

Abstract
Thrombin is a potent mitogen for vascular smooth muscle cells (VSMC) and has been implicated its pathogenic role in vascular remodelling. However, the signalling pathways by which thrombin mediates its mitogenic response are not fully understood.
We have previously reported that thrombin activates p38 mitogen-activated protein kinase (p38 MAPK) by a tyrosine kinase-dependent mechanism, and that p38 MAPK has a role in thrombin-induced mitogenic response in rat VSMC.
In the present study, we examine the involvement of epidermal growth factor (EGF) receptor in thrombin-induced p38 MAPK activation. We found that thrombin induced EGF receptor tyrosine phosphorylation (transactivation) in A10 cells, a clonal VSMC cell line. A selective inhibitor of EGF receptor kinase (AG1478) inhibited the p38 MAPK activation in a dose-dependent manner, whereas it had no effect on the response to platelet-derived growth factor (PDGF). EGF receptor phosphorylation induced by thrombin was inhibited by BAPTA-AM and GF109203X, which suggest a requirement for intracellular Ca2+ increase and protein kinase C.
We next examined the effect of AG1478 on thrombin-induced DNA synthesis. AG1478 inhibited thrombin-induced DNA synthesis in a dose-dependent manner. In contrast, PDGF-induced DNA synthesis was not affected by AG1478.
In conclusion, these data suggest that the EGF receptor transactivation and subsequent p38 MAPK activation is required for thrombin-induced proliferation of VSMC.
Keywords: Thrombin, p38 mitogen-activated protein kinase, DNA synthesis, epidermal growth factor receptor, vascular smooth muscle cell
Introduction
Thrombin is a multifunctional serine protease and stimulation of cells with thrombin leads to protein phosphorylation, gene expression, contractility and proliferation in a variety of cells. These effects are mediated by the thrombin receptor, which has been shown to be as a member of the family of G protein-coupled receptors (GPCRs) (Zhong et al., 1992). In cultured vascular smooth muscle cells (VSMC) thrombin has been reported to act as a growth factor (McNamara et al., 1993). VSMC proliferation is a key step of arterial remodelling in disease of the vasculture, including atherosclerosis, hypertension and restenosis after PTCA (Ross, 1993). Under these pathological conditions, thrombin has been implicated in the regulation of VSMC proliferation (Sarembock et al., 1991; Gibbons & Dzau, 1994). Therefore, the signalling pathways leading to its mitogenic effects will be useful to understand the smooth muscle remodelling and dysfunction.
Mitogen-activated protein kinase (MAPK) cascades are important systems of signal transduction from the cell surface to the nucleus. MAPK has been implicated in the regulation of many cellular responses, such as proliferation, differentiation and apoptosis. In mammalian cells, MAPK is divided into at least three subfamilies: extracellular signal-regulated protein kinase (ERK), c-Jun N-terminal kinase (JNK), and p38 MAPK (Davis, 1994). The ERK cascade has been relatively well-elucidated and found to be activated by GPCRs and tyrosine-kinase receptors (Cobb & Goldsmith, 1995). Two other subfamilies of MAPK, JNK and p38 MAPK, were found to be activated by both stresses and pro-inflammatory cytokines (Kyriakis & Avruch, 1996). Recently, p38 MAPK can also be activated by GPCR agonists, such as carbachol, isoproterenol and angiotensin II, in several kinds of cells (Larsen et al., 1997; Moule et al., 1998; Ushio-Fukai et al., 1998). The mechanisms from GPCR to the activation of p38 MAPK are largely unknown.
In a previous study, we have shown that thrombin activates p38 MAPK by a tyrosine kinase-dependent mechanism and that SB203580, a selective p38 MAPK inhibitor, inhibits thrombin-induced DNA synthesis in VSMC, suggesting a possible role of thrombin in cell proliferation (Kanda et al., 2001). However, the identity of the tyrosine kinase and its physiological significance in the signalling pathway remained unclear.
In the present study, we investigate whether epidermal growth factor (EGF) receptor is involved in the thrombin-induced signalling cascade in rat A10 cells, a clonal VSMC cell line. Moreover, to understand the role of EGF receptor in thrombin-induced growth, we examine the effect of AG1478, a selective EGF receptor kinase inhibitor, on thrombin-induced DNA synthesis in the cells. We report here that thrombin induces EGF receptor transactivation and that AG1478 inhibits thrombin-induced DNA synthesis in VSMC, suggesting a possible role of EGF receptor in cell proliferation.
