

Abstract
We investigated the effect of the p38 kinase inhibitor SB 203580 on airway inflammation induced by aerosolized lipopolysaccharide (LPS) in male Wistar rats. SB 203580 significantly inhibited (ED50=15.8 mg kg−1) plasma levels of TNF-α in rats challenged with LPS (1.5 mg kg−1, i.p.).
Aerosolized LPS induced a peak in TNF-α levels and the initiation of a neutrophilic response in bronchoalveolar lavage (BAL) fluid at the 2 h time point. Furthermore, the 4 h time point was associated with the peak in IL-1β levels and the initial plateau of neutrophilia observed in the BAL fluid.
SB 203580 (100 mg kg−1), had no effect on peak TNF-α levels or the associated neutrophilia in the BAL. Interestingly, the PDE 4 inhibitor RP 73401 (100 mg kg−1) significantly reduced both TNF-α levels and neutrophilic inflammation. However, the BAL fluid from rats pre-treated with either compound significantly inhibited TNF-α release from cultured human monocytes 18 h after LPS treatment (83.6 and 44.5% inhibition, respectively).
Alternatively, SB 203580 (100 mg kg−1) produced dose-related inhibition of BAL IL-1β levels (67.5% inhibition, P<0.01) and BAL neutrophilia (45.9% inhibition, P<0.01) 4 h after LPS challenge.
P38 protein was present in lung tissue and the level of expression was not affected by LPS treatment.
P38 kinase appears to be involved in the release of IL-1β and the sustained neutrophilic response in the BAL fluid. This data may suggest a role for p38 inhibitors in the treatment of airway inflammatory diseases in which neutrophilia is a feature of the lung pathology.
Keywords: Lipopolysaccharide, tumour necrosis factor α, airway inflammation, p38 MAP kinase, neutrophilis, interleukin 1β
Introduction
Aerosol or intranasal administration of LPS induces intense lung inflammation, with macrophage activation and recruitment of neutrophils to the interstitium, alveoli, and the airways of guinea-pigs (Gordon et al., 1991), rats (Ulich et al., 1994; Pauwels et al., 1990), and mice (Harmsen, 1988; Gonçalves de Moraes et al., 1996). This response requires the upregulation of adhesion molecules on circulating leukocytes and the pulmonary vascular endothelium and the expression of endogenous chemotactic factors that draw the marginated leukocytes across the endothelial and epithelial barriers into the air spaces (Tang et al., 1995b; Ulich et al., 1991b; Strieter & Kunkel, 1994). Inhalation challenge studies have also shown that LPS causes neutrophilic inflammation in non-atopic subjects, non-asthmatic atopic subjects, and asthmatic subjects (Sandstrom et al., 1994; Blaski et al., 1996; Nightengale et al., 1998; Michel et al., 1997).
TNF-α and IL-1β have been implicated as mediators of LPS-induced airway inflammation (O'Leary et al., 1996; Ulich et al., 1991a; Wesselius et al., 1995). TNF-α and IL-1β have been shown to amplify pulmonary inflammatory responses by stimulating the release of chemotactic factors from alveolar macrophages and airway epithelial cells and by upregulating the expression of leukocyte and endothelial adhesion molecules (Strieter & Kunkel, 1994). Furthermore, instillation of recombinant TNF-α into the tracheobronchial tree induces chemokine release in bronchoalveolar lining fluid (Koh et al., 1996) and upregulation of intercellular adhesion molecule-1 (ICAM-1) on pulmonary vascular endothelium (Mulligan et al., 1993). ICAM-1 mRNA in whole lung is increased following airway instillation of LPS in the rat. This LPS-induced increase was reduced by 81% after treatment of animals with anti-TNF-α antibody and 37% after treatment with IL-1β antibody (Beck-Schimmer et al., 1997). Exposure to aerosolized or endotracheally administered rTNF-α has been associated with neutrophilic infiltration of the interstitium or alveolar septae (Fuchs et al., 1990; Warren et al., 1989). Administration of TNF-α inhibitors has been reported to reduce LPS-induced lung inflammation in some models (Kolls et al., 1995; Ulich et al., 1993) but not in others (Tang et al., 1995a). In addition, the IL-1 receptor antagonist has been shown to inhibit endotoxin and IL-1 induced acute inflammation (Ulich et al., 1991a).
