

Abstract
The present study examined the effect of a range of doses of chronic nicotine (0.75, 1.5, 3.0 and 30.0 mg kg−1 day−1, s.c., 14 days) upon striatal dopaminergic nerve terminal survival following 6-hydroxydopamine (6-OHDA; 10 μg intrastriatal unilaterally) in rats; and the effects of acute nicotine (1 mg kg−1, s.c.) pretreatment upon striatal neurodegeneration induced by methamphetamine (5 mg kg−1, i.p., three doses at 2 h intervals) in wild-type and α4 nicotinic receptor (nAChR) subunit knockout mice.
In both models of Parkinsonian-like damage, loss of striatal dopaminergic nerve terminals was assessed by [3H]-mazindol autoradiography.
In rats, chronic nicotine infusion delivered by osmotic minipump implanted subcutaneously 7 days prior to intrastriatal 6-OHDA injection produced significant and dose-related protection against 6-OHDA-induced neurodegeneration. Low (0.75 and 1.5 mg kg−1 day−1) but not high (3.0 and 30.0 mg kg−1 day−1) nicotine doses significantly inhibited 6-OHDA-induced degeneration.
In wild-type mice, acute nicotine treatment produced significant inhibition of methamphetamine-induced neurodegeneration. In α4 nAChR subunit knockout mice, acute nicotine treatment failed to inhibit methamphetamine-induced neurodegeneration.
Nicotine is capable of protecting dopaminergic neurons against Parkinsonian-like neurodegeneration in vivo. In rats, this neuroprotective effect is critically dependent upon nicotine dose and is consistent with the activation of nAChRs, as high, desensitizing doses of nicotine fail to be neuroprotective. Further, neuroprotection is absent in α4 nAChR subunit knockout mice. The current results therefore suggest that activation of α4 subunit containing nAChRs constitutes a major component of the neuroprotective effect of nicotine upon Parkinsonian-like damage in vivo.
Keywords: Nicotine, nicotinic receptor, Parkinson's disease, α4 nicotinic receptor subunit, neurodegeneration, knockout, 6-hydroxydopamine, methamphetamine
Introduction
Parkinson's disease (PD) is an age-related neurodegenerative disorder associated with the progressive and persistent degeneration of the dopaminergic nigrostriatal pathway (Levy et al., 1997; Olanow & Tatton, 1999). Recent epidemiological studies have documented an approximate halving of the incidence of PD in cigarette smokers relative to non-smokers (Baron, 1986; Morens et al., 1995). This neuroprotective effect is likely to be due to nicotine, as subsequent in vitro studies demonstrated that pretreatment with nicotine and other nicotinic receptor (nAChR) agonists dose-dependently attenuated dexamethasone-potentiated kainic acid neurotoxicity in primary hippocampal cultures (Semba et al., 1996), and glutamate receptor-mediated excitotoxicity in both primary cortical (Akaike et al., 1994; Kaneko et al., 1997) and striatal cultures (Donnelly-Roberts et al., 1996). Furthermore, the neuroprotective effects of nAChR agonists in vitro are attenuated by non-selective nicotinic antagonists such as mecamylamine, demonstrating that nAChR activation is a step essential to the neuroprotective process (Akaike et al., 1994; Donnelly-Roberts et al., 1996; Semba et al., 1996; Kaneko et al., 1997; Kihara et al., 1997).
Several in vivo studies that have investigated the neuroprotective effects of nicotine upon nigrostriatal degeneration in animal models of Parkinsonism have reported equivocal results. Chronic nicotine infusion at a dose rate of 3.0 mg kg−1 day−1, in combination with acute nicotine injections, significantly inhibited the dopaminergic neuron loss that follows 1-methyl-4-phenyl-2,3,6-tetrahydropyridine (MPTP)-induced lesions of the nigrostriatal system (Janson et al., 1988b); as well as the more progressive loss of nigrostriatal neurons that follows partial mesodiencephalic hemitransection (Janson et al., 1988a; 1991; 1994; Fuxe et al., 1990; Janson & Moller, 1993). Chronic nicotine infusion however failed to attenuate 6-hydroxydopamine (6-OHDA)-induced nigrostriatal lesions (Blum et al., 1996), and actually enhanced MPTP-induced damage when given chronically either intermittently or via infusion (Janson et al., 1992; Hadjiconstantinou et al., 1994; Ferger et al., 1998).
The nAChR is a ligand-gated ion channel composed of five subunits assembled around a central cationic pore. Several nAChR subunits have been identified to date (α2 – α7 and β2 – β4), and while these subunits show a wide and heterogeneous distribution within the mammalian brain (for review, see Wonnacott, 1997; Changeux et al., 1998), two major subtypes appear to predominate. The heteromeric α4/β2 receptor accounts for approximately 90% of high-affinity [3H]-nicotine binding sites centrally, whereas the homomeric α7 receptor forms a high-affinity binding site for [125I]-α-bungarotoxin (for review, see Clarke, 1998). The nigrostriatal system however, which is specifically damaged in PD, expresses a diverse array of nAChR subunits (Elliott et al., 1998) including, but not restricted to, α4/β2 and α7 nAChR subtypes. Further, nicotinic modulation of striatal dopamine release is consistent with a heterogeneous nAChR population on dopaminergic nigrostriatal neurons, although the nAChR subtypes present have not been identified with certainty (see Reuben et al., 2000; Sharples et al., 2000).
Attempts to identify the nAChR mediating neuroprotection in vitro by blockade with selective nAChR antagonists has implicated both major nAChR subtypes: both dihydro-β-erythroidine and α-bungarotoxin, antagonists selective for α4/β2 and α7 nAChRs respectively, block the neuroprotective effects of nicotine in cortical cultures (Donnelly-Roberts et al., 1996; Kaneko et al., 1997; Kihara et al., 1997). The identity of the nAChR subtype(s) mediating neuroprotection in vivo is unknown.