Methods
Materials
Thrombin and lysophosphatidic acid (LPA) were from Sigma. Protein G-sepharose was from Pharmacia. Pertussis toxin was from Funakoshi. p38 MAPK assay kit, anti phospho-ERK antibody, and anti JNK1 antibody were from New England Biolabs. Anti phospho-JNK antibody was from Promega. Platelet-derived growth factor (PDGF) was from Genzyme. EGF was from Upstate Biotechnology. [3H]-Thymidine and enhanced chemiluminescence reagents were from Amersham. Anti p38 MAPK polyclonal antibody and anti phosphotyrosine monoclonal antibody (PY20) were from Santa Cruz Biotechnology. 4-(3-Chloroanilino)-6,7-dimethoxyquinqzoline (AG1478) and 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetra(acetoxymethyl) ester (BAPTA-AM) were from Calbiochem. 2-[1-(3-Dimethylaminopropyl)-1H-indol-3-yl)-maleimide (GF109203X) was from RBI. All other reagents were of analytical grades and obtained from commercial sources.
Cell culture
A10 cells (rat thoracic aortic smooth muscle) were provided by American Type Cell Collection. Cells were cultured at 37°C in a humidified atmosphere of 5% CO2/95% air in 100 mm dishes. The growth medium was Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% foetal bovine serum (FBS), 100 u ml−1 of penicillin, and 100 μg ml−1 of streptomycin. Medium was changed twice a week and cell passages 18 – 24 were used for all experiments.
Cell lysis, immunoprecipitation and immunoblotting
A10 cells were serum-starved for 24 h in serum-free DMEM. The cells were stimulated with agonists for the indicated times at 37°C and lysed in a buffer containing (mM): Tris 50, 1% Triton X-100, NaCl 150, Na4P2O7 10, NaF 20, EDTA 2, PMSF 1, pH 7.4, Na3VO4 1, 10 μg ml−1 aprotinin and 10 μg ml−1 leupeptin. Following incubation on ice for 30 min, the lysed cells were centrifuged at 15,000×g for 20 min at 4°C to precipitate debris. The supernatant was collected and assayed for protein concentration with the Bio-Rad protein assay method. For immunoprecipitation, the supernatant was precleared with protein G sepharose beads and incubated with the appropriate antibody conjugated to sepharose beads overnight at 4°C. The samples were analysed on 12% SDS – PAGE, transferred electrophoretically to polyvinilidene difluoride membranes (15 V, 90 min). After blocking with 5% bovine serum albumin for 1 h at room temperature, membranes were reacted with specific antibodies overnight at 4°C. The blots were washed and then incubated with HRP-conjugated secondary antibodies (1 : 2000 dilution) for 1 h at room temperature. After washing, the signal was detected with chemiluminescence ECL detection kit. The bands were quantified using a densitometer.
p38 MAPK activity assay
p38 MAPK activity in immunoprecipitates was measured using the p38 MAPK assay kit according to the manufacture's instructions. Briefly, p38 MAPK was immunoprecipitated from cell lysates using 2 μg of anti-p38 MAPK antibody conjugated to sepharose beads overnight at 4°C. The immunoprecipitates were washed twice with a lysis buffer and twice with a kinase buffer (mM): (Tris 20, MgCl2 20, NaCl 20, Na3VO4 0.1, DTT 2, pH 7.4). The beads were then suspended in 50 μl of the kinase buffer containing 2 μg GST-ATF-2, 20 μM ATP at 30°C for 30 min. Reactions were stopped by adding 5×Laemmli sample buffer and heating for 5 min. Phosphorylation of ATF-2 was analysed by immunoblotting using phosphospecific ATF-2 antibody (1 : 2000 dilution).
ERK and JNK phosphorylation
ERK and JNK phosphorylation were determined using phospho-specific antibodies. ERK phosphorylation was analysed by immunoblotting using anti phospho-ERK antibody (1 : 2000 dilution), as previously described (Mizuno et al., 1999). JNK was immunoprecipitated using anti-JNK1 antibody as described above and JNK phosphorylation was analysed by immublotting using anti phospho-JNK antibody (1 : 5000 dilution).
