

Abstract
We recently demonstrated that intracellular application of Angiotensin II (Angiotensin IIintr) induces rat aorta contraction independent of plasma membrane Angiotensin II receptors. In this study we investigated the effects of Angiotensin IIintr on cell growth in A7r5 smooth muscle cells.
DNA-synthesis was increased dose-dependently by liposomes filled with Angiotensin II as measured by [3H]-thymidine incorporation at high (EC50=27±6 pM) and low (EC50=14±5 nM) affinity binding sites with increases in Emax of 58±4 and 37±4% above quiescent cells, respectively. Cell growth was corroborated by an increase in cell number.
Extracellular Angiotensin II (10 pM – 10 μM) did not modify [3H]-thymidine incorporation.
Growth effects of Angiotensin IIintr mediated via high affinity sites were inhibited by liposomes filled with 1 μM of the non-peptidergic antagonists losartan (AT1-receptor) or PD123319 (AT2-receptor) or with the peptidergic agonist CGP42112A (AT2-receptor). Emax values were decreased to 30±3, 29±4 and 4±2%, respectively, without changes in EC50. The Angiotensin IIintr effect via low affinity sites was only antagonized by CGP42112A (Emax=11±3%), while losartan and PD123319 increased Emax to 69±4%. Intracellular applications were ineffective in the absence of Angiotensin IIintr.
Neither intracellular nor extracellular Angiotensin I (1 μM) were effective.
The Angiotensin IIintr induced growth response was blocked by selective inhibition of phosphatidyl inositol 3-kinase (PI-3K) by wortmannin (1 μM) and of the mitogen-activated protein kinase (MAPK/ERK) pathway by PD98059 (1 μM) to 61±14 and 4±8% of control, respectively.
These data demonstrate that Angiotensin IIintr induces cell growth through atypical AT-receptors via a PI-3K and MAPK/ERK -sensitive pathway.
Keywords: Intracellular angiotensin II, growth, losartan, PD123319, CGP42112A, PI-3 kinase, MAP kinase, A7r5 cells
Introduction
It has been extensively documented that the renin-angiotensin system is a major factor in the regulation of cardiovascular homeostasis, including blood pressure, mineral balance and tissue remodelling (Weber, 1998). However, the beneficial effects of ACE inhibitors on tissue remodelling appear to be independent, at least in part, of their effects on blood pressure (Linz et al., 1995). In this respect, Angiotensin II can be generated either in the kidney and released in the circulation (circulating Angiotensin II) subsequently activating different plasma membrane receptors or it can be produced in different tissues to exert its effects at the place of production (local Angiotensin II; Danser & Schalekamp, 1996). To date, two different receptors have been cloned; namely the AT1 and AT2-subtype receptor (Griendling et al., 1996). These receptors are differently localized and have different functions, among which is modulation of cellular growth. The AT1-receptor, which is prominent in adult tissues, stimulates cell growth (Matsukada & Ichikawa, 1997). In contrast, the AT2-receptor, which is mostly abundant in foetal tissues, inhibits cell growth and promotes apoptosis (Xoriuchi et al., 1999).
There is growing evidence for intracellular actions of Angiotensin II not related to activation of ‘classical' plasma membrane receptors. We recently reported effects of intracellular Angiotensin II (Angiotensin IIintr) on rat aorta contraction, independent of activation of plasma membrane Angiotensin II receptors (Brailoiu et al., 1999). Intracellular Angiotensin II was reported to increase cytosolic [Ca2+] in vascular smooth muscle cells (Haller et al., 1996; 1999), to inhibit gap conductance in heart muscle (De Mello, 1996) and to affect L-type Ca2+ channel in a specific manner (De Mello, 1998). Such changes in Ca2+ homeostasis are important for cell growth, therefore we addressed the following questions in this investigation: (1) Is there a role for an Angiotensin IIintr receptor in vascular smooth muscle cell growth. (2) Is the receptor similar to the known subtypes based on its pharmacological profile. (3) Can we identify part of its signal transduction pathway leading to cell growth.
Methods
Cell culture
A7r5 vascular smooth muscle cells were grown in 75 cm2 flasks in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% foetal calf serum (FCS), penicillin (50 μg ml−1) and streptomycin (50 units ml−1) at 37°C in a humidified atmosphere (5% CO2). The cells were subcultured at 95% confluency by trypsinization. Cell number was established by counting dispersed cells in a Bürker counter (Schreek, Germany). Experiments were performed in 6 well plates (Costar, 9.6 cm2 well−1) at a density of 105 cells well−1, unless stated otherwise.
Determination of DNA synthesis
To obtain quiescent cells, the medium was replaced with DMEM containing 0.1% foetal calf serum 24 h after plating. The intracellular additions of Angiotensin II by liposomal delivery were performed 1 day after the switch to 0.1% FCS containing DMEM and lasted for another 24 h. Extracellular addition of various compounds happened 1 min prior to liposomal addition. [3H]-thymidine (0.5 μCi, specific activity: 20 Ci mmol−1) was added on each well during the final 3 h of incubation. The medium was withdrawn at the end of the incubation period and the cells were washed twice with ice-cold phosphate buffered saline (PBS). To remove non-genomic [3H]-thymidine, the cells were incubated in the presence of 400 μl trichloroacetic acid for 1 h on ice. Finally, the cells were digested with 1 ml of 1 M NaOH and the incorporated radioactivity was measured in a β-scintillation counter. Quiescent cells incorporated 210±11 d.p.m. well−1 (mean±s.e.mean, n=24), whereas 10% FCS stimulated cells incorporated 1078±71 d.p.m. well−1 (n=24).
