCl) buffer (both containing 0.5 mM CaCl2). (C) CEC to BzATP at rat, human or mouse P2X7 receptors were obtained in the presence of 1 mg ml−1 BSA in either calcium-free sucrose buffer containing 0.1 mM EDTA (Sucrose-EDTA), NaCl buffer (NaCl) or sucrose buffer containing 0.5 mM CaCl2 (sucrose) or 0.1 mM CaCl2 (sucrose 0.1 Ca). (D) The effect of various batches of BSA (Sigma Catalogue numbers shown) or foetal bovine serum (FBS) on responses to 8 μM BzATP at the human P2X7 receptor in sucrose buffer. In all panels, data are the mean±s.e.mean of 3 – 4 experiments and are expressed as a percentage of the maximum response obtained in the absence of BSA. Basal fluorescence values were removed from these data. Lines through data points do not represent lines of best fit.
In cells expressing the rat P2X7 receptor it was not possible to increase reliably BzATP potency above that measured in regular sucrose buffer containing 0.5 mM CaCl2. Thus, when the CaCl2 concentration was reduced to 0.1 mM, there was a marked increase in basal accumulation of ethidium, due to endogenously released ATP activating the rat P2X7 receptor (A.D. Michel, unpublished observation). However, BzATP potency was lower (P<0.05, Student's t-test) in NaCl buffer (5.05±0.07) than in sucrose buffer (6.52±0.08). In the NaCl buffer, BSA effects were less marked than in sucrose buffer (Figures 1D and 2B) and the pA2 value of 0.04±0.10 (corresponding to 0.91 mg ml−1) was markedly less (P<0.05, Student's t-test) than determined in sucrose buffer (pA2=1.28, corresponding to 0.052 mg ml−1).
The pEC50 value of BzATP at the mouse P2X7 receptor was significantly higher (P<0.05, Students t-test) in calcium-free sucrose-EDTA buffer (4.97±0.08) than in sucrose buffer (pEC50 4.18±0.02). In the sucrose-EDTA buffer, 1 mg ml−1 BSA produced a more marked effect on the BzATP CEC than observed with 2 mg ml−1 BSA in the sucrose buffer (Figure 1C).
It was noticeable that the large differences in BzATP potency measured at the various species orthologues in the absence of BSA (see Figure 1A – C) were not observed in the presence of BSA. Indeed, in the presence of 1 mg ml−1 BSA the potency estimates for BzATP were remarkably similar at rat, human and mouse P2X7 receptors. In all cases, threshold concentrations of BzATP were 8 – 16 μM (Figure 2C).
Inhibition of BzATP responses was also observed with other grades/forms of BSA (see Methods) and with FBS (Figure 2D).
Effects of BSA and FBS on nucleotide metabolism
The strong dependence of BSA potency to block BzATP responses upon the potency of BzATP, together with the lack of effect of BSA on responses to ATP, suggested that the effects of BSA were not mediated through the P2X7 receptor. Since serum contains nucleotidase activity (Weisman et al., 1988), it was possible that the effects of BSA or FBS reflected breakdown of BzATP by serum nucleotidases. In the absence of methods to measure the stability of BzATP directly, the stability of ATP was examined to provide an estimate of nucleotidase activity in the BSA or FBS solutions. There was no obvious breakdown of ATP by any of the forms of BSA over a 2 h period at 22°C, although FBS produced a substantial breakdown of ATP (Figure 3A). The time course for the breakdown of ATP by FBS is shown in Figure 3B and illustrates the marked effects of 1 – 10% (v v−1) FBS on 1 and 10 μM solutions of ATP. These experiments were performed in DMEM/F12 to illustrate the instability of ATP solutions in cell growth medium containing FBS. ATP breakdown was approximately 2 – 3 fold more rapid at 37°C (data not shown). A similar time course for the breakdown of ATP by FBS was observed in sucrose buffer (data not shown). The contribution of this effect of FBS to the reduction in BzATP potency is probably minimal as 50% breakdown of ATP required at least 20 min at 22°C whereas solutions of BzATP and FBS were prepared 4 min before addition to the cells.
Figure 3.