DNA synthesis
The assay was performed by measuring the incorporation of [3H]-thymidine into acid-insoluble materials as previously described (Kanda et al., 1999). The cells were seeded in 24-well culture plates, allowed to grow to 70 – 80% confluence, and then growth-arrested by incubation in serum-free DMEM. Growth-arrested cells were exposed to growth factors at the indicated concentrations for 24 h. The cells were then pulse-labelled with 1 μCi ml−1 [3H]-thymidine for 2 h just before the end of the incubation period and were incubated at 4°C for 2 h in 10% trichloroacetic acid. Acid-insoluble material was extracted with 0.1 N NaOH, and incorporated radioactivity was measured by liquid scintillation spectroscopy. All experiments were performed in triplicate.
Statistics
Values are expressed as the arithmetic means±s.d. Statistical analysis of the data was performed by the use of one-way analysis of variance (ANOVA), followed by Scheffe test when F ratios were significant (P<0.05).
Results
Thrombin induced transactivation of EGF receptor in A10 cells
We have previously reported that thrombin activates p38 MAPK through a tyrosine kinase-dependent pathway. To examine the possible involvement of EGF receptor kinase in thrombin-induced signal transduction pathway in VSMC, we first study whether thrombin stimulates tyrosine phosphorylation of EGF receptor in A10 cells. Growth-arrested cells were treated with thrombin for various durations and EGF receptor was immunoprecipitated from the lysates and blotted with anti-phosphotyrosine antibody. Immunoprecipitation of equal amounts of protein were confirmed by immunoblotting with anti-EGF receptor antibodies. As shown in Figure 1, thrombin induced tyrosine phosphorylation of EGF receptor in A10 cells, reaching a rapid maximum effect at 2 min and then decline to almost basal levels at 10 min. This data suggests that thrombin induced transactivation of EGF receptor in A10 cells.
Figure 1.

EGF receptor tyrosine phosphorylation by thrombin in A10 cells. After A10 cells were serum-starved for 24 h, the cells were incubated with thrombin (1 u ml−1) for the indicated times. EGF receptor was immunoprecipitated from cell lysates and analysed by immunoblotting with either anti-phosphotyrosine (PY20) (top panel) or anti EGF antibody (polyclonal) (bottom panel). Averaged data quantified by densitometry, expressed as a fold stimulation in which the basal activity is defined as 1.0. Values represent means±s.d. from three separate experiments. *P<0.05 as compared with the respective control.
Inhibition of EGF receptor kinase blocks thrombin-induced p38 MAPK activation in A10 cells
We next examined the effects of a selective EGF receptor kinase inhibitor, AG1478 (Levitzki & Gazit, 1995), on p38 MAPK activity. As shown in Figure 2A, AG1478 blocked thrombin-induced p38 MAPK activation in a dose-dependent manner, whereas it had no effect on the PDGF-induced p38 MAPK activation. In addition, AG1478 inhibited EGF-induced EGF receptor phosphorylation and p38 MAPK activation (Figure 2B,C). These data suggest that thrombin-induced p38 MAPK activation is mediated through EGF receptor transactivation. To examine whether EGFR transactivation is specific for p38 MAPK, we tested the effects of AG1478 on ERK and JNK, which is another subgroup of the MAPK family. As shown in Figure 3, AG1478 inhibited thrombin-induced both ERK and JNK1 phosphorylation. This data suggests that thrombin-induced ERK and JNK1 activation is mediated through EGF receptor-dependent mechanism. Collectively, these data suggest that the transactivation of EGF receptor plays an important role in thrombin-induced MAPK cascade activation in A10 cells.
Figure 2.

Effects of EGF receptor kinase inhibitor (AG1478) on thrombin-induced p38 MAPK activation in A10 cells. A10 cells were serum-starved for 24 h. (A) the cells were pretreated with indicated concentrations of AG1478 for 30 min and incubated with thrombin (1 u ml−1) or PDGF (1 ng ml−1) for 10 min. (B) The cells were incubated with EGF (100 ng ml−1) for 5 min in the presence or absence of AG1478 (2.5 μM). p38 MAPK activity was measured by immunoprecipitation kinase assay using GST-ATF-2 as a substrate. Phosphorylation of ATF-2 was detected by immunoblotting with the phospho-specific ATF-2 antibodies. (C) The cells were incubated with EGF (100 ng ml−1) for 5 min in the presence or absence of AG1478 (2.5 μM). EGF receptor phosphorylation was analysed as described in the legend of Figure 1. Averaged data quantified by densitometry, expressed as a fold stimulation in which the basal activity is defined as 1.0. Values represent means±s.d. from three separate experiments. *P<0.05 as compared with the respective control.
Figure 3.