Liposomes preparation
Liposomes containing Angiotensin II or Angiotensin I and control liposomes containing 140 mM KCl were prepared as described (Brailoiu et al., 1999) from egg phosphatidyl choline, using 10 mg ml−1 of solution to be incorporated. Dialysis against PBS solution was performed for 4 h in order to remove the non-incorporated compounds. To maintain sterile cell culture conditions the liposomes solution was filtrated (0.2 μm pore size). Liposomes were added to the medium above the cells in a ratio of 1 to 20 (v v−1). If other compounds were delivered intracellularly, they were encapsulated together with Angiotensin II. The amount of Angiotensin II delivered intracellularly was determined using 125I-angiotensin II filled liposomes. The incorporation into liposomes after the filtration step was 7.2±0.2% (n=8) of the initial amount of radioactive Angiotensin II added to the cells. Recovery of incorporated 125I angiotensin II into the cells after incubation for 30 min amounted to 5.6±0.2% (n=8) of the initial amount of radioactive Angiotensin II added to the cells.
Measurement of inositol (1,4,5) trisphosphate (Ins(1,4,5)P3)
Mass measurements of Ins(1,4,5)P3 were performed as described earlier (Sipma et al., 1995), using an isotope dilution ligand binding assay. In brief, samples were assayed in 25 mM Tris/HCl (pH=9.0), 1 mM EDTA, 1 mg bovine serum albumin, D-[inositol-1-3H(N)]-Ins(1,4,5)P3 (21.0 Ci mmol−1, 2000 c.p.m. assay−1) and 1 mg binding protein isolated from beef liver. Bound and free radioactivity was separated by centrifugation. The radioactivity in the pellet was determined by scintillation counting.
Measurement of intracellular Ca2+
Intracellular [Ca2+] was measured using Fura-2 fluorometry as described (Filipeanu et al., 1997). Cells were loaded with 5 μM Fura-2 acetoxymethyl ester at 22°C, for 45 min in the dark. Fluorescence was measured at 37°C.
Chemicals
All cell culture media were purchased from Gibco BRL, phosphatidyl choline type X-E, Angiotensin I, Angiotensin II, and wortmannin from Sigma Chemical Co, CGP 422112A (nicotinic acid-Tyr-(N-benzoylcarbonyl-Arg)-Lys-His-Pro-Ile-OH) from RBI, and PD98059 (2-(2-Amino-3-methoxyphenyl)-4H-1-benzopyran-4-one) from Calbiochem. Fura 2-AM was obtained from Molecular Probes, losartan (2-n- butyl-4-chloro-5-hydroxymethyl-1- [(2′-(1H-tetrazol-5-yl)biphenyl-4-yl)methyl]imidazole) from Merck, Sharpe and Dohme, PD123319 ((s)-1-(4-[dimethylamino]-3-methylphenyl)methyl-5- (diphenylacetyl) -4,5,6,7-tetrahydro-1H -imidazo [4,5-c] pyridine-6-carboxylate) from Park-Davis, [6-3H]-thymidine from Amersham Int, D-[inositol-1-3H(N)]-Ins(1,4,5)P3 from NEN Life Science Products, and all other agents from Merck.
Data analysis
Data are given as mean±s.e.mean. The results of the growth experiments are expressed as percentage of the radioactivity incorporated by control quiescent cells. Independent measurements were performed in at least two different passages. Measurements were normalized against liposomes filled with 10−7 M Angiotensin II, present in every experimental protocol. Statistical significance was tested by one-way ANOVA followed by Bonferroni test. A value of P<0.05 was considered statistically significant. Concentration response curves were fitted and the corresponding parameters calculated using Multifit (Dr J.H. Proost, Department of Pharmacokinetics and Drug Delivery, University Centre for Pharmacy, University of Groningen). Curve fitting was based on the following sigmoidal model: Y=V1+V2×X̂V3/(X̂V3+V4^V3)+V5×X̂V6/(X̂V6+V7^V6). Fitting for a single binding site was performed after omission of the term containing the parameters V5, V6 and V7. To determine if the data were fitted significantly better with one or two binding sites the variance was calculated using the F-test.
Results
Intracellular Angiotensin II stimulates cell growth in quiescent A7r5 cells
Addition of Angiotensin II filled liposomes increases DNA synthesis in a dose-dependent fashion, as measured by [3H]-thymidine incorporation into A7r5 cells (Figure 1). The first observation above the background was obtained with liposomes containing 10 pM Angiotensin II, whereas the maximum effect was reached with liposomes containing 0.1 μM Angiotensin II (doubling of [3H]-thymidine incorporation compared to quiescent cells). Data analysis showed that the increases in [3H]-thymidine incorporation were better described using a model with two binding site kinetics. Parameters of the dose-response curve are given in Table 1. The simplest model with V6=1 was used for further analysis since varying the Hill-coefficient (V6) from 1 to 9 for the second binding site did not significantly alter the fitting results. Neither ‘empty' control liposomes, filled only with 140 mM KCl, nor liposomes filled with the parent peptide Angiotensin I (1 μM) affected [3H]-thymidine incorporation (102.7±3.8%, n=36 and 102.6±0.8%, n=12 of control cells, respectively). The growth stimulating effect of liposomes containing 0.1 μM Angiotensin II was corroborated in experiments showing actual increases in cell number (Table 2).