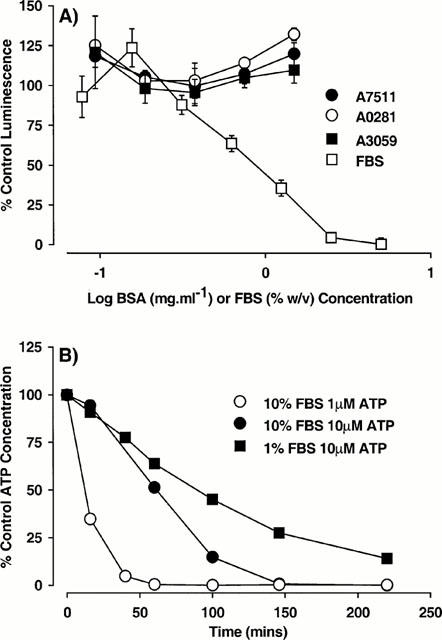
The effect of bovine serum albumin (BSA) and foetal bovine serum (FBS) on ATP solutions. (A) ATP (1 μM) was incubated with the indicated types of BSA (Sigma Catalogue numbers shown) or FBS in sucrose buffer for 2 h at 22°C. A 10 μl aliquot was removed, added to luciferin/luciferase mixture and luminescence recorded. The ATP concentration is expressed as a percentage of that measured at time 0. The data are the mean±s.e.mean of three experiments. (B) ATP (1 or 10 μM) was incubated with 1 or 10% (v v−1) FBS in DMEM/F12 media. At the indicated times, a 10 μl aliquot was withdrawn, added to luciferin/luciferase mixture and luminescence recorded. The ATP concentration is expressed as a percentage of that measured at time 0. In DMEM/F12 media in the absence of FBS the ATP level remained close to 100% throughout the experiment. The data are from a single experiment repeated twice with similar results.
Lipid effects on responses to BzATP
Many agents bind avidly to BSA and there is evidence that high concentrations of ATP can bind to BSA, possible at the fatty acid binding site (Bauer et al., 1992; Takeda et al., 1997). Consequently, BzATP binding to BSA could account for the reduction in BzATP potency produced by BSA. Indeed, several lipids, which bind to BSA, could produce very marked effects on BzATP responses in the presence, but not the absence of BSA. Thus, in cells expressing the human P2X7 receptor, examined in sucrose 0.1 mM CaCl2 buffer, AA had little effect on responses to BzATP in the absence of BSA (Figure 5). However, in the presence of 1 mg ml−1 BSA, AA produced a marked concentration-dependent increase in the potency of BzATP (Figures 4A and 5). Indeed, AA appeared to reverse the effects of BSA on BzATP potency, as the pEC50 for BzATP in the presence of 1 mg ml−1 BSA and 100 μM AA was similar to that determined in the absence of BSA (Figures 4A and 5). The ability of AA to apparently reverse the effects of BSA was affected by the concentration of BSA. Thus, at 0.5 mg ml−1 BSA the effects of AA were more marked (Figures 4B and 5) while at 2 mg ml−1 BSA, only 100 μM AA affected responses to BzATP (Figure 5). DAUDA at concentrations of 25 – 100 μM also produced an increase in BzATP potency in the presence of 0.5 mg ml−1 BSA and the effects of 100 μM DAUDA were comparable to 100 μM AA (data not shown). In the presence of 1 mg ml−1 BSA, AA or DAUDA (both 100 μM) did not affect ATP potency (data not shown).
Figure 5.
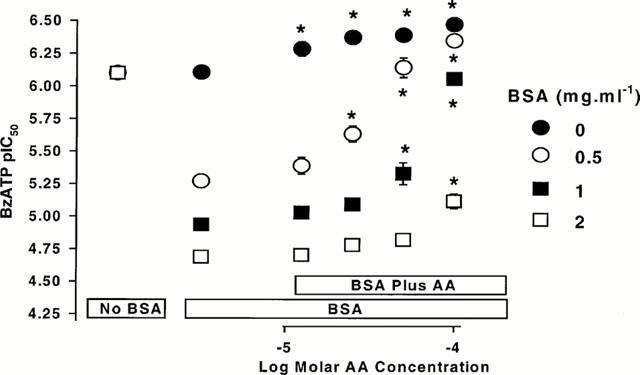
The effect of arachidonic acid (AA) on BzATP-stimulated ethidium accumulation in cells expressing human recombinant P2X7 receptors. pEC50 values for BzATP were determined in sucrose buffer containing 0.1 mM CaCl2 in the absence of bovine serum albumin (BSA) and AA (leftmost data points), in the presence of the indicated concentrations of BSA and absence of AA (second from left set of data points) and in the presence of the indicated concentrations of BSA and AA. Note that at each BSA concentration, a control group (absence of BSA and AA) was included and that these are all in the same location. *Significantly different to pEC50 value determined in the presence of BSA and absence of AA (oneway ANOVA and Dunnett's test). The data are the mean±s.e.mean of four experiments.