Effects of AG1478 on thrombin-induced ERK and JNK phosphorylation in A10 cells. After A10 cells were starved for 24 h, the cells were stimulated with thrombin (1 u ml−1) for 5 min in the presence or absence of AG1478 (2.5 μM). ERK phosphorylation was detected by immunoblotting using anti phospho-specific ERK antibody. JNK was immunoprecipitated from cell lysates with anti JNK1 antibody and JNK phosphorylation was detected by immunoblotting with anti phospho-specific JNK antibody. Averaged data quantified by densitometry, expressed as a fold stimulation in which the basal activity is defined as 1.0. Values represent means±s.d. from three separate experiments. *P<0.05 as compared with the respective control.
EGF receptor phosphorylation by thrombin is mediated via PTX-insensitive G proteins
We next examined the signalling pathways from the thrombin receptor to EGF receptor transactivation. Several G protein-coupled receptor agonists, such as α2A-adrenergic agonist and lysophosphatidic acid (LPA) activate tyrosine kinase and MAP kinase through pertussis toxin (PTX)-sensitive G proteins (DellaRocca et al., 1997). Therefore, we studied whether PTX-insensitive G proteins are involved in EGF receptor transactivation by thrombin. As shown in Figure 4, pretreatment of cells with PTX (100 ng ml−1) for 24 h did not inhibit thrombin-induced EGF receptor tyrosine phosphorylation. In contrast, PTX inhibited LPA-induced EGF receptor tyrosine phosphorylation. These results suggest that PTX-sensitive G proteins do not mediate thrombin-induced EGF receptor transactivation.
Figure 4.
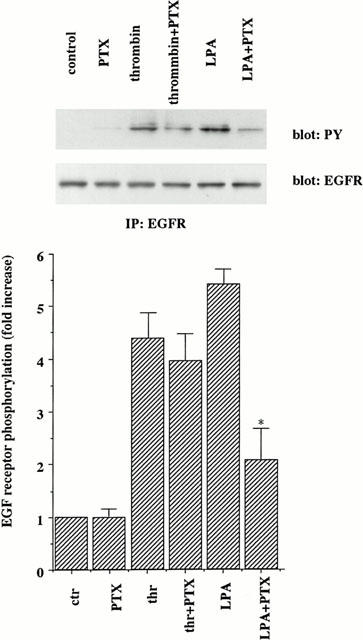
Effects of a pertussis toxin on thrombin-induced EGF receptor tyrosine phosphorylation in A10 cells. A10 cells were starved and incubated with or without pertussis toxin (100 ng ml−1) for 24 h. After preincubation, the cells were stimulated with thrombin (1 u ml−1) or LPA (20 μM) for 10 min. Phosphorylation of EGF receptor was measured as described in the legend of Figure 1. The results shown are representative of three separate experiments. Averaged data quantified by densitometry, expressed as a fold stimulation in which the basal activity is defined as 1.0. Values represent means±s.d. from three separate experiments. *P<0.05 as compared with the respective control.
EGF receptor phosphorylation by thrombin is dependent on protein kinase C and intracellular Ca2+
It has been shown that thrombin activates phospholipase Cβ through Gq in VSMC, to generate inositol 1,4,5-triphosphate and diacylglycerol, which mobilizes intracellular Ca2+ and activates protein kinase C (PKC), respectively. To examine the possible role of PKC and Ca2+ in thrombin-mediated signalling pathways leading to EGF receptor transactivation, we studied the effects of a selective PKC inhibitor, GF109203X (Toullec et al., 1991), an intracellular Ca2+ chelator, BAPTA-AM, and a extracellular Ca2+ chelator, EGTA. As shown in Figure 5, EGF receptor phosphorylation was inhibited by GF109203X and BAPTA-AM, not by EGTA. The inhibitor alone did not affect basal phosphorylation. These results suggest that PKC and intracellular Ca2+ are involved in part in thrombin-induced EGF receptor transactivation.
Figure 5.