Several therapeutic strategies have been employed to control the release of pro-inflammatory cytokines such as IL-1β and TNF-α. Recently attention has focused on the bicyclic pyridinyl imidazole class of compounds such as SB 203580 and SB 202190, which are potent inhibitors of TNF-α and IL-1β release (Lee & Adams, 1995; Lee & Young, 1996; Lee et al., 1993; 1994; Young et al., 1993). The intracellular target of these compounds is the p38 mitogen-activated protein kinase (MAPK, Lee et al., 1994), a key component in cytokine and stress-induced signal transduction pathways (Davis, 1995). Analysis of the inhibitory mechanisms of these compounds indicated that the site of action was primarily at the translational level (Young et al., 1993; Prichett et al., 1995).
Pyridinyl imidazoles such as SB 203580 act by inhibiting p38 kinase activity through competition with ATP (Tong et al., 1997; Young et al., 1997). The anti-inflammatory effects of these compounds can be attributed, in part, to their ability to suppress monocyte/macrophage production of TNF-α and IL-1β (Lee & Young, 1996; Foey et al., 1998; Manthey et al., 1998). However, numerous other anti-inflammatory activities have been elucidated, including suppression of IL-1β-induced prostaglandin H synthase-2 expression by endothelial cells (Ridley et al., 1997), FMLP-induced neutrophil chemotaxis (Zu et al., 1998), and IL-2- and IL-7-induced lymphocyte proliferation (Crawley et al., 1997). Furthermore, in addition to in vitro data, p38 kinase inhibitors such as SB 203580 and SB 220025 have been shown to be efficacious in in vivo models of arthritis and inflammatory angiogenesis (Badger et al., 1996; Jackson et al., 1998).
In this study we investigated the in vivo contribution of the p38 MAPK to LPS-induced airway inflammation using the inhibitor SB 203580. This compound has been shown to be a selective inhibitor of p38 kinase (Cuenda et al., 1995; Lee et al., 1994; Young et al., 1997).
Methods
Animals
Male Wistar rats (150 – 180 g) were purchased from Harlan-Olac (Bicester, U.K.) and housed for at least 5 days before use. Food and water were supplied ad libitum. U.K. Home Office guidelines for animal welfare based on the Animals (Scientific Procedures) Act 1986 were strictly observed.
Systemic LPS-induced TNF-α release in rats
Preliminary experiments were performed in order to determine an appropriate dose of LPS, which induced significant TNF-α release. In subsequent experiments rats were orally dosed with vehicle (1% carboxymethyl cellulose (CMC), 2 ml kg−1) or compound 30 min prior to LPS administration (1.5 mg kg−1, i.p.). Animals were killed using carbon dioxide asphyxiation and heparinized blood samples were collected by cardiac puncture 90 min later. The blood samples were spun at 285 g for 5 min at room temperature and the resulting plasma, removed and stored at −20°C. Rat TNF-α was determined using the rat specific enzyme-linked immunosorbant assay (ELISA) obtained from Genzyme (Cambridge, U.S.A.). The detection limit of this assay was determined to be 10 pg ml−1. There was no detectable cross-reactivity with other cytokines tested at 1 μg ml−1 (rat IL-1β, GRO-β/MIP-2, GRO/KC, MCP-1).