Given the epidemiological data suggesting that nicotine may play an important role in the progression of human PD, coupled with the equivocal data obtained from previous in vitro and in vivo studies, the aims of the current study were 2 fold. Firstly, we investigated the effect of a range of nicotine doses, chronically administered, upon nigrostriatal system survival against 6-OHDA-induced damage in the rat. Secondly, the current study sought to identify the potential nAChR subtype(s) involved in neuroprotection in vivo by examining the effect of deletion of the α4 nAChR subunit gene against methamphetamine-induced damage in the mouse. The results of the current study are consistent with activation of α4 subunit-containing nAChRs as a necessary and major component of the neuroprotective effect of nicotine upon Parkinsonian-like damage in vivo.
Methods
Chronic nicotine treatment experiments
Animals and drug treatment
Female Sprague-Dawley rats (180 – 220 g) were housed under controlled conditions with free access to food and water. All procedures were conducted in accordance with guidelines set out by the Monash University Animal Ethics Committee.
Alzet osmotic minipumps were pre-weighed, filled with saline or (−)-nicotine di-d-tartrate in saline and re-weighed to check for complete filling prior to implantation. Solutions were prepared so as to provide final delivery via minipump of 0, 0.75, 1.5, 3.0 or 30.0 mg kg−1 day−1 nicotine in saline for a total treatment period of 14 days. Rats were anaesthetized (1 midazolam, 10 mg kg−1 : 1 fentanyl, 1.0 mg kg−1 and fluanisone, 20 mg kg−1 : 1 sterile water) (i.p.), and an incision made in the back of the neck. A hollow was made beneath the skin in the interscapular space, and the minipump inserted into this hollow. The incision was then closed and the animal allowed to recover.
Stereotaxic injection of 6-OHDA
Seven days after the implantation of osmotic minipumps, animals underwent stereotaxic surgery to produce lesions of the nigrostriatal system. Within each experimental group, half of the animals received an intrastriatal injection of 6-OHDA with 0.01% ascorbic acid in saline (lesioned animals) while the remaining animals received an intrastriatal injection of 0.01% ascorbic acid in saline (control animals). The surgical procedure for unilateral intrastriatal injection of 6-OHDA is essentially as described by Przedborski and co-workers (1995). Briefly, rats were anaesthetized with sodium pentobarbitone (60 mg kg−1, i.p.) and placed in a Kopf stereotaxic apparatus, where the head was constrained to a tilted skull position (−3.0 mm). An incision was made on the midline of the scalp and a burr hole drilled through the skull at the appropriate coordinates. Through this, an intracerebral injection was delivered into the left striatum using a 30 gauge blunt-tipped cannula. Stereotaxic coordinates for injection were: 0.3 mm anterior and 3.0 mm lateral from Bregma, and 5.2 mm ventral from the cortical surface, according to the atlas of Paxinos & Watson (1986). Lesioned rats received 4 μl of 2.5 μg μl−1 6-OHDA/0.01% (w v−1) ascorbic acid, while control rats received 4 μl of saline/0.01% (w v−1) ascorbic acid. Injections were delivered at a rate of 0.6 μl min−1 and the needle left in position for 10 min following injection before being withdrawn slowly. After sealing the skull, the incision was closed and the animals allowed to recover.
Acute nicotine treatment experiments
Preparation of α4 subunit knockout mice and drug treatments
Knockout mice lacking the α4 nAChR subunit were generated by homologous recombination following partial deletion of the coding sequence contained in exon V; full genotypic and phenotypic characterization was carried out, as described by Ross et al., (2000).
All procedures were carried out in accordance with guidelines approved by the Monash University Animal Ethics Committee. Mice were housed under controlled conditions with free access to food and water. Knockout mice and wild-type littermates (20 – 30 g) were treated according to the methods of Maggio et al. (1997; 1998). Briefly, mice received a series of three injections of methamphetamine (5 mg kg−1, i.p.) at 2 h intervals. Nicotine treated mice received an injection of nicotine (1 mg kg−1, s.c.) 30 min prior to each injection of methamphetamine. Control mice received no treatment.
Preparation of tissues
Seven days after stereotaxic surgery or acute drug treatment, rats and mice respectively were lightly anaesthetized (CO2/O2: 80/20) and decapitated. Brains were rapidly removed and frozen over liquid nitrogen, then stored at −40°C prior to sectioning.
A Reichert Jung cryostat was used to cut consecutive coronal 14 μm sections of striatum at level +0.30 mm from Bregma according to the atlas of Paxinos & Watson (1986). Sections were thaw mounted onto poly-L-lysine coated slides, then stored at −20°C until use.
[3H]-mazindol autoradiography
[3H]-mazindol autoradiography was used to visualize dopaminergic nerve terminals within sections of striatum taken from both rat (chronic treatment) and mouse (acute treatment) brains. All autoradiographic steps were carried out at 4°C to reduce non-specific binding.
Slide mounted sections of striatum were preincubated for 15 min in 50 mM Tris-HCl solution (pH 7.9) containing 120 mM NaCl and 5 mM KCl. Sections were then incubated for 60 min with 4 nM [3H]-mazindol in 50 mM Tris-HCl solution (pH 7.9) containing 300 mM NaCl and 5 mM KCl. Desipramine (DMI; 300 nM) was included in all incubation solutions to prevent non-selective [3H]-mazindol binding at noradrenergic uptake sites. Nomifensine (100 μM), a selective inhibitor of dopamine uptake sites, was used to determine non-specific binding. Sections were washed twice (2×3 min) in ice-cold incubation buffer to remove excess [3H]-mazindol and dried under a stream of cold, dry air.