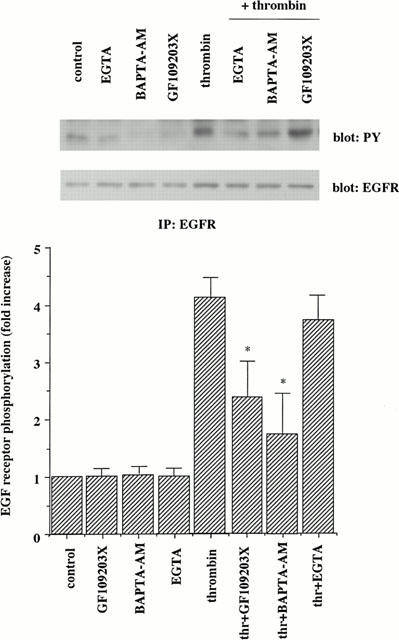
Effects of various inhibitors on thrombin-induced EGF receptor tyrosine phosphorylation in A10 cells. Growth-arrested A10 cells were incubated in the absence or presence of a PKC inhibitor GF109203X (1 μM), an intracellular Ca2+ chelator BAPTA-AM (10 μM), and an extracellular Ca2+ chelator EGTA (1 mM). After preincubation for 30 min, the cells were stimulated with thrombin (1 u ml−1). Phosphorylation of EGF receptor was analysed as described in the legend of Figure 1. The results shown are representative of three separate experiments. Averaged data quantified by densitometry, expressed as a fold stimulation in which the basal activity is defined as 1.0. Values represent means±s.d. from three separate experiments. *P<0.05 as compared with the respective control.
A EGF receptor kinase inhibitor, AG1478, inhibits thrombin-induced DNA synthesis
To further investigate the physiological role of EGF receptor phosphorylation in thrombin-mediated signalling, we examined the effects of AG1478 on thrombin-induced DNA synthesis. As shown in Figure 6, AG1478 inhibited thrombin-induced DNA synthesis in a dose-dependent manner with 61 and 82% inhibition at a 0.25 and 2.5 μM concentration, respectively, whereas it had no effect on basal levels. Equivalent doses of AG1478 did not inhibit PDGF-induced DNA synthesis, providing support for the specificity of thrombin stimulation and lack of cytotoxicity of this compound. These results suggest a possible role of EGF receptor phosphorylation in the proliferative response of A10 cells.
Figure 6.

AG1478 inhibits thrombin-induced DNA synthesis in A10 cells. Growth-arrested A10 cells were starved for 24 h. The cells were then stimulated with thrombin (1 u ml−1, hatched column) or PDGF (100 ng ml−1, closed column) for 24 h in the presence or absence of AG1478. DNA synthesis was measured by [3H]-thymidine incorporation into trichloroacetic acid-precipitate material, as described in Methods. Values represent means±s.d. from three separate experiments performed in triplicate. *P<0.05 as compared with the respective control.
Discussion
We previously reported that thrombin-induced p38 MAPK activation is mediated by a tyrosine kinase and plays a role in mitogenic signalling in A10 cells. In the present study, we show that a selective inhibitor of EGF receptor kinase, AG1478, inhibited thrombin-induced p38 MAPK activation, whereas it had no effect on PDGF-induced activation. Furthermore, AG1478 inhibited thrombin-induced DNA synthesis, suggesting that EGF receptor transactivation and subsequent p38 MAPK activation have an important role of thrombin-mediated mitogenic signalling in VSMC.
p38 MAPK has been found to be involved in inflammation and stresses. Recently, it has been reported that p38 MAPK can be activated by GPCR agonists. However, the signalling pathway from GPCR to p38 MAPK is not fully understood. Since it has been reported that the activation of GPCR requires tyrosine kinase activity (Wan et al., 1996), we focus on tyrosine kinase in GPCR-mediated p38 MAPK activation. We found that EGF receptor kinase is required for thrombin-induced p38 MAPK activation (Figures 1 and 2). Furthermore, the involvement of EGF receptor transactivation was also observed in the activation of ERK and JNK (Figure 3). This observation is supported by the recent findings that several GPCR agonists induce a transactivation of EGF receptor and subsequent ERK activation (Daub et al., 1996; Li et al., 1998). Since p38 MAPK activation by thrombin is inhibited by dominant-negative Ras in A10 cells (Kanda et al., 2001) and Ras has been shown to play a role in EGF-stimulated activation of ERK and JNK (Minden et al., 1994), Ras-mediated signalling pathway might be required for EGF receptor transactivation. c-Src is another tyrosine kinase that has been implicated in the GPCR signalling. It has been recently reported that c-Src is required for p38 MAPK activation mediated by Gq/11 (Nagao et al., 1998). Moreover, in A10 cells, c-Src is activated by thrombin (unpublished data). It is possible that c-Src and EGF receptor cooperatively regulate the p38 MAPK pathway.