Aerosolized LPS-induced cell influx and TNF-α/IL-1β release into rat airway lumen
In preliminary experiments concentration and time dependent relationships for cell influx and TNF-α/IL-1β release into the airway lumen were determined in response to aerosolized LPS. In subsequent experiments rats were orally dosed with vehicle (1% CMC, 2 ml kg−1) or compound 30 min prior to aerosolized LPS (0.3 mg ml−1 for 30 min) using an Ultra-Neb 99 (Sunrise Medical Ltd., Wollaston, U.K.). Bronchoalveolar lavage (BAL) was performed at various time points after LPS challenge to determine TNF-α/IL-1β levels and characterize cell infiltration. To perform the BAL, animals were first killed with sodium pentobarbitone 1 ml kg−1 i.p. and the trachea cannulated. One 10 ml kg−1 aliquot of RPMI 1640 containing 10% (v v−1) FCS and 417 mg l−1 glycyl-L-glutamine (Glutamax™) was delivered through the tracheal cannula and removed after a 30 s interval. This procedure was repeated and samples were then pooled for each animal. Lungs were then either removed and flash frozen with liquid nitrogen and stored at −80°C (for extraction of cytosolic proteins for Western blot analysis) or used to determine the cellular profile within the lung tissue. In these experiments the lungs were removed immediately after BAL, the pulmonary vasculature perfused with RPMI 1640 containing 10% FCS to remove the blood pool of cells and the tissue chopped. Cells were desegregated from the tissue by incubating at 37°C with collagenase (20 u ml−1 for 2 h, then 60 u ml−1 for 1 h) in RPMI/FCS. The recovered cells were filtered (mesh size 70 μM), washed three times and re-suspended in a final volume of 1 ml RPMI 1640/10% FCS.
Quantification of inflammatory cells
Total whole cell counts were obtained in BAL or lung tissue samples by using an automated cell counter (Cobas Argos, Roche ABX Hematologie, Montpellier, France). Cytospins of these samples were prepared by centrifugation of 100 μl aliquots in a cytospin (Shandon, Runcorn, U.K.) at 800×g for 5 min, low acceleration at room temperature. Slides were fixed and stained on a Hema-tek 2000 (Ames Co., Elkhart, U.S.A.) with modified Wrights-Giemsa stain. Four part differential counts on 200 cells per slide were performed following standard morphological criteria and the percentage of eosinophils, macrophages/monocytes, lymphocytes and neutrophils were determined. TNF-α and IL-1β levels were determined by a rat specific ELISA obtained from Genzyme (Cambridge, U.S.A.) and Biosource International (Camarillo, U.S.A.) respectively according to the manufacturer's instructions.
Human monocyte isolation and culture
Peripheral venous blood was drawn from healthy, non-allergic volunteers and layered on a histopaque gradient (ρ=1.077 g ml−1). Peripheral blood mononuclear cells (PBMC) were obtained by density centrifugation (1000×g) for 20 min. PBMCs were collected and washed twice with PBS (Ca2+ and Mg2+ free). A total cell count was performed using a haemocytometer, and a differential cell count to determine the percentage of monocytes in the mononuclear preparation was performed after cytospinning cells (5×105 ml−1) and staining with a Wrights-Giemsa stain. Mononuclear cells were then resuspended in RPMI with 25 mM HEPES, 417 mg l−1 glycyl-L-glutamine (Glutamax™), and 10% (v v−1) FCS and plated 1×104 cells per well in a 96-well microtitre plate. Cells were incubated at 37°C with 5% CO2 for 2 h to induce monocyte adherence. After incubation, the plate was gently shaken and lymphocytes were aspirated off.
LPS-induced TNF-α release from cultured human monocytes
Supernatant from BAL samples (from vehicle- and LPS-treated rats) was diluted 1 : 5, added to monocytes and LPS (10 ng ml−1) was immediately added. TNF-α levels were determined 18 h later by specific human TNF-α ELISA (Genzyme, Cambridge, U.S.A.), according to the manufacturer's instructions.