Figure 4.
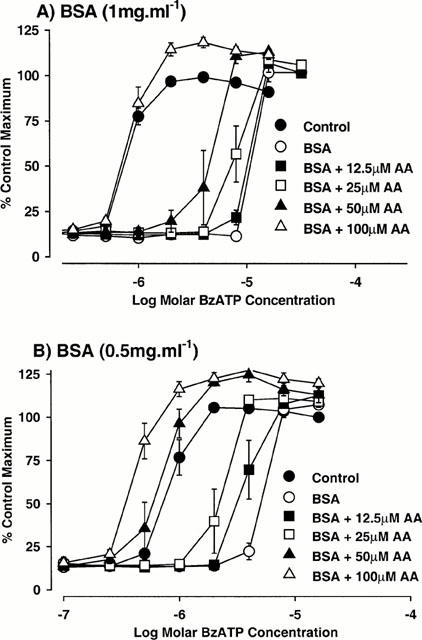
The effect of arachidonic acid (AA) on BzATP-stimulated ethidium accumulation in cells expressing human recombinant P2X7 receptors. Concentration-effect curves (CEC) to BzATP were determined in sucrose buffer containing 0.1 mM CaCl2 in the absence (control) or presence of either (A) 1 mg ml−1 bovine serum albumin (BSA) or (B) 0.5 mg ml−1 BSA and the indicated concentrations of AA. The data are the mean±s.e.mean of four experiments and are expressed as a percentage of the maximum response obtained in the absence of BSA and AA. Basal fluorescence values were not removed from these data. Lines through data points do not represent lines of best fit.
Binding of BzATP to BSA
To investigate the binding of BzATP to BSA, the effect of BzATP on the binding of the fatty acid DAUDA to BSA were examined. BzATP reduced the fluorescence of BSA-DAUDA solutions indicating that BzATP was an inhibitor of DAUDA binding to BSA. The inhibition curves to BzATP were biphasic with 33±1.4% of the DAUDA-BSA fluorescence signal being inhibited by BzATP with relatively high potency (pIC50 5.8±0.07) and the remainder of the fluorescence signal inhibited with lower potency (pIC50 3.5±0.06). ATP produced approximately 10% inhibition of DAUDA-BSA fluorescence at a concentration 300 μM.
Discussion
The main finding of this study was that under certain circumstances BSA or FBS could reduce the potency of BzATP for P2X7 receptors and that this effect primarily reflects serum albumin binding of BzATP. These effects occur at BSA and FBS concentrations often used in the study of P2 receptors. For example, concentrations of 1 mg ml−1 BSA (Vigne et al., 1999; Humphreys & Dubyak, 1996; Wiley et al., 1994) or 10% (v v−1) FBS (Conigrave et al., 1998) have been used in studies on P2 receptors.
The effects of BSA on responses to BzATP appeared, at first, somewhat confusing. Thus, BSA was able to block responses to BzATP at rat and human P2X7 receptors but not mouse P2X7 receptors. There are marked differences in antagonist sensitivity between the species orthologues of the P2X7 receptor (Humphreys et al., 1998; Chessell et al., 1998) and so this may have accounted for the differential effects of BSA. However, BSA effects varied within each species orthologue when ionic conditions were changed and appeared to depend upon agonist potency. Indeed, BSA could only block BzATP responses when the BzATP pEC50 value was greater than approximately 5. This observation, together with the finding that BSA did not block responses to ATP suggested that the effects of BSA were independent of P2X7 receptor blockade.
Serum albumin binding or metabolism of BzATP by BSA represented two alternative explanations for the effects of BSA on BzATP potency. The later explanation was considered as serum is known to contain nucleotidase activity (Weismann et al., 1988). However, metabolism of BzATP seems unlikely as BSA did not affect ATP concentrations, even when incubated for 2 h at 22°C. Interestingly, FBS was able to breakdown ATP solutions as reported previously (Weissman et al., 1988). These observations are of obvious importance when studying effects of ATP (or nucleotide triphosphates in general) in tissue culture studies as nucleotide concentrations will be greatly reduced if solutions are prepared in the presence of FBS and/or exposed to FBS for prolonged periods before use. However, only a few studies describe precautions for overcoming this potential artefact (Cowen et al., 1991).