Figure 1.
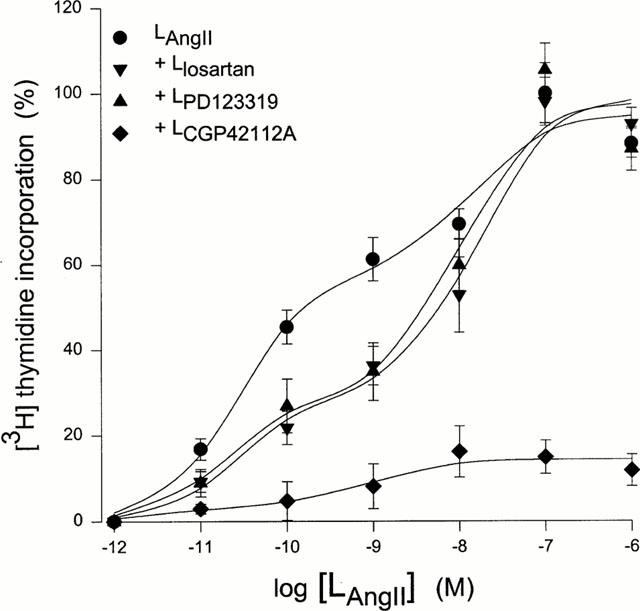
Effects of Angiotensin II filled liposomes on A7r5 cell growth. Effect curves of Angiotensin II filled liposomes alone (n=12) and in the presence of liposomes encapsulating losartan, PD123319 or CGP42112A (all 1 μM, n=12). All results were reported as increases (%) above [3H]-thymidine incorporation in quiescent cells. The maximal value (100% at LAngiotensin II=0.1 μM) corresponds to 448±56 d.p.m. well−1, n=24). Lines were fitted according to the two binding site model given in Table 1.
Table 1.
Dose-response parameters of Angiotensin IIintr induced [3H]-thymidine incorporation

Table 2.
Effect of intracellular applied Angiotensin II on cell number

The growth stimulating effect of Angiotensin II filled liposomes (0.1 μM Angiotensin II) was unchanged by extracellular addition (1 μM) of the nonpeptidergic AT1-type receptor antagonist losartan, the nonpeptidergic AT2-type receptor antagonist PD123319, or the peptidergic AT2-type receptor agonist CGP42112A (97.1±4.0%, 103.9±2.8%, and 91.7±5.6% of control cells, n=18, respectively).
In contrast to intracellular delivered Angiotensin II, extracellular application of Angiotensin II (10 pM to 10 μM) did not change [3H]-thymidine incorporation in quiescent A7r5 cells. A similar amount of radioactivity as in control cells was incorporated after 0.1 μM extracellular Angiotensin II compared to control cells (98.1±3.2%, n=12). A7r5 cells apparently lack functional AT -receptors which is also evident from the inability of extracellular Angiotensin II (10 μM) to change basal Ins(1,4,5)P3 formation (2.3±0.6 vs 2.1±0.8 pmol 105 cells−1, n=12) or basal intracellular Ca2+ concentration (57±6 vs 58±7 nM, n=12), as reported before (Filipeanu et al., 1998a,1998b).
Furthermore, extracellular application of Angiotensin I (1 μM, 102±2.4%, n=12), losartan (1 μM, 95.6±4.7%, n=24), PD123319 (1 μM, 98.7±5.8%, n=16) or CGP42112A (1 μM, 92.9±4.8%, n=18) did not affect [3H]-thymidine incorporation.
Pharmacology of intracellular Angiotensin II
We next attempted to characterize pharmacologically the effects of Angiotensin IIintr. Addition of liposomes filled with losartan (1 μM), PD123319 (1 μM) together with various concentrations of Angiotensin II reduced [3H]-thymidine incorporation (Figure 1). Abolition of the growth stimulating effect of Angiotensin II was obtained by liposomes filled with the AT2-type receptor agonist CGP42112A (1 μM, Figure 1). All three agents substantially reduced stimulation of the high affinity binding site. Non-competitive inhibition in this receptor site is likely involved in view of the decreasing Emax value without changes in EC50. The following rank order of antagonist potencies was obtained: CGP42112A>PD123319=losartan (Figure 2, Table 3). In contrast, at the low affinity binding site these compounds elicited opposite effects. Again strong inhibition was observed for CGP42112A, but losartan and PD123319 significantly increased Emax as compared to control. It is noticeable that maximal values obtained at this site are comparable to the control value at the high affinity binding site (Figure 2, Table 3). To verify the nature of the antagonism by the compounds studied additional experiments were performed at other concentrations of the antagonists across a limited range of Angiotensin II concentrations (Table 3). Although one should be cautious not to over interpret the fitting results of the limited data, it is clear that no evidence of a parallel shift of the log dose-response relationship was observed for both binding sites. The lowest antagonist concentration used (30 nM losartan) was insufficient to induce inhibition. At high antagonist concentrations (e.g. 30 μM PD123319) further increases were not observed in the Emax of the low affinity binding site. This was also concluded from the experiment in which losartan and PD123319 (both 1 μM) were given simultaneously, and from an experiment using losartan (10 μM) or PD123319 (10 μM) at a single dose of Angiotensin II (100 nM), showing maximal [3H]-thymidine incorporation was maintained (98.2±4.1%, n=6 and 109.0±6.5%, n=6 for losartan- and PD123319 filled liposomes, respectively).