Cytosolic proteins extraction
Frozen lung tissue samples were thawed in cold lysis buffer (1% Triton X-100, 1% sodium dodecyl sulphate (SDS), 1.5% deoxycholate, 20 mM Tris-base pH 7.4, 150 mM NaCl, 20 mM EDTA, 2 mM phenylmethyl sulphonyl fluoride (PMSF), 2 mM sodium orthovanadate, 20 μg ml−1 leupeptin, 200 μg ml−1 aprotinin, 10 mM NaF and 20 mM sodium pyrophosphate), homogenized with an Ultra-turrex and centrifuged at 13,000×g for 30 min at 4°C. Supernatant was removed and centrifuged again. Lung supernatant lysates (10 μg of protein) were mixed with sample buffer (62.5 mM Tris-HCl, 20% glycerol, 2% SDS, 10 mM 2-mercaptoethanol, 0.05% bromophenol blue), boiled for 5 min and stored at −70°C until used for Western blot analysis.
Western blotting analysis
Protein samples were separated by SDS-polyacrylamide gel electrophoresis (SDS – PAGE) on 10% acrylamide gel and then transferred to nitrocellulose membranes (Amersham, U.K.) for 1 h at 300 mA in transblotting buffer (0.2 M glycine-HCl, 25 mM Tris-base, and 20% (v v−1) methanol). To block non-specific antibody binding, membranes were incubated for 1 h in blocking buffer (PBS pH 7.4, 0.1% Tween-20) containing 5% (w v−1) non-fat dry milk. Membranes were then incubated overnight at 4°C with the p38 mitogen-activated protein kinase (MAPK) polyclonal antibody (New England Biolabs, Hitchin, U.K.) used at a dilution of 1 : 1000 in blocking buffer where non-fat milk was replaced with 5% BSA. Membranes were washed with blocking buffer for 3×5 min and incubated with 1 : 1500 dilution of alkaline phosphatase-conjugated anti-rabbit secondary antibody, washed and protein detection was carried out using CDPStar™ chemiluminescent reagent. Membranes were drained from excess developing solution and exposed to Kodak X-OMAT-S film.
Materials
SB 203580 [4-(4-fluorophenyl)-2-(4-methylsulphinyl-phenyl)-5-(4-pyridyl)imidazole] and RP 73401 [3-cyclopentyloxy-N-(3,5-dichloro-4-pyridyl)-4-methoxyhenzamide] were synthesized by the Medicinal Chemistry Department at Rhône-Poulenc Rorer (Dagenham, U.K.). SB 203580 was suspended in 1% carboxymethyl cellulose sodium salt (CMC) obtained from BDH Laboratory Supplies (Poole, U.K.). Lipopolysaccharide (LPS) from Escherichia coli serotype 0111:B4 and all other materials were purchased from Sigma (Poole, U.K.) except for Roswell Park Memorial Institute (RPMI) 1640 medium, HEPES, glutamax, phosphate buffered saline (PBS) and foetal calf serum (FCS) all from Gibco (Paisley, U.K.); and sodium pentobarbitone (euthatal) from Rhône Mérieux Ltd., Harlow, U.K.
Data analysis
All the values in the figures and text are expressed as mean±s.e.mean of n observations. Data were compared (in aerosolized LPS treated groups with and without drug treatment at one dose) using the Mann – Whitney U-test for unpaired data. Statistical comparisons were made on data in which multiple comparisons were made using the Kruskal-Wallis test followed by a Dunn's post test. All treatments were compared to vehicle control values, *P<0.05, **P<0.01, ***P<0.001.
Results
Systemic LPS-induced TNF-α release in rats
Preliminary experiments were performed in order to determine an appropriate dose of LPS, which induced significant TNF-α release. LPS evoked a significant increase in plasma TNF-α levels (saline-treated, <10 pg ml-1; LPS-treated, 47.2±6.8 ng ml−1). SB 203580 (10 – 100 mg kg−1, p.o.) significantly inhibited i.p. LPS-induced TNF-α release in a dose-dependent manner (ED50=15.8 mg kg−1; Figure 1). There was no inhibition of TNF-α levels in vehicle-treated rats (Figure 1). The PDE 4 inhibitor RP 73401 (100 mg kg−1, p.o.), which was used as a positive control, also significantly reduced TNF-α levels in rat plasma following intraperitoneal administration of LPS (83.8% inhibition; Figure 1).
Figure 1.