Thrombin is coupled to Gi, Gq, and G12 subclasses of heterotrimeric G proteins (LaMorte et al., 1993; Offermanns et al., 1994). We show that thrombin-induced EGF receptor transactivation is mediated by a pertussis toxin-insensitive G protein, Gq or G12 (Figure 4). It is unclear which G proteins are required for thrombin to activate EGF receptor. Recent studies have shown that the stimulation of Gq by angiotensin II potently causes EGF receptor transactivation in VSMC (Eguchi et al., 1998). In addition, G13, but not G12, has recently been shown to mediate signalling from LPA receptor via EGF receptor to rho (Gohla et al., 1998). Future studies will be needed to identify the subclass of G protein.
The signal transduction pathways from the GPCR to EGF receptor transactivation are poorly understood. Thrombin-induced EGF receptor activation is partially inhibited by a PKC inhibitor (Figure 5). This result suggests that EGF receptor transactivation is mediated by both PKC-dependent and -independent pathways. The role of PKC in EGF receptor transactivation is controversial. Previous studies showed that angiotensin II-induced transactivation is suppressed by inhibitors of PKC in VSMC (Li et al., 1998) and bradykinin-induced transactivation is independent of PKC in COS-7 cells (Adomeit et al., 1999). The discrepancy might be explained by the difference of PKC isoform or cell types. We also found that a Ca2+ chelator, BAPTA-AM, inhibited the thrombin-induced EGF receptor phosphorylation (Figure 4). The role of calcium is further supported in that EGF receptor transactivation by a GPCR agonist, angiotensin II, is a calcium-dependent pathway (Eguchi et al., 1998). Furthermore, a non-receptor tyrosine kinase PYK2 was reported to act as an upstream mediator of the p38 MAPK pathway in response to certain cytotoxic agents (Pandey et al., 1999). Therefore, it remains to be determined whether PYK2 is involved in EGF receptor transactivation and subsequent p38 MAPK pathway.
To further assess the physiological role of EGF receptor in thrombin-mediated signalling, we analysed the DNA synthesis. AG1478 has been used to evaluate the role of the EGF receptor kinase. AG1478 has been shown to be highly selective for EGF receptor kinase and it is unlikely that AG1478 inhibits other kinases (Levitzki & Gazit, 1995). The doses of AG1478 are sufficient to block the EGF receptor kinase (Levitzki & Gazit, 1995) and incomplete to inhibit thrombin-induced DNA synthesis, suggesting that thrombin has an additional mechanism other than EGF receptor transactivation pathway. The alternative pathway may involve p70 S6 kinase. A previous study with bovine tracheal smooth muscle cells showed that thrombin-induced proliferation is dependent on phosphatidylinositol-3 kinase-mediated p70 S6 kinase activation (Scott et al., 1996). Further studies are required for the contribution of the pathway leading to the mitogenic response to thrombin.
In summary, we have shown that EGF receptor transactivation and subsequent p38 MAPK activation plays a role in mediating mitogenic responses in VSMC. Since thrombin has been suggested to play a role in VSMC proliferation leading to atherosclerosis, p38 MAPK may be one of the mediators for the pathogenesis of atherosclerosis.
Acknowledgments
This work was supported in part by a grant from the Smoking Research Foundation to Y. Watanabe.