The alternative explanation, that the effects of BSA on BzATP potency are due to serum albumin binding, seems the most plausible explanation for the present results. Thus, BSA is known to bind a wide range of substances and so reduce their free concentration (Takeda et al., 1997). Indeed, ATP binds to BSA, probably at the same site as fatty acids (Bauer et al., 1992; Takeda et al., 1997). Importantly, lipids such as AA and DAUDA (Wilkinson & Wilton, 1986), which are known to bind to fatty acid binding sites on BSA, exerted marked effects on BzATP potency in the presence, but not the absence, of BSA and were able to reverse the inhibitory effects of BSA on BzATP potency.
It should be noted that AA is known to affect the function of many ion-channels (Meves, 1994) and so the ‘reversal' of BSA effects could have been due to a direct effect upon the P2X7 ion-channel. However, the magnitude of the effect of AA on BzATP potency in the presence of BSA greatly exceeded that observed at other ion channels, such as those for glutamate, where changes in potency of 2 – 3 fold are observed (Miller et al., 1992; Nishikawa et al., 1994). Furthermore, it seems unlikely that the effects of AA observed in this study were due to direct actions at the P2X7 receptor since similar effects were not observed when using ATP.
More convincing evidence for the binding of BzATP to BSA was provided by demonstrating that BzATP could inhibit DAUDA binding to BSA. BzATP produced a biphasic inhibition of DAUDA binding to BSA. A high affinity interaction, with an IC50 of 1 μM, and a low affinity interaction with an IC50 of 300 μM were detected. The reason for the biphasic inhibition of DAUDA binding to BSA by BzATP is not known. However, it has been reported that DAUDA labels several sites on BSA, including a site for billirubin (Wilton, 1990), and so BzATP may selectively interact with just one of these sites.
In contrast to BzATP, ATP was relatively weak (IC50 >300 μM) at blocking DAUDA binding. This is consistent with previous estimates of the affinity of ATP for BSA which suggested a Ki of 120 μM at pH 7.4 (Takeda et al., 1997). The low potency of ATP for binding to BSA may explain why its potency at P2X7 receptors was less affected by BSA than BzATP. Thus, the concentration of BSA in a 1 mg ml−1 solution is approximately 13 μM and so could bind a similar concentration of nucleotide. However, since ATP only activated P2X7 receptors at concentrations of 30 – 100 μM, it is likely that the effects of depleting these solutions by up to a maximum of 13 μM would be minimal.
In the case of BzATP the higher affinity binding to BSA (IC50 ∼1 μM), coupled with the high potency at P2X7 receptors, probably accounts for the much greater effects of BSA on BzATP potency. Thus, given the relatively high potency for BzATP binding to BSA, a 13 μM solution of BSA could bind, and so deplete, a similar concentration of BzATP. Indeed, in the presence of 13 μM BSA (1 mg ml−1), BzATP only activated P2X7 receptors when concentrations exceeded 8 – 16 μM (Figure 2c). The potential for BSA binding to affect BzATP potency at P2X7 receptors increased with increasing potency of BzATP. Similar effects of BSA should also occur at other P2 receptors and may be particularly marked at P2X1 and P2X3 receptors where BzATP potency estimates of 1 – 100 nM have been reported (Bianchi et al., 1999).
Finally, the effects of BSA can seriously affect estimates of potency and selectivity used in classifying P2 receptors. For example, in the presence of 1 mg ml−1 BSA, the potency of BzATP for rat, human and mouse receptors was similar despite the marked differences in potency observed in the absence of BSA. Furthermore, since potency estimates for ATP and BzATP were differentially affected by BSA, the difference in potency between ATP and BzATP is considerably reduced in the presence of BSA.
In conclusion, this study highlights an important effect of BSA on BzATP potency at P2X7 receptors which can alter rank orders of agonist potency. These effects are due primarily to BSA binding of BSA and are also observed with FBS, which in addition can metabolise nucleotides. Finally, the dramatic effects of AA and other lipids on BzATP responses in the presence of BSA could, perhaps wrongly, be interpreted as representing major effects of the lipids on P2X7 receptor function.