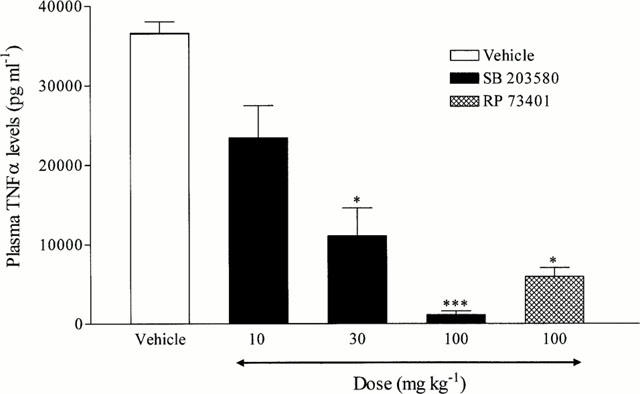
Effect of SB 203590 and RP 73401 on intraperitoneal (i.p.) LPS-induced TNF-α release in rat plasma. Wistar rats were orally (p.o.) dosed with vehicle (1% CMC, 2 ml kg−1) or compound (SB 203580, 10 – 100 mg kg−1; RP 73401, 100 mg kg−1) 30 min prior to administration of LPS (1.5 mg kg−1, i.p). Plasma was collected 90 min later and TNF-α levels determined by ELISA. Results are expressed as mean±s.e.mean of eight animals. Statistical significance was assessed using Kruskal-Wallis with a Dunn s post test (*P<0.05, ***P<0.001).
Time course of inflammatory cell recruitment and cytokine expression after inhalational LPS challenge
These studies were performed in order to identify the biological profile of the airway inflammatory response following aerosolized LPS in terms of the cytokine release and neutrophilia observed. In these experiments there was a clear time-dependent neutrophil infiltration into the airway lumen in response to aerosolized LPS (1 mg ml−1 for 30 min). BAL neutrophilia after aerosolized LPS was evident at 60 min, reached a plateau at 2 – 6 h with a further increase at 8 h and returned toward basal levels by 24 – 48 h (Figure 2a).
Figure 2.
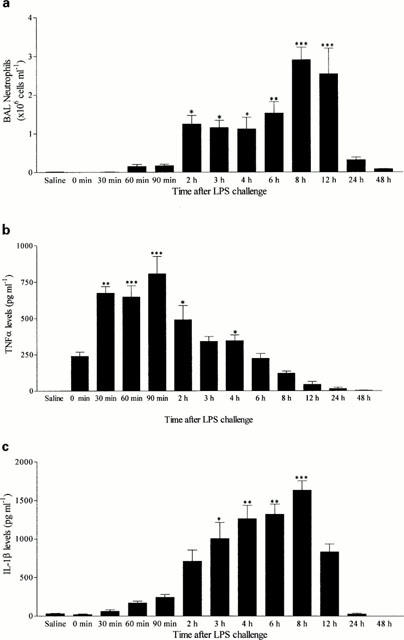
Time Course of neutrophil recruitment (a) and TNF-α (b) and IL-1β (c) protein levels after aerosolized LPS challenge. Male Wistar rats were exposed to aerosolized LPS (1 mg ml−1 for 30 min) and BAL fluid collected at various time points later (0 – 48 h). Neutrophil influx was determined in the BAL samples by differential cell counting and cytokine levels by ELISA. Results are expressed as mean±s.e.mean of 6 – 10 animals. Statistical significance was assessed using Kruskal-Wallis with a Dunn's post test (*P<0.05, **P<0.01, ***P<0.001).
In the same samples, TNF-α levels were increased very rapidly after aerosolized LPS (1 mg ml−1 for 30 min). The highest levels of TNF-α were found in BAL samples collected 30 – 90 min after aerosolized LPS (which coincided with the start of the neutrophilic response) and were no longer significant 24 h after LPS exposure (Figure 2b). In addition, there was a time-dependent increase in IL-1β levels following aerosolized LPS administration which peaked 4 – 8 h after aerosolized LPS (which was temporally associated with the initial plateau in neutrophilia at 2 – 6 h and was still increasing at 8 h which was the peak of neutrophilia) and was no longer significant 24 h after exposure (Figure 2c).
