

Abstract
Nitric oxide (NO) donors are known to induce both delayed cardioprotection and myocardial heat stress protein (HSP) expression. Moreover, heat stress (HS), which also protects myocardium against ischaemic damages, is associated with a NO release. Therefore, we have investigated the implication of NO in HS-induced resistance to myocardial infarction, in the isolated rat heart model.
Rats were divided in six groups (n=10 in each group), subjected or not to heat stress (42°C internal temperature, 15 min) and treated or not with nitro-L-arginine-methylester (L-NAME) a non-selective inhibitor of NO synthase isoforms, or L-N6-(1-imino-ethyl)lysine (L-NIL), a selective inhibitor of the inducible NO synthase. Twenty-four hours after heat stress, their hearts were isolated, retrogradely perfused, and subjected to a 30-min occlusion of the left coronary artery followed by 120 min of reperfusion.
Infarct-to-risk ratio was significantly reduced in HS (18.7±1.6%) compared to Sham (33.0±1.7%) hearts. This effect was abolished in L-NAME-treated (41.7±3.1% in HS+L-NAME vs 35.2±3.0% in Sham+L-NAME) and L-NIL-treated (36.1±3.4% in HS+L-NIL vs 42.1±4.6% in Sham+L-NIL) groups. Immunohistochemical analysis of myocardial HSP 27 and 72 showed an HS-induced increase of these proteins, which was not modified by L-NAME pretreatment.
We conclude that NO synthases, and in particular the inducible isoform, appear to play a role in the heat stress-induced cardioprotection, independently of HSP 27 and 72 levels. Further investigations are required to elucidate the precise role of HSPs in this adaptive response.
Keywords: Heat stress, infarct size, nitric oxide synthases, heat stress protein
Introduction
Heat stress (HS) is known to protect the myocardium against ischaemia-reperfusion injury by preserving mechanical function (Currie et al., 1988) and reducing myocardial necrosis (Donnelly et al., 1992; Yellon et al., 1992; Joyeux et al., 1997). Mechanisms involved in these protective effects have been explored and different potential end-effectors have been identified. Among them, cardiac heat stress proteins (HSPs) have been proposed. In particular, a direct correlation between the amount of HSP 72 expression and the degree of HS-induced ischaemic tolerance has been observed in the rat (Hutter et al., 1994) and in the rabbit (Marber et al., 1994). However, the precise mechanisms underlying HSP synthesis and the signal transduction pathways associated with the development of this HS-induced cardioprotection remain to be determined.
It has been observed that HS sharply increases the NO production in different rat organs and that this effect precedes the increase in HSP 72 synthesis (Malyshev et al., 1995). Moreover, it seems that the increased HSP 72 expression, induced 24 h after HS, is attenuated when NO synthesis is inhibited (Malyshev et al., 1995).
On the other hand, the HS-induced cardioprotection resembles that observed during the second window of protection following ischaemic preconditioning (Marber et al., 1993; Yellon & Baxter, 1995). Mediators under investigation for their role in ischaemic preconditioning may therefore provide a potential mechanism for HS-induced protection (Parratt & Szekeres, 1995; Richard et al., 1996). Hence, NO has been shown to be implicated in the delayed cardioprotection following ischaemic preconditioning in vivo in the rabbit (Bolli et al., 1997; Qiu et al., 1997). Furthermore, it has been observed in conscious rabbits that NO donor administration induces protection against myocardial ischaemia-reperfusion injury 24 h later, thus mimicking the late phase of ischaemic preconditioning (Takano et al., 1998).
Therefore, the aim of the present study was to examine the role of NO as a trigger of the infarct size-reducing effect conferred by heat stress in the isolated rat heart. For that, two different NO synthase (NOS) inhibitors were used: the non-selective NOS inhibitor, nitro-L-arginine-methylester (L-NAME), and the selective inducible NOS (iNOS) inhibitor, L-N6-(1-imino-ethyl)lysine (L-NIL).
Methods
Experimental treatment groups
Male Wistar rats (280 – 340 g) were used for these studies. This investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication no 85-23, revised 1985).
This study was conducted in two parts. In the first part, rats were submitted to either heat stress (HS groups) or anaesthesia without hyperthermia (Sham groups). Prior to this procedure, the animals received either L-NAME or L-NIL as previously described (Lagneux et al., 2000). Subsequently, all animals were allowed to recover for 24 h. In the second part, ischaemia (30 min)-reperfusion (120 min) was performed in isolated hearts. Six experimental groups (n=10 in each group) were studied:
Group Sham – rats were lightly anaesthetized with pentobarbitone sodium (25 mg kg−1, i.p.);
Group Sham+L-NAME – animals were pretreated with L-NAME 80 mg l−1 added to the drinking water during 48 h before sham HS and with an injection (50 mg kg−1, i.p.) 10 min prior to sham HS;
Group Sham+L-NIL – animals were pretreated with four injections of L-NIL (3 mg kg−1, i.p.) 36, 24, 12 h and 10 min prior to sham HS;
In Groups HS, HS+L-NAME and HS+L-NIL, rats were similarly treated prior to undergoing heat stress. The experimental protocol is summarized in Figure 1.