Abbreviations
- AA
arachidonic acid
- BSA
bovine serum albumin
- BzATP
2′-& 3′-O-(4-benzoylbenzoyl)-ATP
- CEC
concentration-effect curves
- DAUDA
11-((5-dimethylaminonaphthalene-1-sulphonyl)amino)undecanoic acid
- DMEM/F12
Dulbecco's Modified Eagle's Medium and Hams F12
- FBS
foetal bovine serum
References
- BAUER M., BAUMANN J., TROMMER W.E. ATP binding to bovine serum albumin. FEBS Letts. 1992;313:288–290. doi: 10.1016/0014-5793(92)81211-4. [DOI] [PubMed] [Google Scholar]
- BIANCHI B.R., LYNCH K.J., TOUMA E., NIFORATOS W., BURGARD E.C., ALEXANDER K.M., PARK H.S., YU H., METZGER R., KOWALUK E., JARVIS M.F., VAN BIESEN T. Pharmacological characterization of recombinant human and rat P2X receptor subtypes. Eur. J. Pharmacol. 1999;376:127–138. doi: 10.1016/s0014-2999(99)00350-7. [DOI] [PubMed] [Google Scholar]
- BOYER J.L., COOPER C.L., HARDEN T.K. [32P]3′-O-(4-benzoyl)benzoyl ATP as a photoaffinity label for a phospholipase C-coupled P2Y-purinergic receptor. J. Biol. Chem. 1990;265:13515–13520. [PubMed] [Google Scholar]
- BOYER J.L., HARDEN T.K. Irreversible activation of phospholipase C-coupled P2Y-purinergic receptors by 3′-O-(4-benzoyl)benzoyl adenosine 5′-triphosphate. Mol. Pharmacol. 1989;36:831–835. [PubMed] [Google Scholar]
- CHESSELL I.P., SIMON J., HIBELL A.D., MICHEL A.D., BARNARD E.A., HUMPHREY P.P.A. Cloning and functional characterisation of the mouse P2X7 receptor. FEBS Letts. 1998;439:26–30. doi: 10.1016/s0014-5793(98)01332-5. [DOI] [PubMed] [Google Scholar]
- COMMUNI D., ROBAYE B., BOEYNAEMS J-M. Pharmacological characterization of the human P2Y11 receptor. Br. J. Pharmacol. 1999;128:1199–1206. doi: 10.1038/sj.bjp.0702909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CONIGRAVE A.D., LEE J.Y., VAN DER WEYDEN L., JIANG L., WARD P., TASEVSKI V., LUTTRELL B.M., MORRIS M.B. Pharmacological profile of a novel cyclic AMP-linked P2 receptor on undifferentiated HL-60 leukemia cells. Br. J. Pharmacol. 1998;124:1580–1585. doi: 10.1038/sj.bjp.0701985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COWEN D.S., BERGER M., NUTTLE L., DUBYAK G.R. Chronic treatment with P2-purinergic receptor agonists induces phenotypic modulation of the HL-60 and U937 human myelogenous leukemia cell lines. J. Leukoc. Biol. 1991;50:109–122. doi: 10.1002/jlb.50.2.109. [DOI] [PubMed] [Google Scholar]
- ERB L., LUSTIG K.D., AHMED A.H., GONZALEZ F.A., WEISMAN G.A. Covalent incorporation of 3′-O-(4-benzoyl)benzoyl-ATP into a P2 purinoceptor in transformed mouse fibroblasts. J. Biol. Chem. 1990;265:7424–7431. [PubMed] [Google Scholar]
- EVANS R.J., LEWIS C., BUELL G., VALERA S., NORTH R.A., SURPRENANT A. Pharmacological characterization of heterologously expressed ATP-gated cation channels (P2x purinoceptors) Mol. Pharmacol. 1995;48:178–183. [PubMed] [Google Scholar]
- GONZALEZ F.A., AHMED A.H., LUSTIG K.D., ERB L., WEISMAN G.A. Permeabilization of transformed mouse fibroblasts by 3′-O-(4-benzoyl)benzoyl adenosine 5′-triphosphate and the desensitization of the process. J. Cell Physiol. 1989;139:109–115. doi: 10.1002/jcp.1041390116. [DOI] [PubMed] [Google Scholar]
- HUMPHREYS B.D., DUBYAK G.R. Induction of the P2z/P2X7 nucleotide receptor and associated phospholipase D activity by lipopolysaccharide and IFN-gamma in the human THP-1 monocytic cell line. J. Immunol. 1996;157:5627–5637. [PubMed] [Google Scholar]
- HUMPHREYS B.D., VIRGINIO C., SURPRENANT A., RICE J., DUBYAK G.R. Isoquinolines as antagonists of the P2X7 nucleotide receptor: High selectivity for the human versus rat receptor homologues. Mol. Pharmacol. 1998;54:22–32. doi: 10.1124/mol.54.1.22. [DOI] [PubMed] [Google Scholar]