Figure 2.
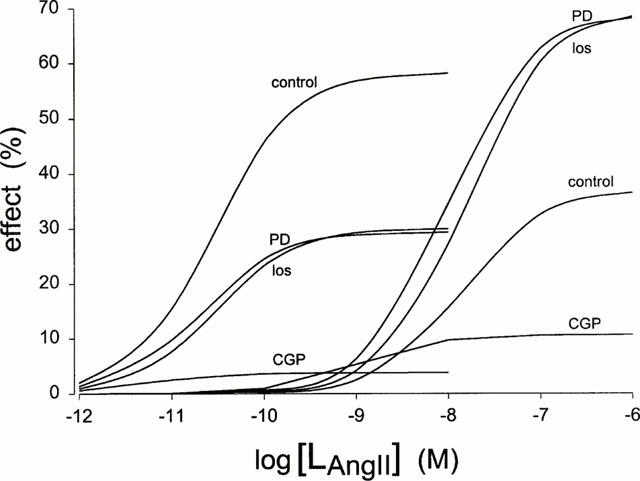
Contribution of the different binding sites to growth induced by intracellular Angiotensin II in the absence or presence of antagonists. Dose-response curves were plotted using the data of Figure 1 according to the model as given in Table 1. Angiotensin II filled liposomes were in the absence (control) or in the presence of liposomes encapsulating losartan (los), PD123319 (PD) or CGP42112A (CGP). Values obtained for Emax and EC50 are presented in Table 3.
Table 3.
Effect of LAngiotensin II on [3H]-thymidine incorporation in the presence of various agents on dose-response parameters

In the absence of Angiotensin II filled liposomes, losartan-, PD123319-, and CGP4112A-filled liposomes did not modify basal [3H]-thymidine incorporation into quiescent cells (n=12; 99.1±2.9%, 101.5±2.4%, 98.3±5.5% of control, respectively).
Signal transduction of intracellular Angiotensin II effects
Extracellular signals often use multiple pathways to modify cell growth. In order to gain insight in the mechanism involved in growth stimulation by Angiotensin IIintr two of these pathways were tested. Intracellular Angiotensin II induced cell growth was totally abolished by inhibition of the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) pathway by co-incubation of the cells with extracellular PD98059 (1 μM, Figure 3). Growth stimulation was also reduced, but to a lesser extent by inhibition of the phosphatidyl inositol 3-kinase (PI-3K) pathway with wortmannin (1 μM, Figure 3).
Figure 3.
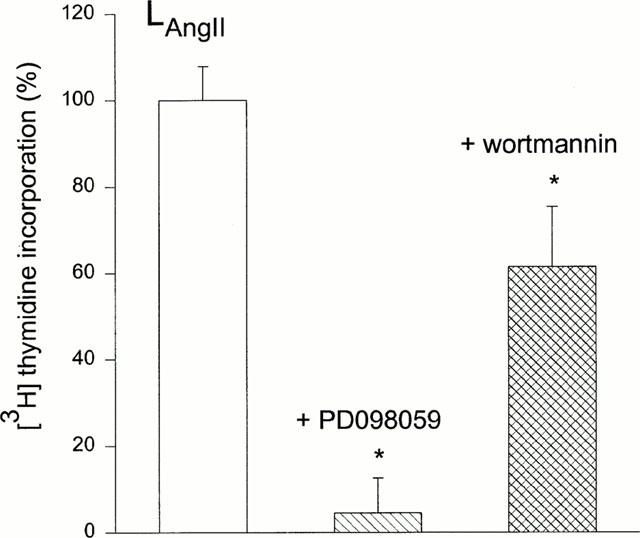
Involvement of MAPK and PI-3K in Angiotensin II induced cell growth. Cell growth induced by Angiotensin filled liposomes (LAngiotensin II, 0.1 μM) was inhibited by simultaneous extracellular treatment with PD98059 (1 μM, n=16) or wortmannin (1 μM, n=16).