Figure 1.
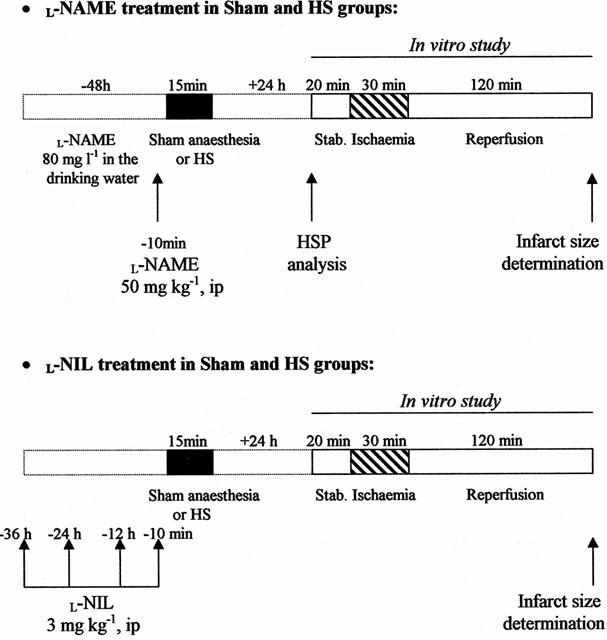
Description of the treatments and experimental protocols in rats subjected to sham anaesthesia (Sham groups) or heat stress (HS groups).
Heat stress protocol
Heat stress was achieved by placing anaesthetized (with 25 mg kg−1 i.p. sodium pentobarbitone) rats in an environmental chamber under an infrared light. Their body temperature, recorded with a rectal probe, was increased to 42±0.2°C for 15 min. Sham control animals were anaesthetized only. All rats were allowed to recover for 24 h.
Ischaemia-reperfusion protocol
Twenty-four hours after heat stress, rats were anaesthetized with 60 mg kg−1 i.p. sodium pentobarbitone and treated with heparin (1000 u kg−1, i.p.). The heart was rapidly excised and immediately immersed in 4°C Krebs-Henseleit buffer solution (mM: NaCl 118.0, KCl 4.7, CaCl2 1.8, KH2PO4 1.2, MgSO4 1.2, NaHCO3 25.2 and glucose 11.0). The aortic stump was then cannulated and the heart perfused retrogradely using the Langendorff technique at a constant pressure (75 mmHg) with oxygenated Krebs-Henseleit buffer. A water-filled balloon, coupled to a pressure transducer (Statham), was inserted into the left ventricular cavity via the left atrium for pressure recordings. Left ventricular end-diastolic pressure (LVEDP) was adjusted between 8 – 10 mmHg. Myocardial temperature was measured by a thermoprobe inserted into the left ventricle and was maintained constant close to 37°C. For temporary occlusion of the left coronary artery (LCA), a 3/0 silk suture (Mersilk W546, Ethicon) was placed around the artery a few millimetres distal to the aortic root. After 20 min of stabilization, regional ischaemia was induced by tightening the snare around the LCA for 30 min. Thereafter the heart was reperfused for 120 min. Coronary flow (CF) was measured throughout the ischaemia-reperfusion procedure, by collecting the effluent. Heart rate (HR) and left ventricular developed pressure (LVDP=difference between left ventricular systolic pressure and LVEDP) were continuously recorded on a polygraph (Windograph, Gould Instrument). At the end of the reperfusion period, the coronary artery ligature was retied and unisperse blue (Ciba-Geigy) dye was slowly infused through the aorta to delineate the myocardial risk zone. After removal of the right ventricle and connective tissues, the heart was frozen and then sectioned into 2 mm transverse sections from apex to base (6 – 7 slices per heart). Following defrosting, the slices were incubated at 37°C with 1% triphenyltetrazolium chloride in phosphate buffer (pH 7.4) for 10 – 20 min and fixed in 10% formaldehyde solution to distinguish clearly stained viable tissue and unstained necrotic tissue. Left ventricular infarct zone (I) was determined using a computerized planimetric technique (Minichromax, Biolab) and expressed as a percentage of the risk zone (R) and of the left ventricle (LV). It can be noticed that in this model, infarct size evolution is incomplete after 2 h reperfusion and it is possible that our results would vary using a longer reperfusion duration leading to the ultimate extent of necrosis.