Once dry, radiolabelled sections were apposed to Hyperfilm-3H and exposed for 21 days to allow an image of striatal dopaminergic nerve terminal density to develop on the film. Following the exposure period, films were developed for 5 min in Phenisol X-ray developer, rinsed briefly in a weak solution of stopbath and fixed in Hypam X-ray fixer for 10 min.
Analysis of autoradiograms
Computer-assisted densitometry (MCID system, St. Catherine's, Ontario, Canada) was used to quantify the optical density of film images. The system was calibrated using [3H]-standards, so that optical density measurements were made in nCi mm−2. Specific binding was determined by subtracting the non-specific binding image from that of total binding, and was measured in the entire striatum.
Data analysis
The mean optical density and standard error of the mean were determined from independent measurements taken in at least three consecutive coronal sections of striatum for each animal. For chronic treatment studies, six to eighteen animals were contained within each treatment group; for acute experiments, three to eight animals were contained within each treatment group. One-way analysis of variance with a Dunnett's post-hoc test, was used for comparisons within treatment groups. In all cases, probability levels of P<0.05 were considered statistically significant.
Materials
Alzet osmotic minipumps (model 2002, Alza Corp., U.S.A.); 6-OHDA (RBI); (−)-nicotine di-d-tartrate (RBI); nomifensine maleate (RBI); desipramine (RBI); [3H]-mazindol (24 Ci mmol−1; DuPont); Phenisol X-ray developer and Hypam X-ray fixer (Ilford, Australia); Hyperfilm-3H and [3H]-standards (Amersham, Australia).
Results
Chronic treatment experiments
Effect of 6-OHDA lesion upon nigrostriatal dopaminergic nerve terminals
Intrastriatal 6-OHDA injection caused a marked and significant loss of dopaminergic nerve terminals, as assessed by specific [3H]-mazindol autoradiography, within the 6-OHDA-injected striatum relative to vehicle-injected controls. This loss accounted for approximately 65% of dopaminergic nerve terminals (see Figure 1). No significant loss of striatal dopaminergic terminals was detected following an intrastriatal injection of vehicle (data not shown). Although a loss of approximately 25% of [3H]-mazindol binding was detected contralateral to the 6-OHDA lesion (specific [3H]-mazindol binding=1.20±0.07 nCi mm−2) when compared with the contralateral striatum of vehicle-injected controls (specific [3H]-mazindol binding=0.91±0.12 nCi mm−2), this difference was not statistically significant (unpaired t-test, n.s.). Regardless, all comparisons in the neuroprotection studies are only made between ipsilateral injected striata.
Figure 1.

Levels of dopaminergic nerve terminals, measured as density of specific [3H]-mazindol binding (nCi mm−2) within the striatum of chronically treated rats. Each column represents the mean and s.e.mean of averaged triplicates or quadruplicates of the density of specific [3H]-mazindol binding measured in the injected striatum of 3 – 9 rats. Differences in dopaminergic nerve terminal survival were assessed in striata taken from animals treated chronically with saline and injected with vehicle (saline+vehicle), versus those taken from animals treated chronically with saline or nicotine 0.75, 1.5, 3.0 and 30.0 mg kg−1 day−1, and injected with 6-OHDA. One-way ANOVA with a Dunnett's post-hoc test for multiple comparisons with a control (saline+vehicle) was used to statistically assess differences in the survival of striatal dopaminergic nerve terminals, where P<0.05 was considered statistically significant. *(P<0.05), **(P<0.01).
Effect of chronic nicotine upon 6-OHDA-induced loss of dopaminergic nerve terminals
Chronic nicotine treatment caused a dose-related, neuroprotective effect upon 6-OHDA-induced loss of striatal dopaminergic nerve terminals, as assessed by [3H]-mazindol autoradiography (see Figure 1). In animals treated with either of the lower doses of nicotine (0.75 and 1.5 mg kg−1 day−1) and subsequently injected with 6-OHDA, striatal [3H]-mazindol binding levels were not significantly different from those measured in control animals treated with saline and subsequently injected with vehicle. For both of these low nicotine doses, striatal terminal survival was maintained at greater than 75% of control levels. In animals treated with either of the higher nicotine doses (3.0 and 30.0 mg kg−1 day−1) however, a significant loss of terminals was still associated with 6-OHDA-injected striata. For all treatment groups, there was no systematic effect of chronic nicotine treatment upon [3H]-mazindol binding levels, as measured in the contralateral striatum of animals injected intrastriatally with vehicle (specific [3H]-mazindol binding (nCi mm−2): saline=1.20±0.07; nicotine 0.75 mg kg−1 day−1=1.17±0.02; nicotine 1.5 mg kg−1 day−1=1.22±0.11; nicotine 3.0 mg kg−1 day−1=1.07±0.09; nicotine 30.0 mg kg−1 day−1=0.99±0.07; one-way ANOVA, n.s.).
Acute nicotine experiments
Effect of methamphetamine upon nigrostriatal dopaminergic nerve terminals
Methamphetamine treatment caused a marked and significant loss of dopaminergic nerve terminals, assessed by specific [3H]-mazindol autoradiography, within the striatum of both wild-type and knockout mice (see Figure 2). This loss of dopaminergic terminals accounted for approximately 50% of terminals in wild-type mice, and approximately 40% of terminals in knockout mice, relative to respective control animals.
Figure 2.