Discussion
In the present study, we demonstrate that Angiotensin IIintr stimulates cell growth in quiescent A7r5 cells. Although ‘classical' plasma membrane Angiotensin II receptors are involved in growth and apoptotic processes underlying cardiovascular remodelling (Dzau & Horiuchi, 1998) our results suggest additional targets for Angiotensin II at sites which have not previously been recognized. The growth response was characterized by the presence of two distinct binding sites for Angiotensin IIintr. The high affinity site (picomolar range) was sensitive to intracellular delivered antagonists in the rank order of potencies, CGP42112A>PD123319=losartan. In contrast, only the peptidergic antagonist CGP42112A could inhibit the low affinity site (nanomolar range). The other compounds even increased the maximal effect to the value obtained by stimulation of the high affinity site, indicating that both sites are possibly closely involved in the growth response. Also important is the observation that all three compounds are ineffective in the absence of Angiotensin IIintr, showing that occupation of the receptor site by Angiotensin II is needed for their action. Although the nature and physiological significance of these distinct sites needs further investigation, it is unlikely that ‘classical' AT1- or AT2-receptors (De Gasparo et al., 1998) mediate the Angiotensin IIintr effect on [3H]-thymidine incorporation in view of the different potencies and the lack of competitive inhibition observed, and the growth inhibitory action of both CGP42112A and PD123319. These compounds were described as agonist and antagonist of the anti-proliferative AT2-receptor subtype, respectively (Timmermans et al., 1992; De Gasparo et al., 1998). Antagonist activity of CGP42112A on AT1- or AT2-receptors has only been reported for extracellular Angiotensin II mediated phospholipase A2 activation (Lokuta et al., 1994). The presence of an unusual type of receptor in A7r5 cells becomes also apparent from the lack of the growth response by extracellular addition of Angiotensin II and the ineffectiveness of extracellular addition of AT1-or AT2-receptor antagonists. No functional plasma membrane AT-receptors seems to be present in A7r5 cells, also in view of the absence of other cellular responses to extracellular Angiotensin II like Ins(1,4,5)P3 formation and cytosolic [Ca2+] elevation. Therefore, the growth response elicited by Angiotensin IIintr is likely to be mediated by atypical AT-receptors with distinct pharmacology from AT1- or AT2-receptors.
Liposomes filled with the parent peptide Angiotensin I did not affect DNA synthesis. In contractility studies of adult rat aorta, we observed that both Angiotensin I and Angiotensin II filled liposomes induced contraction (Brailoiu et al., 1999). Either the contractile and growth responses are not intimately related or different pharmacological profiles of Angiotensin IIintr-receptors are present in different cell types. Differences in pharmacological receptor profiles among different cell types is supported by recent observations that Angiotensin IIintr inhibited inward Ca2+ current in rat cardiac myocytes, but stimulated this current in hamster cardiac myocytes (De Mello, 1998).
The presence of various atypical angiotensin receptors was reported previously (Noble et al., 1993; 1996; Smith, 1995; Regitz-Zagrosek et al., 1996; Li et al., 1998; Moriuchi et al., 1998). The pharmacological profile of one of those atypical angiotensin receptors resembles the profile obtained in A7r5 cells. Although observed in another species, this receptor mediates microvascular network formation in chick embryo, has a low affinity for losartan and PD123319 and is antagonized by CGP42112A (Noble et al., 1993; 1996). Further studies are necessary to elucidate if the receptors activated by Angiotensin IIintr, as observed by us, are related to one of those atypical receptors, and to establish their existence and binding profiles in other tissues.
Extracellular Angiotensin II induces several effects commonly evoked by growth factor receptor stimulation, such as tyrosine phosphorylation or activation of the Ras/ERK pathway ultimately leading to protein synthesis and cell cycle progression (Berk, 1999; Eguchi et al., 1999; Inagami et al., 1999). Stimulation of plasma membrane AT1 -receptors activate the MAPK cascade in vascular smooth muscle cells other than A7r5 cells (Ge & Anand-Srivastava, 1998; Li et al., 1998; Moriuchi et al., 1998) and this pathway is inhibited by PD98059 (Servant et al., 1996; Ushio-Fukai et al., 1998). Our experiments showed that inhibition of this pathway by PD98059 effectively blocked the growth response to Angiotensin IIintr administration. Activation of the MAPK cascade can be achieved via the PI-3K pathway, but a redundant pathway stimulates MAPK when large numbers of receptors are activated (Duckworth & Cantley, 1997). Interestingly, wortmannin only partially inhibited our Angiotensin IIintr effect, a finding also reported for extracellular Angiotensin II induced growth (Berk, 1999) and for other stimuli or cell types activated (Balla et al. 1998; Gutkind, 1998). This indicates that a strong signal is evoked by the Angiotensin IIintr mediated stimulation, comparable to activation of large number of ‘classical' plasma membrane receptors.
The obvious physiological candidate to stimulate the Angiotensin IIintr-receptor is Angiotensin II. Intracellular trafficking of Angiotensin II might be important for directing Angiotensin II to certain cellular locations to fully express its biological response. Several studies have demonstrated that Angiotensin II is internalized into the cells via an AT1-but not AT2-mediated process (Anderson et al., 1993; Hein et al., 1997). Intracellular pools of Angiotensin II were noticed in cardiomyocytes (Sadoshima et al., 1993) and recently angiotensin peptides, ACE-activity and AT1-receptors were detected in a renal endosomal fraction (Imig et al., 1999). The functional targets for Angiotensin IIintr are still unclear, but nuclear binding-proteins were reported for Angiotensin II (Booz et al., 1992; Tang et al., 1992; Jimenez et al., 1994). Interaction of Angiotensin IIintr with proteins at the cytosolic side of the plasma membrane also occurs in view of the results of Angiotensin IIintr on Ca2+ channels and gap junctions (De Mello 1996; 1998; Haller et al., 1996; 1999). This is possibly only a secondary related phenomenon, since the MAP-kinase pathway shown to be activated by Angiotensin IIintr in the present paper, modulates the opening of L-type Ca2+ channels in cardiomyocytes (Murata et al., 1999).