Immunohistochemical analysis of myocardial HSP 27 and 72
To determine myocardial HSP 27 and 72 expression, additional animals (n=4 in each group) were submitted to HS or sham HS, treated or not with L-NAME. Twenty-four hours later, animals were re-anaesthetized and treated with heparin as described above before the hearts were quickly excised. Hearts were also fixed in 10% paraformaldehyde, embedded in paraffin and cut in 5 μm sections which were stained with a monoclonal mouse anti-HSP 70 (dilution 1 : 100; Stressgen) or anti-HSP 27 (dilution 1 : 50; Neomarkers) antibody. The fixed antibodies were exposed for 30 min to a rabbit streptevidin-biotin-peroxydase complex (dilution 1 : 600; Dako, LSAB Denmark) and the peroxydase reaction was developed using the AEC chromogen (AEC kit, Sigma France). The slides were counter-stained with Mayer's hematoxylin and observed.
Statistical analysis
The data are presented as mean±s.e.mean. Comparisons in CF, HR and LVDP were performed using two-way repeated measures ANOVA. Infarct size was analysed by a one-way ANOVA. Post hoc comparisons were done using Tukey tests. P values ⩽0.05 were considered significant.
Exclusion criteria
Only hearts with CF within 8 – 15 ml min−1 and LVDP >70 mmHg at the end of the stabilization period were included in this study. The efficiency of coronary occlusion was indicated by a decrease in CF >30%. All hearts which developed ventricular fibrillation (VF) during ischaemia-reperfusion and did not revert spontaneously within 2 min were defibrillated by a gentle mechanical stimulation. Finally, the risk zone determined at the end of the ischaemia-reperfusion procedure had to represent 40 – 60% of the LV (Joyeux et al., 1997).
Materials
L-NIL and L-NAME were purchased from Sigma (France). All other reagents were of analytical reagent quality.
Results
Haemodynamic data
Table 1 summarizes CF, HR and LVDP data recorded in the six experimental groups during the stabilization and ischaemia-reperfusion periods. Twenty-four hours after heat stress or sham anaesthesia, there was no statistically significant difference in hemodynamic performance between the six groups.
Table 1.
Haemodynamic data
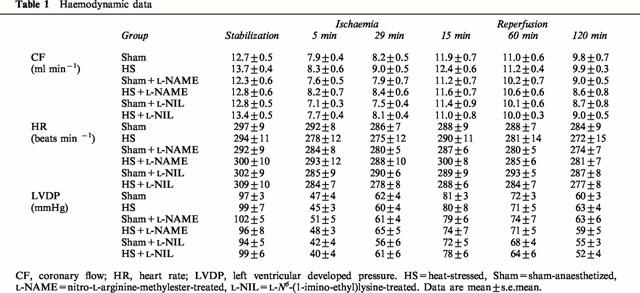
Infarct data
Figure 2 represents infarct-to-risk ratio (I/R) of the six experimental groups. Heat stress significantly reduced I/R from 33.0±1.7% in Sham to 18.7±1.6% in HS (P⩽0.05). This infarct size-reducing effect of heat stress was abolished in L-NAME-treated (41.7±3.1%) and L-NIL-treated (36.1±3.4%) groups. In non-heat stressed rats, treatment with L-NAME (35.2±3.0%) and L-NIL (42.1±4.6%) had no effect on infarct size (vs 33.0±1.7% in Sham group). Similar results were observed with the I/LV ratio of the six groups (data not shown). Myocardial risk size, expressed as a percentage of the left ventricle (R/LV), ranged between 40 – 50% and was not different between the various groups. Therefore, differences in infarct size did not result from variability in the risk zone.
Figure 2.
Infarct size (I) expressed as a percentage of the risk zone (R) in isolated rat hearts subjected to 30-min coronary occlusion followed by 120-min reperfusion. Rats were treated with either nitro-L-arginine-methylester (L-NAME) or L-N6-(1-imino-ethyl)lysine (L-NIL) prior to undergoing heat stress (HS) or sham anaesthesia (Sham). *P⩽0.05.
HSP 72 and 27 immunohistochemical analysis
Immunohistochemical analysis of myocardial HSP 27 (Figure 3) and 72 (Figure 4) expression showed a marked increase of these proteins following heat stress. L-NAME-pretreatment did not modify the heat stress-induced increase of HSP 27 and 72 (groups HS+L-NAME (D) vs Sham+L-NAME (C), Figures 3 and 4). Since L-NAME is a non-selective inhibitor of NOS isoforms, the analysis with L-NIL, a selective inhibitor of the iNOS, was not performed.
Figure 3.