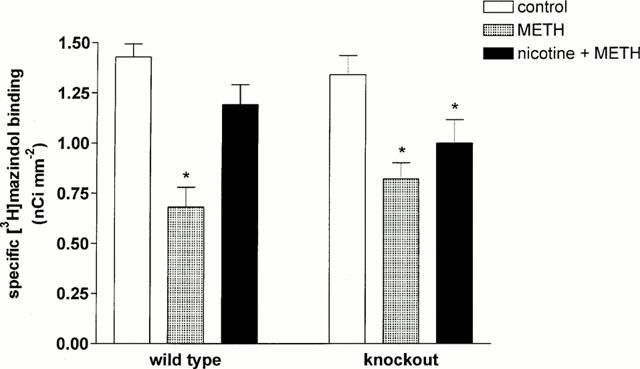
Levels of dopaminergic nerve terminals, measured as density of specific [3H]-mazindol binding (nCi mm−2) within the striatum of wild-type and α4 nAChR subunit knockout mice treated with methamphetamine (5 mg kg−1; three injections i.p.; METH) and nicotine (1 mg kg−1; s.c.) plus methamphetamine (nicotine+METH). Each column represents the mean and s.e.mean of averaged triplicates or quadruplicates of the density of specific [3H]-mazindol binding measured in striata taken from 3 – 8 mice. Differences in dopaminergic nerve terminal survival were assessed by one-way ANOVA with a Dunnett's post-hoc test for multiple comparisons with a control, in wild-type and knockout mice, respectively, where P<0.05 was considered statistically significant. * (P<0.05).
Effect of acute nicotine upon methamphetamine-induced loss of dopaminergic nerve terminals
Acute nicotine treatment significantly protected against methamphetamine-induced loss of striatal dopaminergic nerve terminals in wild-type mice but not in α4 nAChR subunit knockout mice (see Figure 2). In wild-type mice, nicotine treatment maintained dopaminergic nerve terminal levels at greater than 80% of control levels. Nicotine treatment, however, failed to significantly attenuate methamphetamine-induced loss of dopaminergic terminals in knockout mice. Although an increase in [3H]-mazindol binding levels (approximately 15% relative to control animals) was detected in striata of knockout mice treated with both nicotine and methamphetamine, this increase was not statistically significant, and levels remained significantly below those measured in control animals.
Discussion
Nicotine has been investigated previously as a neuroprotective agent both in vitro and in vivo. The current study demonstrates that both chronic and acute nicotine treatment protect against 6-OHDA and methamphetamine-induced neurodegeneration in vivo, in rats and mice, respectively. The neuroprotective effect of chronic nicotine in vivo was critically dependent upon the dose administered, where low (0.75 and 1.5 mg kg−1 day−1), but not high (3.0 and 30.0 mg kg−1 day−1) doses protected against 6-OHDA-induced nigrostriatal degeneration. These results are consistent with the possibility that it is the activation of nAChRs, rather than receptor desensitization, that mediates the neuroprotective effect of nicotine in vivo. An active role for nAChRs in neuroprotection is further supported by the loss of neuroprotection observed in α4 nAChR subunit knockout mice, demonstrating that nAChRs containing the α4 subunit contribute strongly to protection of dopaminergic neurons by nicotine in vivo.
Intrastriatally injected 6-OHDA is thought to damage nigrostriatal neurons by promoting the production of hydroxyl radicals: this rapidly destroys the nerve terminal (Gerlach & Riederer, 1996; Lotharius et al., 1999), and produces a gradual retrograde loss of cell bodies in the substantia nigra pars compacta (SNpc). Such damage mimics the progressive and incomplete nigrostriatal degeneration that characterizes early human PD (Berger et al., 1991; Sauer & Oertel, 1994; Przedborski et al., 1995; Lee et al., 1996). In comparison, excitatory amino acids have been directly implicated in the mechanism of methamphetamine-induced nigrostriatal toxicity (Sonsalla et al., 1989; 1991).
Both 6-OHDA and methamphetamine treatments produced significant nigrostriatal damage, where striatal dopaminergic nerve terminal survival was used as a measure of nigrostriatal damage, and was assessed by [3H]-mazindol autoradiography. This method has been shown previously to be a useful tool for quantifying striatal dopaminergic populations, where DMI-insensitive [3H]-mazindol binding sites represent dopamine uptake sites (Javitch et al., 1984; 1985). Further, the loss of [3H]-mazindol binding following intrastriatal 6-OHDA correlates well with cell body loss from the SNpc (Przedborski et al., 1995; Lee et al., 1996), and with the degree of striatal dopamine depletion following both 6-OHDA (Ryan & Loiacono, unpublished observations) and methamphetamine treatments (Maggio et al., 1997; 1998).
The effect of nicotine upon nigrostriatal neuron survival was investigated at four dose levels: 0.75, 1.5, 3.0 and 30.0 mg kg−1 day−1. Doses of 0.75, 1.5 and 3.0 mg kg−1 day−1 are likely to achieve brain nicotine concentrations of 0.2 to at least 0.5 μM (Rowell & Li, 1997). This range approximates the EC50 reported for nicotine-evoked dopamine release in striatal synaptosomes and slices (0.16 – 0.5 μM) (Grady et al., 1992; 1994; Whiteaker et al., 1995; Clarke & Reuben, 1996; Wonnacott, 1997), and falls within the range that is maximally protective against neurotoxic damage in vitro (approximately 0.1 – 5 μM) (Donnelly-Roberts et al., 1996; Semba et al., 1996). The dose of 3.0 mg kg−1 day−1 was of particular interest, as it is widely cited as a dose that reflects plasma nicotine concentrations in smokers, where smokers demonstrate a diminished incidence of PD (Baron, 1986; Morens et al., 1995).