Abbreviations
- DMEM
Dulbecco's modified Eagle's medium
- EGF
epidermal growth factor
- ERK
extracellular signal-regulated kinase
- GPCR
G protein-coupled receptor
- JNK
c-Jun N-terminal kinase
- LPA
lysophosphatidic acid
- MAPK
mitogen-activated protein kinase
- PDGF
platelet-derived growth factor
- PKC
protein kinase C
- PTX
pertussis toxin
- VSMC
vascular smooth muscle cells
References
- ADOMEIT A., GRANESS A., GROSS S., SEEDORF K., WETZKER R., LIEBMANN C. Bradykinin B2 receptor-mediated mitogen-activated protein kinase activation in COS-7 cells requires dual signalling via both protein kinase C pathway and epidermal growth factor receptor transactivation. Mol. Cell. Biol. 1999;9:5289–5297. doi: 10.1128/mcb.19.8.5289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COBB M.H., GOLDSMITH E.J. How MAP kinases are regulated. J. Biol. Chem. 1995;270:14843–14846. doi: 10.1074/jbc.270.25.14843. [DOI] [PubMed] [Google Scholar]
- DAVIS R.J. MAPKs: new JNK expands the group. Trends Biochem. Sci. 1994;19:470–473. doi: 10.1016/0968-0004(94)90132-5. [DOI] [PubMed] [Google Scholar]
- DELLAROCCA G.J., VANBIESEN T., DAAKA Y., LUTTRELL D.K., LUTTRELL L.M., LEFKOWITZ R.J. Ras-dependent mitogen-activated protein kinase activation by G protein-coupled receptors. J. Biol. Chem. 1997;272:19125–19132. doi: 10.1074/jbc.272.31.19125. [DOI] [PubMed] [Google Scholar]
- DAUB H., WEISS F.U., WALLASCH C., ULLRICH A. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature. 1996;379:557–560. doi: 10.1038/379557a0. [DOI] [PubMed] [Google Scholar]
- EGUCHI S., NUMAGUCHI K., IWASAKI H., MATSUMOTO T., YAMAKAWA T., UTSUNOMIYA H., MOTLEY E.D., KAWAKATSU H., OWADA K.M., HIRATA Y., MARUMO F., INAGAMI T. Calcium-dependent epidermal growth factor receptor transactivation mediates the angiotensin II-induced mitogen-activated protein kinase activation in vascular smooth muscle cells. J. Biol. Chem. 1998;273:8890–8896. doi: 10.1074/jbc.273.15.8890. [DOI] [PubMed] [Google Scholar]
- GIBBONS G.H., DZAU V.J. The emerging concept of vascular remodeling. N. Engl. J. Med. 1994;330:1431–1438. doi: 10.1056/NEJM199405193302008. [DOI] [PubMed] [Google Scholar]
- GOHLA A., HARHAMMER R., SCHULTZ G. The G-protein G13 but not G12 mediates signalling from lysophosphatidic acid receptor via epidermal growth factor receptor to rho. J. Biol. Chem. 1998;273:4653–4659. doi: 10.1074/jbc.273.8.4653. [DOI] [PubMed] [Google Scholar]
- KANDA Y., NISHIO E., WATANABE Y. Differential regulation of Na+/H+ exchange and DNA synthesis in vascular smooth muscle cells. Eur. J. Pharmcol. 1999;371:69–74. doi: 10.1016/s0014-2999(99)00162-4. [DOI] [PubMed] [Google Scholar]
- KANDA Y., NISHIO E., KUROKI Y., MIZUNO K., WATANABE Y.Thrombin activates p38 mitogen-activated protein kinase in vascular smooth muscle cells Life Sci. 2001(in press) [DOI] [PubMed]
- KYRIAKIS J.M., AVRUCH J. Sounding the alarm: protein kinase cascades activated by stress and inflammation. J. Biol. Chem. 1996;271:24313–24316. doi: 10.1074/jbc.271.40.24313. [DOI] [PubMed] [Google Scholar]
- LAMORTE V.J., HAROOTUNIAN A.T., SPIEGEL A.M., TSIEN R.Y., FERAMISCO J.R. Mediation of growth factor induced DNA synthesis and calcium mobilization by Gq and Gi2. J. Cell. Biol. 1993;121:91–99. doi: 10.1083/jcb.121.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LARSEN J.K., YAMBOLIEV I.A., WEBER L.A., GERTHOFFER W.T. Phosphorylation of the 27-kDa heat shock protein via p38 MAP kinase and MAPKAP kinase in smooth muscle. Am. J. Physiol. 1997;273:930–940. doi: 10.1152/ajplung.1997.273.5.L930. [DOI] [PubMed] [Google Scholar]
- LEVITZKI A., GAZIT A. Tyrosine kinase inhibition: an approach to drug development. Science. 1995;267:1782–1788. doi: 10.1126/science.7892601. [DOI] [PubMed] [Google Scholar]