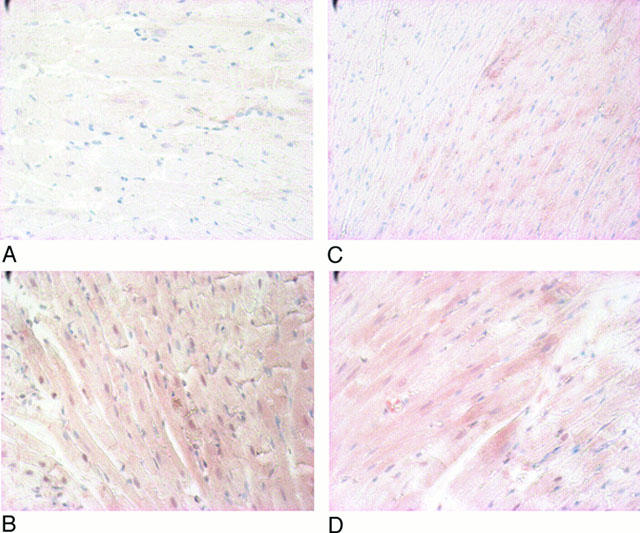
Immunohistochemical analysis of myocardial HSP 27 in hearts from Sham (A), HS (B), Sham+L-NAME (C) and HS+L-NAME (D) groups. HS=heat-stressed, Sham=sham-anaesthetized, L-NAME=nitro-L-arginine-methylester-treated.
Figure 4.
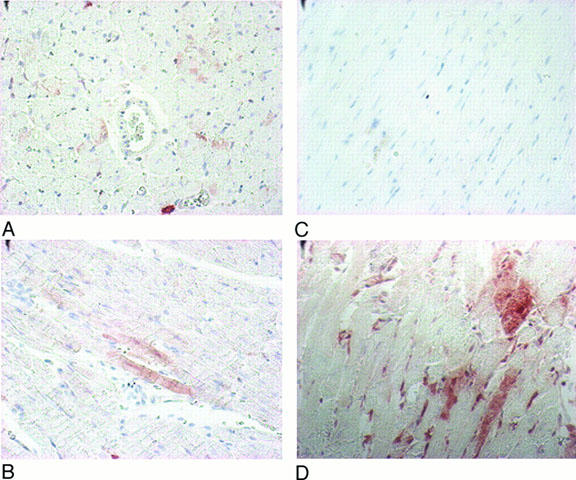
Immunohistochemical analysis of myocardial HSP 72 in hearts from Sham (A), HS (B), Sham+L-NAME (C) and HS+L-NAME (D) groups. HS=heat-stressed, Sham=sham-anaesthetized, L-NAME=nitro-L-arginine-methylester-treated.
Discussion
This study provides the first demonstration of the implication of NO in the heat stress-induced delayed cardioprotection. We observed that prior heat stress significantly reduced infarct size in the isolated rat heart subjected to an ischaemia-reperfusion sequence, in accordance with previous studies (Donnelly et al., 1992; Marber et al., 1993; Joyeux et al., 1997). This myocardial ischaemic tolerance was abolished by the administration of both L-NAME and L-NIL prior to heat stress. The use of both inhibitors allowed the investigation of the role of the different NOS isoforms. While L-NAME is unselective, L-NIL is a selective inhibitor of iNOS (Moore et al., 1994). In our study, the treatment applied had to satisfy two points. First, NOS isoforms had to be inhibited at the moment of heat stress. This point has been assessed by Schwartz et al. (1997). They have demonstrated that following similar L-NAME and L-NIL treatment, corresponding NOS isoforms were inhibited since LPS-induced hypotension was corrected. By the same manner, LPS-induced increase in both urinary excretion of nitrates-nitrites and cyclic GMP level was abolished (Schwartz et al., 1997; Lortie et al., 2000). Secondly, treatments had to be reversible since in this study we have investigated the role of NO as a trigger of the heat stress-induced cardioprotection and not as a mediator during the ischaemia-reperfusion sequence. We have previously verified this point in vivo in the rat (Lagneux et al., 2000). This study showed that 24 h after the end of treatment, mean arterial blood pressure of anaesthetized rats was within the normal range, and that the hypotension induced by bradykinin, which is triggered by NO production (Davisson et al., 1996), was comparable to that of control animals.
It seems that HS induces NO production, since Malyshev et al. (1995) have observed a sharp transient increase in NO generation 1 h after HS in different organs of the rat and notably the myocardium. Our study shows that the non-selective NOS inhibitor, L-NAME, completely abolished the HS-induced protection against myocardial infarction in rat heart. Thus, NO formation seems to play an essential role in this cardioprotective phenomenon. We also demonstrate that the selective iNOS inhibitor, L-NIL, is as effective as L-NAME, providing the first evidence that NO produced by iNOS is involved in HS-induced cardioprotection.