The four nicotine doses used by the current study fall into two ranges; a low range (⩽1.5 mg kg−1 day−1) and a high range (⩾3.0 mg kg−1 day−1). Doses of approximately 2.4 mg kg−1 day−1 and higher have been shown to upregulate nAChRs centrally (Rowell & Li, 1997): this is more than likely to occur as a result of desensitization block (see Wonnacott, 1990). One recent functional study for example showed that while nicotine infused at a dose of 4.0 mg kg−1 day−1 desensitized nicotine-stimulated striatal dopamine release, a lower dose of 1.0 mg kg−1 day−1 showed no such desensitization of function (Benwell & Balfour, 1997). It should be noted however that an enhancement of function has also been associated with nAChR upregulation in vivo (see Marshall et al., 1997).
Evidence from in vitro studies strongly implicates nAChR activation as an essential step in neuroprotection, as neuroprotection is blocked by nAChR antagonists (Akaike et al., 1994; Donnelly-Roberts et al., 1996; Semba et al., 1996; Kaneko et al., 1997; Kihara et al., 1997). Data from the present study is consistent with the possibility that it is the activation of nAChRs, rather than desensitization, that underlies the neuroprotective effect of nicotine in vivo. While low nicotine doses (⩽1.5 mg kg−1 day−1) significantly protected striatal dopaminergic terminals, higher nicotine doses (⩾3.0 mg kg−1 day−1) failed to significantly attenuate 6-OHDA-induced damage. Consistent with the current results, a prior study that infused nicotine at a dose of 3.0 mg kg−1 day−1 failed to find any effect of this treatment upon 6-OHDA-induced neurodegeneration (Blum et al., 1996). The results of the current study are in contrast with those of several prior studies examining the effect of nicotine treatment upon Parkinsonian-like damage in vivo. Such studies have suggested that it is a desensitization of nAChRs on nigrostriatal neurons that underlies neuroprotection by nicotine, resulting in a decrease in the firing of nigrostriatal neurons (Grenhoff et al., 1991), and a decrease in nigrostriatal dopamine (Fuxe et al., 1990) and glucose (Owman et al., 1989) utilization. Importantly, these prior studies used nicotine at a dose rate of 3.0 mg kg−1 day−1: this dose was not significantly protective in the current study. As nAChR regulation and function in vivo are affected not only by nicotine dose but also the duration (acute versus chronic) and method (continuous versus intermittent) of treatment (see Marshall et al., 1997; Rowell & Li, 1997), these factors may potentially influence the neuroprotective capacity of nicotine in vivo.
We sought to resolve whether it is the activation or desensitization of nAChRs that underlies neuroprotection in vivo, by examining the neuroprotective capacity of nicotine in a nAChR knockout mouse model. We chose to examine the α4 nAChR subunit knockout mouse, as the α4/β2 nAChR is the most common within the mammalian brain. While acute nicotine attenuated methamphetamine-induced nigrostriatal damage in wild-type mice, no significant neuroprotection was detected in mice lacking the α4 subunit. A contribution from α4-containing nAChRs therefore appears to be essential if a significant degree of neuroprotection is to be achieved: this implies that the activation of nAChRs, and more specifically, α4-containing nAChRs, contribute significantly to the neuroprotective process in vivo. The current results do not rule out the involvement of additional nAChR subtypes in neuroprotection in vivo: although failing to reach statistical significance, a trend towards increased dopaminergic neuron survival following nicotine treatment was observed in α4 knockout mice. This effect may be due to the involvement of additional nAChR subtype(s) in vivo: the expression of multiple nAChR subunits and modulation of dopaminergic function by a heterogeneous nAChR population within nigrostriatal neurons (see Reuben et al., 2000; Sharples et al., 2000) adds strength to this possibility. Further, the wide distribution of nAChRs, particularly α4 subunit-containing nAChRs, implies that the effect of systemically administered nicotine need not be restricted to direct actions upon the nigrostriatal system, but may involve nAChRs at several sites within the brain.
Conclusion
The results of the current study are consistent with the view that nAChR activation mediates the neuroprotective effects of nicotine in vivo: firstly because low, potentially activating nicotine doses are neuroprotective whereas higher, potentially desensitizing nicotine doses fail to significantly protect nigrostriatal neurons; and secondly, because neuroprotection is abolished in α4 nAChR subunit knockout mice. Taken together, these results suggest that the activation of α4 subunit-containing nAChRs comprises a major component of the neuroprotective effect of nicotine upon Parkinsonian-like damage in vivo.
Acknowledgments
This research was supported by a grant from the Smoking and Health Research Foundation of Australia (R.E. Loiacono), and by a grant from the Australian National Health and Medical Research Council (J. Drago). R. Ryan is supported by an Australian Postgraduate Award. J. Drago is a Logan Research Fellow at Monash University. Part of the work contained in this manuscript was performed in the Department of Pharmacology at the University of Melbourne. The authors are grateful to Professor R.J. Summers for critical reading of the manuscript. This study is not intended to promote cigarette smoking, and has not been supported by any tobacco company.