Dose-dependent effect of LPS inhalational challenge on neutrophil recruitment
In concentration response studies, LPS concentrations above 0.1 mg ml−1 (aerosolized over 30 min) produced significant BAL neutrophilia 2 h post-challenge (Figure 3). Further studies utilized LPS concentrations of 0.3 mg ml−1 aerosolized for 30 min and analysed the effect of drugs on the initial neutrophilia seen at 2 h (associated with peak levels of TNF-α in BAL) and the established plateau of neutrophilia observed at 4 h (associated with peak levels of IL-1β in BAL).
Figure 3.

Effect of aerosolized LPS exposure on neutrophil accumulation in the BAL fluid. Male Wistar rats were exposed to various concentrations of aerosolized LPS (0.001 – 10 mg ml−1 for 30 min) and BAL fluid collected 2 h later. Neutrophil influx was determined in the BAL samples by differential cell counting. Results are expressed as mean±s.e.mean of five or six animals. Statistical significance was assessed using Kruskal-Wallis with a Dunn's post test (**P<0.01, ***P<0.001).
Effect of SB 203580 and RP 73401 on aerosolized LPS-induced TNF-α and neutrophil infiltration into the airway lumen 90 min post challenge
Rats were orally dosed with compound (100 mg kg−1) or vehicle (1% CMC, 2 ml kg−1) 30 min prior to exposure with aerosolized LPS (0.3 mg ml−1 for 30 min). SB 203580 had no impact on BAL TNF-α levels (Figure 4a) or neutrophil infiltration into the airway lumen (Figure 4b). However, the PDE 4 inhibitor RP 73401 (100 mg kg−1), which was used as a positive control, significantly reduced both BAL TNF-α levels (Figure 4a) and neutrophil infiltration (Figure 4c).
Figure 4.
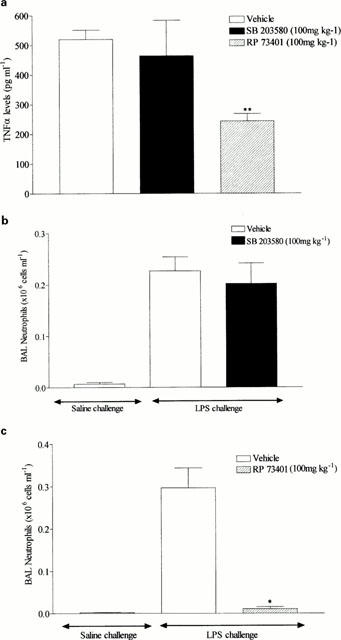
Effect of SB 203580 (100 mg kg−1, p.o.) on TNF-α levels (a) and neutrophil infiltration (b) into the BAL fluid induced by aerosolized LPS (0.3 mg ml−1 for 30 min). In these experiments the PDE 4 inhibitor, RP 73401 (100 mg kg−1, p.o.), was used as a positive control and its effects investigated on TNF-α levels (a) and neutrophil infiltration (c) into the BAL fluid induced by aerosolized LPS. Wistar rats were orally dosed with vehicle (1% CMC, 2 ml kg−1) or compound 30 min prior to aerosolized LPS and BAL fluid collected 2 h later. Neutrophils were determined by differential cell counting and TNF-α levels by ELISA. Results are expressed as mean±s.e.mean of eight animals. Statistical significance was assessed using Kruskal-Wallis with Dunn's post test (*P<0.05, **P<0.01).
Given the lack of effect of SB 203580 on LPS-induced neutrophilia and TNF-α levels in BAL fluid, we performed experiments to determine whether active drug substance was present in the BAL fluid after oral dosing. BAL fluid from rats orally dosed with SB 203580 or RP 73401 (100 mg kg−1) was added to cultured human monocytes (10,000 cells per well) followed by the addition of LPS (2.5 ng per well). The results depicted in Figure 5 show that there was a significant inhibition of TNF-α release from human monocytes 18 h post-LPS stimulation by BAL fluid obtained from rats treated with RP 73401 (44.5% inhibition) or SB 203580 (83.6% inhibition).