NO has also been shown to be involved in other cardioprotective phenomena. Indeed, Bolli's group has recently presented convincing evidence that NO is a trigger of the delayed protection conferred by ischaemic preconditioning in the conscious rabbit (Bolli et al., 1998). Pretreatment with a NOS inhibitor during the initial ischaemic stimulus blocked protection (Qiu et al., 1997), and conversely a NO donor in lieu of ischaemia induced delayed cardioprotection (Takano et al., 1998; Banerjee et al., 1999). Moreover, iNOS seems to be the principal NOS isoform involved in this cardioprotective phenomenon, since a selective iNOS inhibitor completely abolished the delayed protection induced by the ischaemic preconditioning in vivo in the rabbit (Imagawa et al., 1999). This is in accordance with a study from Guo et al. (1999) which demonstrates in vivo in the mouse that the late phase of ischaemic preconditioning is associated with a selective up-regulation of myocardial iNOS. NO seems to also trigger the delayed protective effect of monophosphoryl lipid A (MLA) in the isolated rat heart, since co-administration of NOS inhibitors and MLA abolished the preservation of ventricular function induced by MLA alone (Tosaki et al., 1998; György et al., 1999).
Our immunohistochemical analysis showed an increase in myocardial HSP 27 and 72 synthesis induced 24 h after heat stress, which was not modified by the blockade of all NOS isoforms. It seems thus that the heat stress-induced cardioprotection does not appear to be related to induction of HSP 27 and 72 synthesis, since pretreatment with L-NAME abolished myocardial ischaemic tolerance while it had no effect on the increase in myocardial HSP levels. Several studies point to a relation between HSP 27 and 72 induction and cardioprotection. Hence, Marber et al. (1993) have observed that prior hyperthermia induces a high level of myocardial HSP 72 expression along with the enhanced myocardial tolerance to ischaemic injury. Moreover, the level of HSP 72 has been directly correlated to the degree of heat stress-induced cardioprotection in the rat (Hutter et al., 1994) and in the rabbit (Marber et al., 1994). Furthermore, improved functional recovery or reduced infarct size has been observed in transgenic mouse and rat hearts overexpressing HSP 72 and subjected to an ischaemia-reperfusion sequence (Marber et al., 1995; Plumier et al., 1995; Hutter et al., 1996; Suzuki et al., 1997). By the same manner, it has been shown that the overexpression of HSP 27 or 72 protects rat cardiomyocytes against ischaemia insult (Martin et al., 1997; Mestril et al., 1996).
Our study shows that protection of myocardium can be blocked independently of the level of HSP 27 and 72 induction. This finding is in agreement with previous studies showing that protein kinase C (PKC) inhibition or α1-adrenoceptor blockade abolished the cardioprotection conferred by heat stress with no effect on myocardial HSP 72 synthesis (Joyeux et al., 1997; 1998a). One possible explanation is that NOS inhibition, as PKC inhibition or α1-adrenoceptor blockade, could alter the phosphorylation and/or the functional state of HSPs thus rendering them ineffective in protecting the myocardium. Further experiments are required to explore this hypothesis.
Although HSPs are widely studied as primary effectors of heat stress-induced protection, other mediators can be evoked (Joyeux et al., 1999). Hence, ATP-sensitive potassium (KATP) channel opening appears to mediate the heat stress-induced delayed cardioprotection in the rat (Joyeux et al., 1998b) and in the rabbit (Hoag et al., 1997; Pell et al., 1997). Moreover, some physiological effects of NO seem to be due to the KATP channel activation. For example, peripheral vascular vasodilatory response have been found to involve specific KATP channels (Champion \& Kadowitz, 1997) and it seems that NO could potentiate the KATP channel current in isolated guinea-pig ventricular cells (Shinbo \& Iijima, 1997). A recent study on rabbit ventricular myocytes suggests that NO could act directly as a mitochondrial KATP channel opener (Sasaki et al., 2000). Furthermore, it has been observed that KATP channel blockers abolish the ability of the NO donor to protect cultured myocytes against ischaemic injury (Stambaugh et al., 1999). thus, it could be hypothesized that the NO-dependent opening of KATP channels mediates HS-induced cardioprotection.
In summary, this study provides the first demonstration of the implication of NO as trigger of resistance to myocardial infarction induced by heat stress in the isolated rat heart, since both L-NAME and L-NIL pretreatments abolished the heat stress-induced cardioprotection. Although iNOS appears to play a role in this cardioprotective phenomenon, the role of the other isoforms remains to be determined. Finally, NO appears to mediate this cardioprotection by a mechanism independent of HSP 27 and 72 induction. Further investigations are required to clarify the signal transduction pathways which co-ordinate the heat stress response and the potential role of HSP 27 and 72, and of other stress-inducible proteins, in mediating adaptative cytoprotection.