In conclusion, these data demonstrate that intracellular delivered Angiotensin II induces cell growth in A7r5 cells. Atypical AT-receptors are involved in view of the ineffectiveness of extracellular addition and the rank order of antagonist potencies obtained by intracellular application. The Angiotensin IIintr induced growth response is mediated via a PI-3K and MAPK/ERK – sensitive pathway. Angiotensin IIintr actions, inaccessible for common treatment, might open new views in the understanding and treatment of cardio-vascular related diseases.
Acknowledgments
Catalin M. Filipeanu is a recipient of an Ubbo Emmius fellowship from Groningen University Institute of Drug Exploration (GUIDE). Disposition of the Multifit software by Hans Proost and additional help with the fitting procedures is greatly appreciated.
Abbreviations
- Angiotensin IIintr
intracellular angiotensin II
- CGP42112A
nicotinic acid-Tyr-(N-benzoylcarbonyl-Arg)-Lys-His-Pro-Ile-OH
- DMEM
Dulbecco's Modified Eagle's Medium
- ERK
extracellular signal-regulated kinase
- FCS
foetal calf serum
- Ins(1,4,5)P3
inositol 1,4,5-trisphosphate
- losartan
(2-n- butyl-4-chloro-5-hydroxymethyl-1-[(2′-(1H-tetrazol-5-yl)biphenyl-4-yl)methyl]imidazole)
- MAPK
mitogen-activated protein kinase
- PI-3K
phosphatidyl inositol 3-kinase
- PD98059
2-(2-Amino-3-methoxyphenyl)-4H-1-benzopyran-4-one
- PD123319
(s)-1-(4-[dimethylamino]-3-methylphenyl)methyl-5-(diphenylacetyl)-4,5,6,7-tetrahydro-1H-imidazo[4,5-c]pyridine-6-carboxylate
References
- ANDERSON K.M., MURAHASHI T., DOSTAL D.E., PEACH M.J. Morphological and biochemical analysis of Angiotensin II internalization in cultured rat aortic smooth muscle cells. Am. J. Physiol. 1993;264:C179–C188. doi: 10.1152/ajpcell.1993.264.1.C179. [DOI] [PubMed] [Google Scholar]
- BALLA T., VARNAI P., TIAN Y., SMITH R.D. Signaling events activated by angiotensin II receptors: what goes before and after the calcium signals. Endocr. Res. 1998;24:335–344. doi: 10.3109/07435809809032613. [DOI] [PubMed] [Google Scholar]
- BERK B.C. Angiotensin II signal transduction in vascular smooth muscle: pathways activated by specific tyrosine kinases. J. Am. Soc. Nephrol. 1999;10:S62–S68. [PubMed] [Google Scholar]
- BOOZ G.W., CONRAD K.M., HESS A.L., SINGER H.A., BAKER K.M. Angiotensin-II-binding sites on hepatocyte nuclei. Endocrinology. 1992;130:3641–3649. doi: 10.1210/endo.130.6.1597161. [DOI] [PubMed] [Google Scholar]
- BRAILOIU E., FILIPEANU C.M., TICA A., TOMA C.P., DE ZEEUW D., NELEMANS S.A. Contractile effects by intracellular angiotensin II via receptors with a distinct pharmacological profile(in rat aorta. Br. J. Pharmacol. 1999;126:1133–1138. doi: 10.1038/sj.bjp.0702421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DANSER A.H., SCHALEKAMP M.A. Is there an internal cardiac renin-angiotensin system. Heart. 1996;76:28–32. doi: 10.1136/hrt.76.3_suppl_3.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE GASPARO M., CATT K.J., INAGAMI T.The IUPHAR Compendium of Receptor Characterization and Classification. Angiotensin receptors 199880–86.1th edition
- DE MELLO W.C. Renin-Angiotensin system and cell communication in the falling heart. Hypertension. 1996;27:1267–1272. doi: 10.1161/01.hyp.27.6.1267. [DOI] [PubMed] [Google Scholar]
- DE MELLO W.C. Intracellular Angiotensin regulates the inward calcium current in cardiac myocytes. Hypertension. 1998;32:976–982. doi: 10.1161/01.hyp.32.6.976. [DOI] [PubMed] [Google Scholar]
- DUCKWORTH B.C., CANTLEY L.C. Conditional inhibition of the mitogen-activated protein kinase cascade by wortmannin. Dependence on signal strength. J. Biol. Chem. 1997;272:27665–27670. doi: 10.1074/jbc.272.44.27665. [DOI] [PubMed] [Google Scholar]
- DZAU V.J., HORIUCHI M. Vascular remodeling–the emerging paradigm of programmed cell death (apoptosis): the Francis B. Parker lectureship. Chest. 1998;114:91S–99S. doi: 10.1378/chest.114.1_supplement.91s-a. [DOI] [PubMed] [Google Scholar]