Figure 5.

Effect of BAL fluid from rats orally dosed with SB 203580 (100 mg kg−1) and RP 73401 (100 mg kg−1) on LPS-induced TNF-α release from human monocytes. Supernatant from BAL samples of treated rats was added to cultured human monocytes (10,000 cells per well) together with LPS (2.5 ng per well). TNF-α levels were determined in the cell supernatant by ELISA 18 h later. Results are expressed as mean±s.e.mean of four animals. Statistical significance was assessed using Kruskal-Wallis with a Dunn's post test (*P<0.05, **P<0.01).
Effect of SB 203580 and dexamethasone on aerosolized LPS-induced IL-1β and neutrophil infiltration into the airway lumen 4 h post-challenge
SB 203580 produced dose-related inhibition of IL-1β levels in the BAL at 4 h after aerosolized LPS exposure that reached statistical significance at 100 mg kg−1 (67.5% inhibition; Figure 6a). SB 203580 also produced dose-related inhibition of the temporally associated neutrophil infiltration into the BAL fluid (at 100 mg kg−1, 45.9% inhibition; Figure 6b) but this inhibitory effect did not reach significance in the lung tissue (at 100 mg kg−1, 21.4% inhibition; Figure 6c) at the same time point. The synthetic glucocorticoid, dexamethasone (1 mg kg−1, p.o.) used as a positive control, significantly inhibited IL-1β and neutrophil levels in the BAL fluid following LPS exposure (99.3 and 81.9% inhibition, respectively; Figure 6a,b). Furthermore, dexamethasone significantly inhibited neutrophil accumulation into the lung tissue following LPS exposure (64.3% inhibition; Figure 6c).
Figure 6.
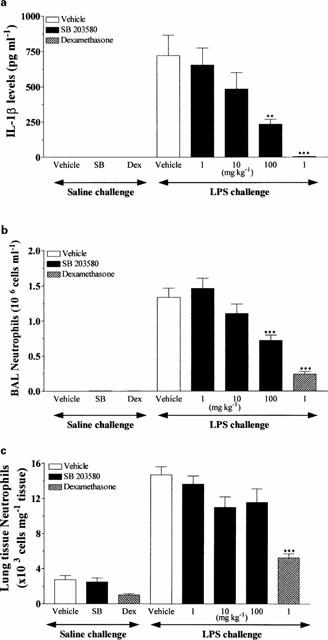
Effects of SB 203580 (1 – 100 mg kg−1, p.o.) and dexamethasone (1 mg kg−1, p.o.) on IL-1β levels (a) and neutrophil infiltration into the BAL fluid (b) and lung tissue (c) induced by aerosolized LPS (0.3 mg ml−1 for 30 min). Wistar rats were orally dosed with vehicle (1% CMC, 2 ml kg−1) or compound 30 min prior to aerosolized LPS and BAL fluid collected 4 h later. Neutrophils were determined by differential cell counting and IL-1β levels by ELISA. Results are expressed as mean±s.e.mean of eight animals. Statistical significance was assessed using Kruskal-Wallis with Dunn's post test (**P<0.01, ***P<0.001).
Effect of LPS inhalation on p38 protein expression in rat lung tissue
Rat lung tissue was flash frozen in liquid nitrogen and assayed for p38 protein expression using Western blot at various time intervals up to 60 min after LPS inhalation (0.3 mg ml−1 for 30 min). Expression of the p38 protein occurred at every time point measured and expression was at a constant level which did not vary with treatment (Figure 7).
Figure 7.

Effect of LPS on p38 protein expression in rat lung. Expression of p38 kinase protein in rat lung tissue at different time points (0, lanes 1, 2; 10 min, lanes 3, 4; 30 min, lanes 5, 6; 60 min, lanes 7, 8) after saline (lanes 1, 3, 5, 7) or LPS (lanes 2, 4, 6, 8) inhalation (0.3 mg ml−1 for 30 min).