Abbreviations
- CF
coronary flow
- HR
heart rate
- HS
heat stress
- HSP
heat stress protein
- I
infarct zone
- iNOS
inducible nitric oxide synthase
- KATP channel
ATP-sensitive potassium channel
- LV
left ventricle
- LVDP
left ventricular developed pressure
- LVEDP
left ventricular end-diastolic pressure
- L-NAME
nitro-L-arginine-methylester
- L-NIL
L-N6-(1-imino-ethyl)lysine
- MLA
monophosphoryl lipid A
- NO
nitric oxide
- NOS
nitric oxide synthase
- PKC
protein kinase C
- R
risk zone
- VF
ventricular fibrillation
References
- BANERJEE S., TANG X.L., QIU Y., TAKANO H., MANCHIKALAPUDI S., DAWN B., SHIRK G., BOLLI R. Nitroglycerin induces late preconditioning against myocardial stunning via a PKC-dependant pathway. Am. J. Physiol. 1999;277:H2488–H2494. doi: 10.1152/ajpheart.1999.277.6.H2488. [DOI] [PubMed] [Google Scholar]
- BOLLI R., DAWN B., TANG X.L., QIU Y., PING P., XUAN Y.T., JONES W.K., TAKANO H., GUO Y., ZHANG J. The nitric oxide hypothesis of late preconditioning. Bas. Res. Cardiol. 1998;93:325–338. doi: 10.1007/s003950050101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOLLI R., MANCHIKALAPUDI S., TANG X.L., TAKANO H., QIU Y., GUO Y., ZHANG Q., JADOON A.K. The protective effect of late preconditioning against myocardial stunning in conscious rabbits is mediated by nitric oxide synthase. Evidence that nitric oxide acts both as a trigger and as a mediator of the late phase of ischemic preconditioning. Circ. Res. 1997;81:1094–1107. doi: 10.1161/01.res.81.6.1094. [DOI] [PubMed] [Google Scholar]
- CHAMPION H.C., KADOWITZ P.J. NO release and the opening of K+ATP channels mediate vasodilator responses to histamine in the cat. Am. J. Physiol. 1997;273:H928–H937. doi: 10.1152/ajpheart.1997.273.2.H928. [DOI] [PubMed] [Google Scholar]
- CURRIE R.W., KARMAZYN M., KLOC M., MAILER K. Heat-shock response is associated with enhanced postischemic ventricular recovery. Circ. Res. 1988;63:543–549. doi: 10.1161/01.res.63.3.543. [DOI] [PubMed] [Google Scholar]
- DAVISSON R.L., BATES J.N., JOHNSON A.K., LEWIS S.J. Use-dependent loss of acetylcholine- and bradykinin-mediated vasodilation after nitric oxide synthase inhibition. Evidence for preformed stores of nitric oxide-containing factors in vascular endothelial cells. Hypertension. 1996;28:354–360. doi: 10.1161/01.hyp.28.3.354. [DOI] [PubMed] [Google Scholar]
- DONNELLY T.J., STEVENS R.E., VISSERN F.L.J., WELCH W.J., WOLFE C.L. Heat shock protein induction in rat hearts. A role for improved myocardial salvage after ischemia and reperfusion. Circulation. 1992;85:769–778. doi: 10.1161/01.cir.85.2.769. [DOI] [PubMed] [Google Scholar]
- GUO Y., JONES W.K., XUAN Y.T., TANG X.L., BAO W., WU W.J., HAN H., LAUBACH V.E., PING P., YANG Z., QIU Y., BOLLI R. The late phase of ischemic preconditioning is abrogated by targeted disruption of the inducible NO synthase gene. Proc. Natl. Acad. Sci. 1999;96:11507–11512. doi: 10.1073/pnas.96.20.11507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GYÖRGY K., MULLER B., VÉGH A., KLESCHOV A.L., STOCLET J.C. Triggering role of nitric oxide in the delayed protective effect of monophosphoryl A in rat heart. Br. J. Pharmacol. 1999;127:1892–1898. doi: 10.1038/sj.bjp.0702725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOAG J.B., QIAN Y.Z., NAYEEM M.A., D'ANGELO M., KUKREJA R.C. ATP-sensitive potassium channel mediates delayed ischemic protection by heat stress in rabbit heart. Am. J. Physiol. 1997;273:H2458–H2464. doi: 10.1152/ajpheart.1997.273.5.H2458. [DOI] [PubMed] [Google Scholar]
- HUTTER J.J., MESTRIL R., TAM E.K., SIEVERS R.E., DILLMANN W.H., WOLFE C.L. Overexpression of heat shock protein 72 in transgenic mice decreases infarct size in vivo. Circulation. 1996;94:1408–1411. doi: 10.1161/01.cir.94.6.1408. [DOI] [PubMed] [Google Scholar]