Abbreviations
- DMI
desmethylimipramine
- MPTP
1-methyl-4-phenyl-2,3,6-tetrahydropyridine
- nAChR
neuronal nicotinic receptor
- PD
Parkinson's disease
- 6-OHDA
6-hydroxydopamine
- SNpc
substantia nigra pars compacta
References
- AKAIKE A., TAMURA Y., SHIMOHAMA S., KIMURA J. Nicotine-induced protection of cultured cortical neurons against N-methyl-D-aspartate receptor-mediated glutamate cytotoxicity. Brain Res. 1994;644:181–187. doi: 10.1016/0006-8993(94)91678-0. [DOI] [PubMed] [Google Scholar]
- BARON J.A. Cigarette smoking and Parkinson's Disease. Neurology. 1986;36:1490–1496. doi: 10.1212/wnl.36.11.1490. [DOI] [PubMed] [Google Scholar]
- BENWELL M.E.M., BALFOUR D.J.K. Regional variation in the effects of nicotine on catecholamine overflow in rat brain. Eur. J. Pharmacol. 1997;325:13–20. doi: 10.1016/s0014-2999(97)00101-5. [DOI] [PubMed] [Google Scholar]
- BERGER K., PRZEDBORSKI S., CADET J.L. Retrograde degeneration of nigrostriatal neurons induced by intrastriatal 6-hydroxydopamine injection in rats. Brain Res. 1991;26:301–307. doi: 10.1016/0361-9230(91)90242-c. [DOI] [PubMed] [Google Scholar]
- BLUM M., WU G., MUDO G., BELLUARDO N., ANDERSSON K., AGNATI L.F., FUXE K. Chronic continuous infusion of (−)nicotine reduces basic fibroblast growth factor messenger RNA levels in the ventral midbrain of the intact but not of the 6-hydroxydopamine-lesioned rat. Neurosci. 1996;70:169–177. doi: 10.1016/0306-4522(95)00364-o. [DOI] [PubMed] [Google Scholar]
- CHANGEUX J.P., BERTRAND D., CORRINGER J.P., DEHAENE S., EDELSTEIN S., LENA C., LE NOVERE N., MARUBIO L., PICIOTTO M., ZOLI M. Brain nicotinic receptors: structure and regulation, role in learning and reinforcement. Brain Res. Rev. 1998;26:198–216. doi: 10.1016/s0165-0173(97)00040-4. [DOI] [PubMed] [Google Scholar]
- CLARKE P.B.S.Functional anatomy of nicotinic acetylcholine receptors in mammalian brain Neuronal Nicotinic Receptors: Pharmacology and Therapeutic Opportunities 1998Wiley-Liss; 127–139.eds. Arneric, S.P. & Brioni, J.D. pp [Google Scholar]
- CLARKE P.B., REUBEN M. Release of [3H]-noradrenaline from rat hippocampal synaptosomes by nicotine: mediation by different nicotinic receptor subtypes from striatal [3H]-dopamine release. Br. J. Pharmacol. 1996;117:595–606. doi: 10.1111/j.1476-5381.1996.tb15232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DONNELLY-ROBERTS D.L., XUE I.C., ARNERIC S.P., SULLIVAN J.P. In vitro neuroprotective properties of the novel cholinergic channel activator (ChCA), ABT-418. Brain Res. 1996;719:36–44. doi: 10.1016/0006-8993(96)00063-7. [DOI] [PubMed] [Google Scholar]
- ELLIOTT K.J., JONES J.M., SACAAN A.I., LLOYD G.K., COREY-NAEVE J. 6-hydroxydopamine lesion of rat nigrostriatal dopaminergic neurons differentially affects nicotinic acetylcholine receptor subunit mRNA expression. J. Mol. Neurosci. 1998;10:251–260. doi: 10.1007/BF02761778. [DOI] [PubMed] [Google Scholar]
- FERGER B., SPRATT C., EARL C.D., TEISMANN P., OERTEL W.H., KUCHINSKY K. Effects of nicotine on hydroxyl free radical formation in vitro and on MPTP-induced neurotoxicity in vivo. Naunyn-Schmiedeberg's Arch. Pharmacol. 1998;358:351–359. doi: 10.1007/pl00005264. [DOI] [PubMed] [Google Scholar]
- FUXE K., JANSON A.M., JANSSON A., ANDERSSON K., ENCROTH P., AGNATI L.F. Chronic nicotine treatment increases dopamine levels and reduces dopamine utilisation in substantia nigra and in surviving forebrain dopaminergic nerve terminal systems after partial di-mesencephalic hemitransection. Naunyn-Schmiedeberg's Arch. Pharmacol. 1990;341:171–181. doi: 10.1007/BF00169727. [DOI] [PubMed] [Google Scholar]
- GERLACH M., RIEDERER P. Animal models of Parkinson's disease: an empirical comparison with the phenomenology of the disease in man. J. Neural. Transm. 1996;103:987–1041. doi: 10.1007/BF01291788. [DOI] [PubMed] [Google Scholar]
- GRADY S.R., MARKS M.J., COLLINS A.C. Desensitization of nicotine-stimulated [3H]dopamine release from mouse striatal synaptosomes. J. Neurochem. 1994;62:1390–1398. doi: 10.1046/j.1471-4159.1994.62041390.x. [DOI] [PubMed] [Google Scholar]
- GRADY S., MARKS M.J., WONNACOTT S., COLLINS A.C. Characterization of nicotinic receptor-mediated [3H]dopamine release from synaptosomes prepared from mouse striatum. J. Neurochem. 1992;59:848–856. doi: 10.1111/j.1471-4159.1992.tb08322.x. [DOI] [PubMed] [Google Scholar]
- GRENHOFF J., JANSON A.M., SVENSSON T.H., FUXE K. Chronic continuous nicotine treatment causes decreased burst firing of nigral dopamine neurons in rats partially hemitransected at the meso-diencephalic junction. Brain Res. 1991;2:347–351. doi: 10.1016/0006-8993(91)90646-d. [DOI] [PubMed] [Google Scholar]