- LI X., LEE J.W., GRAVES M., EARP H.S. Angiotensin II stimulates ERK via two pathways in epithelial cells: protein kinase C suppresses a G-protein coupled receptor-EGF receptor transactivation pathway. EMBO J. 1998;17:2574–2583. doi: 10.1093/emboj/17.9.2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCNAMARA C.A., SAREMBOCK I.J., GIMPLE L.W., FENTON J.W., II, COUGHLIN S.R., OWENS G.K. Thrombin stimulates proliferation of cultured rat aortic smooth muscle cells by a proteolytically activated receptor. J. Clin. Invest. 1993;91:94–98. doi: 10.1172/JCI116206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MINDEN A., LIN A., MACMAHON M., LANGE-CARTER C., DERIJARD B., DAVIS R.J., JOHNSON G.L., KARIN M. Differential activation of ERK and JNK mitogen-activated protein kinases by Raf-1 and MEKK. Science. 1994;266:1719–1723. doi: 10.1126/science.7992057. [DOI] [PubMed] [Google Scholar]
- MIZUNO K., KANDA Y., KUROKI Y., TOMIYAMA K., WATANABE Y. Phosphorylation of extracellular signal-regulated kinases 1 and 2 in 3T3-L1 adipocytes by stimulation of β3-adrenoceptor. Eur. J. Pharmacol. 1999;385:63–69. doi: 10.1016/s0014-2999(99)00733-5. [DOI] [PubMed] [Google Scholar]
- MOULE S.K., DENTON R.M. The activation of p38 MAPK by the beta-adrenergic agonist isoproterenol in rat epididymal fat cells. FEBS Lett. 1998;439:287–290. doi: 10.1016/s0014-5793(98)01392-1. [DOI] [PubMed] [Google Scholar]
- NAGAO M., YAMAUCHI J., KAZIRO Y., ITOH H. Involvement of protein kinase C and src family tyrosine kinase in Gαq/11-induced activation of c-Jun N-terminal kinase and p38 mitogen-activated protein kinase. J. Biol. Chem. 1998;273:22892–22898. doi: 10.1074/jbc.273.36.22892. [DOI] [PubMed] [Google Scholar]
- OFFERMANNS S., LAUGWITZ K., SPICHER K.L., SCHULTZ G. G protein of the G12 family are activated via thromboxane A2 and thrombin receptors in human platelets. Proc. Natl. Acad. Sci. U.S.A. 1994;91:504–508. doi: 10.1073/pnas.91.2.504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PANDEY P., AVRAHAM S., KUMAR S., NAKAZAWA A., PLACE A., GHANEM L., RANA A., KUMAR V., MAJUMDER P.K., AVRAHAM H., DAVIS R.J., KHARBANDA S. Activation of p38 mitogen-activated protein kinase by PYK2/related adhesion focal tyrosine kinase-dependent mechanism. J. Biol. Chem. 1999;274:10140–10144. doi: 10.1074/jbc.274.15.10140. [DOI] [PubMed] [Google Scholar]
- ROSS R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- SAREMBOCK I.J., GERTZ S.D., GIMPLE L.W., OWEN R.M., POWERS E.R., ROBERTS W.C. Effectiveness of recombinant desulphatohirudin in reducing restenosis after balloon angioplasty of atherosclerotic femoral arteries in rabbits. Circulation. 1991;84:232–243. doi: 10.1161/01.cir.84.1.232. [DOI] [PubMed] [Google Scholar]
- SCOTT P.H., BELHAM C.M., AL-HAFIDH J., CHILVERS E.R., PEACOCK A.J., GOULD G.W., PLEVIN R. A regulatory role for cAMP in phosphatidylinositol 3-kinase/p70 ribosomal S6 kinase-mediated DNA synthesis in platelet-derived-growth-factor-stimulated bovine airway smooth-muscle cells. Biochem J. 1996;318:965–971. doi: 10.1042/bj3180965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TOULLEC D., PIANETTI P., COSTE H., BELLEVERGUE P., GRAND-PERRET T., AJAKANE M., BAUDET V., BOISSIN P., BOURSIER E., LORIOLLE F., DUHAMEL L., VHARON D., KIRILOYSKY J. The bisindolylmaleimide GF 109023X is a potent and selective inhibitor of protein kinase C. J. Biol. Chem. 1991;266:15771–15781. [PubMed] [Google Scholar]
- USHIO-FUKAI M., ALEXANDER R.W., AKERS M., GRIENDLING K.K. p38 Mitogen-activated protein kinase is a critical component of the redox-sensitive signalling pathways activated by angiotensin II. J. Biol. Chem. 1998;273:15022–15029. doi: 10.1074/jbc.273.24.15022. [DOI] [PubMed] [Google Scholar]
- WAN Y., KUROSAKI T., HUANG X.Y. Tyrosine kinases in activation of the MAP kinase cascade by G-protein-coupled receptors. Nature. 1996;380:541–544. doi: 10.1038/380541a0. [DOI] [PubMed] [Google Scholar]
- ZHONG C., HAYZEN D.J., CORSON M.A., RUNGE M.S. Molecular cloning of the rat vascular smooth muscle thrombin receptor. J. Biol. Chem. 1992;267:16975–16979. [PubMed] [Google Scholar]