- HUTTER M.M., SIEVERS R.E., BARBOSA V., WOLFE C.L. Heat-shock protein induction in rat hearts. A direct correlation between the amount of heat-shock protein induced and the degree of myocardial protection. Circulation. 1994;89:355–360. doi: 10.1161/01.cir.89.1.355. [DOI] [PubMed] [Google Scholar]
- IMAGAWA J., YELLON D.M., BAXTER G.F. Pharmacological evidence that inducible nitric oxide synthase is a mediator of delayed preconditioning. Br. J. Pharmacol. 1999;126:701–708. doi: 10.1038/sj.bjp.0702368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JOYEUX M., BAXTER G.F., THOMAS D.L., RIBUOT C., YELLON D.M. Protein kinase C is involved in resistance to myocardial infarction induced by heat stress. J. Mol. Cell. Cardiol. 1997;29:3311–3319. doi: 10.1006/jmcc.1997.0556. [DOI] [PubMed] [Google Scholar]
- JOYEUX M., GODIN-RIBUOT D., DEMENGE P., YELLON D.M., RIBUOT C. Infarct size-reducing effect of heat stress and α1 Adrenoceptors in Rats. Br. J. Pharmacol. 1998a;125:645–650. doi: 10.1038/sj.bjp.0702137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JOYEUX M., GODIN-RIBUOT D., RIBUOT C. Resistance to myocardial infarction induced by heat stress and the effect of ATP-sensitive potassium channel blockade in the rat isolated heart. Br. J. Pharmacol. 1998b;123:1085–1088. doi: 10.1038/sj.bjp.0701710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JOYEUX M., GODIN-RIBUOT D., YELLON D.M., DEMENGE P., RIBUOT C. Heat stress response and myocardial protection. Fund. Clin. Pharmacol. 1999;13:1–10. doi: 10.1111/j.1472-8206.1999.tb00314.x. [DOI] [PubMed] [Google Scholar]
- LAGNEUX C., GODIN-RIBUOT D., DEMENGE P., RIBUOT C. Nitric oxide and its role in the induction of kinin B(1)-receptors after heat stress in the rat. Immunopharmacology. 2000;48:43–49. doi: 10.1016/s0162-3109(00)00178-8. [DOI] [PubMed] [Google Scholar]
- LORTIE M.J., ISHIZUKA S., SCHWARTZ D., BLANTZ R.C. Bioactive products of arginine in sepsis: tissue and plasma composition after LPS and iNOS blockade. Am. J. Physiol. Cell Physiol. 2000;278:C1191–C1199. doi: 10.1152/ajpcell.2000.278.6.C1191. [DOI] [PubMed] [Google Scholar]
- MALYSHEV I.Y., MANUKHINA E.B., MIKOYAN V.D., KUBRINA L.N., VANIN A.F. Nitric oxide is involved in heat-induced HSP70 accumulation. FEBS Lett. 1995;370:159–162. doi: 10.1016/0014-5793(95)00801-f. [DOI] [PubMed] [Google Scholar]
- MARBER M.S., LATCHMAN D.S., WALKER J.M., YELLON D.M. Cardiac stress protein elevation 24 hours after brief ischemia or heat stress is associated with resistance to myocardial infarction. Circulation. 1993;88:1264–1272. doi: 10.1161/01.cir.88.3.1264. [DOI] [PubMed] [Google Scholar]
- MARBER M.S., MESTRIL R., CHI S.H., SAYEN R., YELLON D.M. Overexpression of the rat inducible 70-kD heat stress protein in a transgenic mouse increases the resistance of the heart to ischemic injury. J. Clin. Invest. 1995;95:1446–1456. doi: 10.1172/JCI117815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARBER M.S., WALKER J.M., LATCHMAN D.S., YELLON D.M. Myocardial protection after whole body heat stress in the rabbit is dependent on metabolic substrate and is related to the amount of the inducible 70-kD heat stress protein. J. Clin. Invest. 1994;93:1087–1094. doi: 10.1172/JCI117059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARTIN J.L., MESTRIL R., HILAL-DANDAN R., BRUNTON L.L., DILLMANN W.H. Small heat shock proteins and protection against ischemic injury in cardiac myocytes. Circulation. 1997;96:4343–4348. doi: 10.1161/01.cir.96.12.4343. [DOI] [PubMed] [Google Scholar]
- MESTRIL R., GIORDANO F.J., CONDE A.G., DILLMANN W.H. Adenovirus-mediated gene transfer of a heat shock protein 70 (hsp 70i) protects against simulated ischemia. J. Mol. Cell. Cardiol. 1996;28:2351–2358. doi: 10.1006/jmcc.1996.0228. [DOI] [PubMed] [Google Scholar]
- MOORE W.M., WEBBER R.K., JERONE G.M., TJOENG F.S., MISKO T.P., CURRIE M.G. L-N6-(1-imminoethyl)lysine: a selective inhibitor of inducible nitric oxide synthase. J. Med. Chem. 1994;37:3886–3888. doi: 10.1021/jm00049a007. [DOI] [PubMed] [Google Scholar]