- HADJICONSTANTINOU M., HUBBLE J.P., WEMLINGER T.A., NEFF N.H. Enhanced MPTP neurotoxicity after treatment with isoflurophate or cholinergic agonists. J. Pharmacol. Exp. Ther. 1994;270:639–644. [PubMed] [Google Scholar]
- JANSON A.M., FUXE K., AGNATI L.F., KITAYAMA I., HARFSTRAND A., ANDERSSON K., GOLDSTEIN M. Chronic nicotine treatment counteracts the disappearance of tyrosine-hydroxylase-immunoreactive nerve cell bodies, dendrites and terminals in the mesostriatal dopamine system of the male rat after partial hemistransection. Brain Res. 1988a;455:332–345. doi: 10.1016/0006-8993(88)90092-3. [DOI] [PubMed] [Google Scholar]
- JANSON A.M., FUXE K., GOLDSTEIN M. Differential effects of acute and chronic nicotine treatment on MPTP-(1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) induced degeneration of nigrostriatal dopamine neurons in the black mouse. Clin. Investig. 1992;70:232–238. doi: 10.1007/BF00184656. [DOI] [PubMed] [Google Scholar]
- JANSON A.M., FUXE K., SUNDSTROM E., AGNATI L.F., GOLDSTEIN M. Chronic nicotine treatment partly protects against the 1-methyl-4-phenyl-2,3,6-tertrahydropyridine-induced degeneration of nigrostriatal dopamine neurons in the black mouse. Acta Physiol. Scand. 1988b;132:589–591. doi: 10.1111/j.1748-1716.1988.tb08372.x. [DOI] [PubMed] [Google Scholar]
- JANSON A.M., HEDLUND P.B., FUXE K., VON EULER G. Chronic nicotine treatment counteracts dopamine D2 receptor upregulation induced by a partial meso-diencephalic hemitransection in the rat. Brain Res. 1994;655:25–32. doi: 10.1016/0006-8993(94)91593-8. [DOI] [PubMed] [Google Scholar]
- JANSON A.M., MEANA J.J., GOINY M., HERRERA-MARSCITZ M. Chronic nicotine treatment counteracts the decrease in extracellular dopamine induced by a unilateral transection at the mesodiencephalic junction in rats: a microdialysis study. Neurosci. Lett. 1991;134:88–92. doi: 10.1016/0304-3940(91)90515-u. [DOI] [PubMed] [Google Scholar]
- JANSON A.M., MOLLER A. Chronic nicotine treatment counteracts nigral cell loss induced by a partial mesodiencephalic hemitransection: an analysis of the total number and mean volume of neurons and glia in substantia nigra of the male rat. Neurosci. 1993;57:931–941. doi: 10.1016/0306-4522(93)90039-i. [DOI] [PubMed] [Google Scholar]
- JAVITCH J.A., BLAUSTEIN R.O., SNYDER S.H. [3H]mazindol binding associated with neuronal dopamine and norepinephrine uptake sites. Mol. Pharmacol. 1984;26:35–44. [PubMed] [Google Scholar]
- JAVITCH J.A., STRITTMATTER S.M., SNYDER S.H. Differential visualisation of dopamine and norepinephrine uptake sites in rat brain using [3H]mazindol autoradiography. J. Neurosci. 1985;5:1513–1521. doi: 10.1523/JNEUROSCI.05-06-01513.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KANEKO S., MAEDA T., KUME T., KOCHIYAMA H., AKAIKE A., SHIMOHAMA S., KIMURA J. Nicotine protects cultures cortical neurons against glutamate-induced cytotoxicity via α7-neuronal receptors and neuronal CNS receptors. Brain Res. 1997;765:135–140. doi: 10.1016/s0006-8993(97)00556-8. [DOI] [PubMed] [Google Scholar]
- KIHARA T., URUSHITANI M., SAWADA H., KIMURA J., KUME T., MAEDA T., AKAIKE A. Stimulation of α4β2 nicotinic acetylcholine receptors inhibits β-amyloid toxicity. Brain Res. 1997;792:331–334. doi: 10.1016/s0006-8993(98)00138-3. [DOI] [PubMed] [Google Scholar]
- LEE C.S., SAUER H., BJORKLUND A. Dopaminergic neuronal degeneration and motor impairments following axon terminal lesion by intrastriatal 6-hydroxydopamine in the rat. Neurosci. 1996;72:641–653. doi: 10.1016/0306-4522(95)00571-4. [DOI] [PubMed] [Google Scholar]
- LEVY R., HAZRATI L.-N., HERRERO M.-T., VILA M., HASSANI O.-K., MOUROUX M., RUBERG M., ASENSI H., AGID J., FEGER J., OBESO J.A., PARENT A., HIRSCH E.C. Re-evaluation of the functional anatomy of the basal ganglia in normal and parkinsonian states. Neurosci. 1997;76:335–343. doi: 10.1016/s0306-4522(96)00409-5. [DOI] [PubMed] [Google Scholar]
- LOTHARIUS J., DUGAN L.L., O'MALLEY K.L. Distinct mechanisms underlie neurotoxin-mediated cell death in cultured dopaminergic neurons. J. Neurosci. 1999;19:1284–1293. doi: 10.1523/JNEUROSCI.19-04-01284.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAGGIO R., RIVA M., VAGLINI F., FORNAI F., MOLTENI R., ARMOGIDA M., RACAGNI G., CORSINI G.U. Nicotine prevents experimental parkinsonism in rodents and induces striatal increase of neurotrophic factors. J. Neurochem. 1998;71:2439–2446. doi: 10.1046/j.1471-4159.1998.71062439.x. [DOI] [PubMed] [Google Scholar]