- MEVES H. Modulation of ion channels by arachidonic acid. Prog. Neurobiol. 1994;43:175–186. doi: 10.1016/0301-0082(94)90012-4. [DOI] [PubMed] [Google Scholar]
- MICHEL A.D., CHESSELL I.P., HUMPHREY P.P.A. Ionic effects on human recombinant P2X7 receptor function. Naunyn-Schmiedeberg's Arch. Pharmacol. 1999;359:102–109. doi: 10.1007/pl00005328. [DOI] [PubMed] [Google Scholar]
- MICHEL A.D., KAUR R., CHESSELL I.P., HUMPHREY P.P.A. Antagonist effects on human P2X7 receptor-mediated cellular accumulation of YO-PRO-1. Br. J. Pharmacol. 2000;130:513–520. doi: 10.1038/sj.bjp.0703368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MILLER B., SARANTIS M., TRAYNELIS S.F., ATTWELL D. Potentiation of NMDA receptor currents by arachidonic acid. Nature. 1992;355:722–725. doi: 10.1038/355722a0. [DOI] [PubMed] [Google Scholar]
- NISHIKAWA M., KIMURA S., AKAIKE N. Facilitatory effect of docosahexaenoic acid on N-methyl-D-aspartate response in pyramidal neurones of rat cerebral cortex. J. Physiol. 1994;475:83–93. doi: 10.1113/jphysiol.1994.sp020051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RASSENDREN F., BUELL G., VIRGINIO C., COLLO G., NORTH R.A., SURPRENANT A. The permeabilizing ATP receptor, P2X7: cloning and expression of a human cDNA. J. Biol. Chem. 1997;272:5482–5486. doi: 10.1074/jbc.272.9.5482. [DOI] [PubMed] [Google Scholar]
- STEINBERG T.H., NEWMAN A.S., SWANSON J.A., SILVERSTEIN S.C. ATP4- permeabilizes the plasma membrane of mouse macrophages to fluorescent dyes. J. Biol. Chem. 1987;262:8884–8888. [PubMed] [Google Scholar]
- SURPRENANT A., RASSENDREN F., KAWASHIMA E., NORTH R.A., BUELL G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7) Science. 1996;272:735–738. doi: 10.1126/science.272.5262.735. [DOI] [PubMed] [Google Scholar]
- TAKEDA S., MIYAUCHI S., NAKAYAMA H., KAMO N. Adenosine 5′-triphosphate binding to bovine serum albumin. Biophys. Chem. 1997;69:175–183. doi: 10.1016/s0301-4622(97)00084-7. [DOI] [PubMed] [Google Scholar]
- VIGNE P., HECHLER B., GACHET C., BREITTMAYER J.P., FRELIN C. Benzoyl ATP is an antagonist of rat and human P2Y1 receptors and of platelet aggregation. Biochem. Biophys. Res. Commun. 1999;256:94–97. doi: 10.1006/bbrc.1999.9558. [DOI] [PubMed] [Google Scholar]
- WEISMAN G.A., LUSTIG K.D., LANE E., HUANG N-N., BELZER I., FRIEDBERG I. Growth inhibition of transformed mouse fibroblasts by adenine nucleotides occurs via generation of extracellular adenosine. J. Biol. Chem. 1988;263:12367–12372. [PubMed] [Google Scholar]
- WILEY J.S., CHEN J.R., SNOOK M.B., JAMIESON G.P. The P2Z-purinoceptor of human lymphocytes: actions of nucleotide agonists and irreversible inhibition by oxidized ATP. Br. J. Pharmacol. 1994;112:946–950. doi: 10.1111/j.1476-5381.1994.tb13172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WILKINSON T.C., WILTON D.C. Studies on fatty acid-binding proteins. The detection and quantification of the protein from rat liver by using a fluorescent fatty acid analogue. Biochem. J. 1986;238:419–424. doi: 10.1042/bj2380419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WILLIAMS N., COLEMAN P.S. Exploring the adenine nucleotide binding sites on mitochondrial F1-ATPase with a new photoaffinity probe, 3′-O-(4-benzoyl)benzoyl adenosine 5′-triphosphate. J. Biol. Chem. 1982;257:2834–2841. [PubMed] [Google Scholar]
- WILTON D.C. The fatty acid analogue 11-(dansylamino)undecanoic acid is a fluorescent probe for the bilirubin-binding sites of albumin and not for the high-affinity fatty acid-binding sites. Biochem. J. 1990;270:163–166. doi: 10.1042/bj2700163. [DOI] [PMC free article] [PubMed] [Google Scholar]