- PARRATT J.R., SZEKERES L. Delayed protection of the heart against ischaemia. Trends Pharmacol. Sci. 1995;16:351–355. doi: 10.1016/s0165-6147(00)89069-0. [DOI] [PubMed] [Google Scholar]
- PELL T.J., YELLON D.M., GOODWIN R.W., BAXTER G.F. Myocardial ischemic tolerance following heat stress is abolished by ATP-sensitive potassium channel blockade. Cardiovasc. Drugs Ther. 1997;11:679–686. doi: 10.1023/a:1007791009080. [DOI] [PubMed] [Google Scholar]
- PLUMIER J.C., ROSS B.M., CURRIE R.W., ANGELIDIS C.E., KAZLARIS H., KOLLIAS G., PAGOULATOS G.N. Transgenic mice expressing the human heat shock protein 70 have improved post-ischemic myocardial recovery. J. Clin. Invest. 1995;95:1854–1860. doi: 10.1172/JCI117865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QIU Y., RIZVI A., TANG X.L., MANCHIKALAPUDI S., TAKANO H., JADOON A.K., WU W.J., BOLLI R. Nitric oxide triggers late preconditioning against myocardial infarction in conscious rabbits. Am. J. Physiol. 1997;273:H2931–H2936. doi: 10.1152/ajpheart.1997.273.6.H2931. [DOI] [PubMed] [Google Scholar]
- RICHARD V., KAEFFER N., THUILLEZ C. Delayed protection of the ischemic heart - from pathophysiology to therapeutic applications. Fund. Clin. Pharmacol. 1996;10:409–415. doi: 10.1111/j.1472-8206.1996.tb00595.x. [DOI] [PubMed] [Google Scholar]
- SASAKI N., SATO T., OHLER A., O'ROURKE B., MARBAN E. Activation of mitochondrial ATP-dependent potassium channels by nitric oxide. Circulation. 2000;101:439–445. doi: 10.1161/01.cir.101.4.439. [DOI] [PubMed] [Google Scholar]
- SCHWARTZ D., MENDONCA M., SCHWARTZ I., XIA Y., SATRIANO J., WILSON C.B., BLANTZ R.C. Inhibition of constitutive nitric oxide synthase (NOS) by nitric oxide generated by inducible NOS after lipopolysaccharide administration provokes renal dysfunction in rats. J. Clin. Invest. 1997;100:439–448. doi: 10.1172/JCI119551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHINBO A., IIJIMA T. Potentiation by nitric oxide of the ATP-sensitive K+ current induced by K+ channel openers in guinea-pig ventricular cells. Br. J. Pharmacol. 1997;120:1568–1574. doi: 10.1038/sj.bjp.0701069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STAMBAUGH K., ELLIOTT G.T., JACOBSON K.A., LIANG B.T. Additive effects of late preconditioning produced by monophosphoryl lipid A and the early preconditioning mediated by adenosine receptors and KATP channel. Circulation. 1999;99:3300–3307. doi: 10.1161/01.cir.99.25.3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUZUKI K., SAWA Y., KANEDA Y., ICHIKAWA H., SHIRAKURA R., MATSUDA H. In vivo gene transfection with heat shock protein 70 enhances myocardial tolerance to ischemia-reperfusion injury in rat. J. Clin. Invest. 1997;99:1645–1650. doi: 10.1172/JCI119327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAKANO H., TANG X.L., QIU Y., GUO Y., FRENCH B.A., BOLLI R. Nitric oxide donors induce late preconditioning against myocardial stunning and infarction in conscious rabbits via an antioxidant-sensitive mechanism. Circ. Res. 1998;83:73–84. doi: 10.1161/01.res.83.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TOSAKI A., MAULIK N., ELLIOTT G.T., BLASIG I.E., ENGELMAN R.M., DAS D.K. Preconditioning of rat heart with monophosphoryl lipid A: A role for nitric oxide. J. Pharmacol. Exp. Ther. 1998;285:1274–1279. [PubMed] [Google Scholar]
- YELLON M.D., BAXTER G.F. A ‘second window of protection' or delayed preconditining phenomenon: future horizons for myocardial protection. J. Mol. Cell. Cardiol. 1995;27:1023–1034. doi: 10.1016/0022-2828(95)90071-3. [DOI] [PubMed] [Google Scholar]
- YELLON M.D., PASINI E., CARONONI A., MARBER M.S., LATCHMAN D.S., FERRARI R. The protective role of heat stress in the ischemic and reperfused rabbit myocardium. J. Mol. Cell. Cardiol. 1992;24:895–907. doi: 10.1016/0022-2828(92)91102-b. [DOI] [PubMed] [Google Scholar]