- MAGGIO R., RIVA M., VAGLINI F., FORNAI F., RACAGNI G., CORSINI G.U. Striatal increase of neurotrophic factors as a mechanism of nicotine protection in experimental parkinsonism. J. Neural Transm. 1997;104:1113–1123. doi: 10.1007/BF01273324. [DOI] [PubMed] [Google Scholar]
- MARSHALL D.L., REDFERN P.H., WONNACOTT S. Presynaptic nicotinic modulation of dopamine release in the three ascending pathways studied by in vivo microdialysis: comparison of naive and chronic nicotine-treated rats. J. Neurochem. 1997;68:1511–1519. doi: 10.1046/j.1471-4159.1997.68041511.x. [DOI] [PubMed] [Google Scholar]
- MORENS D.M., GRANDINETTI A., REED D., WHITE L.R., ROSS G.W. Cigarette smoking and protection from Parkinson's Disease: false association or etiologic clue. Neurology. 1995;45:1041–1051. doi: 10.1212/wnl.45.6.1041. [DOI] [PubMed] [Google Scholar]
- OLANOW C.W., TATTON W.G. Etiology and pathogenesis of Parkinson's Disease. Ann. Rev. Neurosci. 1999;22:123–144. doi: 10.1146/annurev.neuro.22.1.123. [DOI] [PubMed] [Google Scholar]
- OWMAN C.H., FUXE K., JANSON A.M., KAHRSTROM J. Chronic nicotine treatment eliminates asymmetry in striatal glucose utilisation following unilateral transection of the mesostriatal dopamine pathway in rats. Neurosci. Lett. 1989;102:279–283. doi: 10.1016/0304-3940(89)90092-x. [DOI] [PubMed] [Google Scholar]
- PAXINOS G., WATSON C. The rat brain in stereotaxic coordinates 1986Academic Press, London; (2nd ed.) [Google Scholar]
- PRZEDBORSKI S., LEVIVIER M., JIANG H., FERRERIA M., JACKSON-LEWIS V., DONALDSON D., TOGASAKI D.M. Dose-dependent lesions of the dopaminergic nigrostriatal pathway induced by intrastriatal injection of 6-hydroxydopamine. Neurosci. 1995;67:631–647. doi: 10.1016/0306-4522(95)00066-r. [DOI] [PubMed] [Google Scholar]
- REUBEN M., BOYE S., CLARKE P.B.S. Nicotinic receptors modulating somatodendritic and terminal dopamine release differ pharmacologically. Eur. J. Pharmacol. 2000;393:39–49. doi: 10.1016/s0014-2999(00)00004-2. [DOI] [PubMed] [Google Scholar]
- ROSS S.A., WONG J.Y.F., CLIFFORD J.J., KINSELLA A., MASSALAS J.S., HORNE M.K., SCHEFFER I.E., KOLA I., WADDINGTON J.L., BERKOVIC S.F., DRAGO J. Phenotypic characterisation of an α4 neuronal nicotinic acetylcholine receptor subunit knockout mouse. J. Neurosci. 2000;20:6431–6441. doi: 10.1523/JNEUROSCI.20-17-06431.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROWELL P.P., LI M. Dose-Response Relationship for Nicotine-Induced Up-Regulation of Rat Brain Nicotinic Receptors. J. Neurochem. 1997;68:1982–1989. doi: 10.1046/j.1471-4159.1997.68051982.x. [DOI] [PubMed] [Google Scholar]
- SAUER H., OERTEL W.H. Progressive degeneration of nigrostriatal dopamine neurons following intrastriatal terminal lesions with 6-hydroxydopamine: a combined retrograde tracing and immunocytochemical study in the rat. Neurosci. 1994;59:401–415. doi: 10.1016/0306-4522(94)90605-x. [DOI] [PubMed] [Google Scholar]
- SEMBA J., MIYOSHI R., KITO S. Nicotine protects against dexamethasone potentiation of kainic acid-induced neurotoxicity in cultured hippocampal neurons. Brain Res. 1996;735:335–338. doi: 10.1016/0006-8993(96)00926-2. [DOI] [PubMed] [Google Scholar]
- SHARPLES C.G.V., KAISER S., SOLIAKOV L., MARKS M.J., COLLINS A.C., WASHBURN M., WRIGHT E., SPENCER J.A., GALLAGHER T., WHITEAKER P., WONNACOTT S. UB-165: A novel nicotinic agonist with subtype selectivity implicates the α4/β2* subtype in the modulation of dopamine release from rat striatal synaptosomes. J. Neurosci. 2000;20:2783–2791. doi: 10.1523/JNEUROSCI.20-08-02783.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SONSALLA P.K., NICKLAS W.J., HEIKKILA R.E. Role for excitatory amino acids in methamphetamine-induced nigrostriatal dopaminergic toxicity. Science. 1989;243:398–400. doi: 10.1126/science.2563176. [DOI] [PubMed] [Google Scholar]
- SONSALLA P.K., RIORDAN D.E., HEIKKILA R.E. Competitive and noncompetitive antagonists at N-methyl-D-aspartate receptors protect against methamphetamine-induced dopaminergic damage in mice. J. Pharmacol. Exp. Therap. 1991;256:506–512. [PubMed] [Google Scholar]
- WHITEAKER P., GARCHA H.S., WONNACOTT S., STOLERMAN I.P. Locomotor activation and dopamine release produced by nicotine and isoarecolone in rats. Br. J. Pharmacol. 1995;116:2097–2105. doi: 10.1111/j.1476-5381.1995.tb16417.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WONNACOTT S. The paradox of acetylcholine receptor upregulation by nicotine. Trends in Pharmacol. Sci. 1990;11:216–219. doi: 10.1016/0165-6147(90)90242-z. [DOI] [PubMed] [Google Scholar]
- WONNACOTT S. Presynaptic nicotinic ACh receptors. Trends in Neural Sci. 1997;20:92–98. doi: 10.1016/s0166-2236(96)10073-4. [DOI] [PubMed] [Google Scholar]