- EGUCHI S., IWASAKI H., UENO H., FRANK G.D., MOTLEY E.D., EGUCHI K., MARUMO F., HIRATA Y., INAGAMI T. Intracellular signalling of angiotensin II-induced p70 S6 kinase phosphorylation at Ser(411) in vascular smooth muscle cells. Possible requirement of epidermal growth factor receptor, Ras, extracellular signal-regulated kinase, and Akt. J. Biol. Chem. 1999;274:36843–36851. doi: 10.1074/jbc.274.52.36843. [DOI] [PubMed] [Google Scholar]
- FILIPEANU C.M., DE ZEEUW D., NELEMANS S.A. Delta9-Tetrahydrocannabinol activates [Ca2+]i increases partly sensitive to capacitative store refilling. Eur. J. Pharmacol. 1997;336:R1–R3. doi: 10.1016/s0014-2999(97)01254-5. [DOI] [PubMed] [Google Scholar]
- FILIPEANU C.M., HENNING R.H., DE ZEEUW D., NELEMANS S.A.Functional evidence for a role of intracellular Angiotensin II in A7r5 cells Br. J. Pharmacol. 1998a123141P(Abstract) [Google Scholar]
- FILIPEANU C.M., NELEMANS S.A., HENNING R.H., BRAILOIU E., DE ZEEUW D.Intracellular angiotensin II effects in vascular smooth muscle cells J. Am. Soc. Nephrol. 1998b9422A(Abstract) [Google Scholar]
- GE C., ANAND-SRIVASTAVA M.B. Involvement of phosphatidylinositol 3-kinase and mitogen-activated protein kinase pathways in AII-mediated enhanced expression of Gi proteins in vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 1998;251:570–575. doi: 10.1006/bbrc.1998.9505. [DOI] [PubMed] [Google Scholar]
- GRIENDLING K.K., LASSEGUE B., ALEXANDER R.W. Angiotensin receptors and their therapeutic implications. Annu. Rev. Pharmacol. Toxicol. 1996;36:281–306. doi: 10.1146/annurev.pa.36.040196.001433. [DOI] [PubMed] [Google Scholar]
- GUTKIND J.S. The pathways connecting G protein-coupled receptors to the nucleus through divergent mitogen-activated protein kinase cascades. J. Biol. Chem. 1998;273:1839–1842. doi: 10.1074/jbc.273.4.1839. [DOI] [PubMed] [Google Scholar]
- HALLER H., LINDSCHAU C., ERDMANN B., QUASS P., LUFT F.C. Effects of intracellular Angiotensin II in vascular smooth muscle cells. Circ. Res. 1996;79:765–772. doi: 10.1161/01.res.79.4.765. [DOI] [PubMed] [Google Scholar]
- HALLER H., LINDSCHAU C., QUASS P., LUFT F.C. Intracellular actions of angiotensin II in vascular smooth muscle cells. J. Am. Soc. Nephrol. 1999;10:S75–S83. [PubMed] [Google Scholar]
- HEIN L., MEINEL L., PRATT R.E., DZAU V.J., KOBILKA B.K. Intracellular trafficking of Angiotensin II and its AT1 and AT2 receptors: evidence for selective sorting of receptor and ligand. Mol. Endocrinol. 1997;11:1266–1277. doi: 10.1210/mend.11.9.9975. [DOI] [PubMed] [Google Scholar]
- IMIG J.D., NAVAR G.L., ZOU L.X., O'REILLY K.C., ALLEN P.L., KAYSEN J.H., HAMMOND T.G., NAVAR L.G. Renal endosomes contain angiotensin peptides, converting enzyme, and AT(1A) receptors. Am. J. Physiol. 1999;277:F303–F311. doi: 10.1152/ajprenal.1999.277.2.F303. [DOI] [PubMed] [Google Scholar]
- INAGAMI T., EGUCHI S., NUMAGUCHI K., MOTLEY E.D., TANG H., MATSUMOTO T., YAMAKAWA T. Cross-talk between angiotensin II receptors and the tyrosine kinases and phosphatases. J. Am. Soc. Nephrol. 1999;10:S57–S61. [PubMed] [Google Scholar]
- JIMENEZ E., VINSON G.P., MONTIEL M. Angiotensin II (AII)-binding sites in nuclei from rat liver: partial characterization of the mechanism of AII accumulation in nuclei. J. Endocrinol. 1994;143:449–453. doi: 10.1677/joe.0.1430449. [DOI] [PubMed] [Google Scholar]
- LI X., SHAMS M., ZHU J., KHALIG A., WILKES M., WHITTLE M., BARNES N., AHMED A. Cellular localization of AT1 receptor mRNA and protein in normal placenta and its reduced expression in intrauterine growth. Angiotensin II stimulates the release of vasorelaxants. J. Clin. Invest. 1998;101:442–454. doi: 10.1172/JCI119881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LINZ W., WIEMER G., GOHLKE P., UNGER T., SCHOLKENS B.A. Contribution of kinins to the cardiovascular actions of angiotensin-converting enzyme inhibitors. Pharmacol. Rev. 1995;47:25–49. [PubMed] [Google Scholar]
- LOKUTA A.J., COOPER C., GAA H.E., ROGERS T.B. Angiotensin II stimulates the release of phospholipid-derived second messengers through multiple receptor subtypes in heart cells. J. Biol. Chem. 1994;269:4832–4838. [PubMed] [Google Scholar]
- MATSUSAKA T., ICHIKAWA I. Biological functions of angiotensin and its receptors. Annu. Rev. Physiol. 1997;59:395–412. doi: 10.1146/annurev.physiol.59.1.395. [DOI] [PubMed] [Google Scholar]
- MORIUCHI R., SHIBATA S., HIMENO A., JOHREN O., HOE K.L., SAAVEDRA J.M. Molecular cloning and pharmacological characterization of an atypical gerbil angiotensin II type-1 receptor and its mRNA expression in brain and peripheral tissues. Brain. Res. Mol. Brain. Res. 1998;60:234–246. doi: 10.1016/s0169-328x(98)00187-9. [DOI] [PubMed] [Google Scholar]
- MURATA M., FUKUDA K., ISHIDA H., MIYOSHI S., KOURA T., KODAMA H., NAKAZAWA H.K., OGAWA S. Leukemia inhibitory factor, a potent cardiac hypertrophic cytokine, enhances L-type Ca2+ current and [Ca2+]i transient in cardiomyocytes. J. Mol. Cell. Cardiol. 1999;31:237–245. doi: 10.1006/jmcc.1998.0866. [DOI] [PubMed] [Google Scholar]
- LE NOBLE F.A., KESSELS-VAN WYLICK L.C., HACKING W.J., SLAAF D.W., OUDE EGBRINK M.G., STRUIJKER-BOUDIER H.A. The role of angiotensin II and prostaglandins in arcade formation in a developing microvascular network. J. Vasc. Res. 1996;33:480–488. doi: 10.1159/000159187. [DOI] [PubMed] [Google Scholar]
- LE NOBLE F.A., SCHREURS N.H., VAN STRAATEN H.W., SLAAF D.W., SMITS J.F., ROGG H., STRUIJKER-BOUDIER H.A. Evidence for a novel angiotensin II receptor involved in angiogenesis in chick embryo chorioallantoic membrane. Am. J. Physiol. 1993;264:R460–R465. doi: 10.1152/ajpregu.1993.264.2.R460. [DOI] [PubMed] [Google Scholar]
- REGITZ-ZAGROSEK V., NEUSS M., WARNECKE C., HOLZMEISTER J., HILDEBRANDT A.G., FLECK E. Subtype 2 and atypical angiotensin receptors in the human heart. Basic. Res. Cardiol. 1996;91:73–77. doi: 10.1007/BF00795366. [DOI] [PubMed] [Google Scholar]
- SADOSHIMA J., XU Y., SLAYTER H.S., IZUMO S. Autocrine release of angiotensin II mediates stretch-induced hypertrophy of cardiac myocytes in vitro. Cell. 1993;75:977–984. doi: 10.1016/0092-8674(93)90541-w. [DOI] [PubMed] [Google Scholar]
- SERVANT M.J., GIASSON E., MELOCHE S. Inhibition of growth factor-induced protein synthesis by a selective MEK inhibitor in aortic smooth muscle cells. J. Biol. Chem. 1996;271:16047–16052. doi: 10.1074/jbc.271.27.16047. [DOI] [PubMed] [Google Scholar]
- SIPMA H., DUIN M., HOITING B., DEN HERTOG A., NELEMANS A. Regulation of histamine- and UTP-induced increases in Ins(1,4,5)P3, Ins(1,3,4,5)P4 and Ca2+ by cyclic AMP in DDT1 MF-2 cells. Br. J. Pharmacol. 1995;114:383–390. doi: 10.1111/j.1476-5381.1995.tb13238.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SMITH R.D. Identification of atypical (non-AT1, non-AT2) angiotensin binding sites with high affinity for angiotensin I on IEC-18 rat intestinal epithelial cells. FEBS. Lett. 1995;373:199–202. doi: 10.1016/0014-5793(95)01039-h. [DOI] [PubMed] [Google Scholar]
- TANG S.S., ROGG H., SCHUMACHER R., DZAU V.J. Characterization of nuclear angiotensin-II-binding sites in rat liver and comparison with plasma membrane receptors. Endocrinology. 1992;131:374–380. doi: 10.1210/endo.131.1.1612017. [DOI] [PubMed] [Google Scholar]
- TIMMERMANS P.B., CHIU A.T., HERBLIN W.F., WONG P.C., SMITH R.D. Angiotensin II receptor subtypes. Am. J. Hypertens. 1992;5:406–410. doi: 10.1093/ajh/5.6.406. [DOI] [PubMed] [Google Scholar]
- USHIO-FUKAI M., ALEXANDER R.W., AKERS M., GRIENDLING K.K. p38 Mitogen-activated protein kinase is a critical component of the redox-sensitive signaling pathways activated by angiotensin II. Role in vascular smooth muscle cell hypertrophy. J. Biol. Chem. 1998;273:15022–15029. doi: 10.1074/jbc.273.24.15022. [DOI] [PubMed] [Google Scholar]
- WEBER M.A. Unsolved problems in treating hypertension: rationale for new approaches. Am. J. Hypertension. 1998;11:145S–149S. doi: 10.1016/s0895-7061(98)00191-5. [DOI] [PubMed] [Google Scholar]
- XORIUCHI M., HAMAI M., CUI T.X., IWAI M., MINOKOSHI Y. Cross talk between angiotensin II type 1 and type 2 receptors: cellular mechanism of angiotensin type 2 receptor-mediated cell growth inhibition. Hypertens. Res. 1999;22:67–74. doi: 10.1291/hypres.22.67. [DOI] [PubMed] [Google Scholar]