Discussion
In recent years steps have been taken to delineate the intracellular signalling cascades in cells that mediate inflammation. Much attention has been given to the mitogen-activated protein kinase (MAPK) superfamily due to their consistent activation by pro-inflammatory cytokines, and to their role in nuclear signalling. This superfamily includes extracellular-regulated kinases (ERKs, also known as p42/p44), the c-Jun N-terminal kinases (JNKs, also known as stress-activated protein kinases or SAPK) and the p38 MAP kinases (also known as cytokine suppressive binding protein; CSBP) (Karin, 1998). ERKs are activated by growth factors and mitogenic stimuli whereas p38 and JNK are regulated by stress-inducing signals (like UV irradiation and osmotic shock) and proinflammatory cytokines (Karin, 1998). Interest in the p38 family has been particularly intense following the discovery that p38 inhibitors are anti-inflammatory in in vivo models of arthritis and inflammatory angiogenesis (Lee et al. 1994; Badger et al., 1996; Jackson et al., 1998). It has been shown in vitro that p38 kinase is induced by LPS and plays a key role in LPS-induced signal transduction pathways leading to cytokine synthesis (Lee & Young, 1996; Lee et al., 1994). However the role and the contribution of p38 kinase in airway inflammation in vivo is less clear. In this study, we have investigated the role of p38 kinase in the LPS inhalational challenge model of airway inflammation in the rat using the selective kinase inhibitor SB 203580.
LPS inhalation induced a marked increase in neutrophil recruitment in airway tissue and lumen. The observed kinetics of neutrophil recruitment was similar to that described in the Sprague-Dawley or Lewis rats after intratracheal LPS instillation (Ulich et al., 1991; Yi et al., 1996; Miller-Larsson et al., 1999) and in BALB/c mice after LPS inhalation (Gonçalves de Moraes et al., 1996; Wohlford-Lenane et al., 1999). LPS inhalation was followed by a time-dependent release of TNF-α into the BAL with a peak increase situated around 30 – 90 min after challenge in agreement with published data obtained in rats and mice (Gonçalves de Moraes et al., 1996; O'Leary et al., 1996; Miller-Larsson et al., 1999; Wohlford-Lenane et al., 1999). Thirty to 90 min after the end of LPS exposure, TNF-α concentration reached peak levels at a time where there was no significant neutrophilia observed in BAL fluid. Therefore, TNF-α synthesis precedes the influx of neutrophils and this could suggest a causal relationship. However, this assumption may not be justified given that the role of other important neutrophil chemoattractants such as IL-8 and LTB4 has not been investigated in this study. Several lines of evidence suggest the involvement of TNF-α as a mediator of LPS-induced airway inflammation (O'Leary et al., 1996; Ulich et al., 1991b; 1993; Wesselius et al., 1995; Koh et al., 1996; Fuchs et al., 1990; Kolls et al., 1995). The effect of TNF-α has been shown to be mediated through the 55-kDa type 1 TNF receptor (TNFR1). It has been demonstrated that TNFR1-deficient mice exposed to LPS aerosol, showed a persistent reduction in neutrophil recruitment to the air spaces of the lungs, in comparison with wild-type animals, that was associated with depressed chemokine levels in BAL fluid (Skerrett et al., 1999). Ulich et al. (1993) found that co-administration of human soluble TNFR1 reduced by 50 – 60% the number of bronchoalveolar neutrophils recovered 6 h after intratracheal injection of LPS in rats but did not affect the number of neutrophils 4 or 12 h after LPS challenge. Ulich et al. (1994) also reported that co-administration of soluble human TNFR2 with intratracheal LPS reduced neutrophil recruitment by up to 40% 6 h later, whereas coinjection of a dimeric construct of human TNFR2 linked to the Fc fragment of IgG did not influence neutrophil recruitment in response to intratracheal LPS.
Beside TNF-α, LPS inhalation also induced IL-1β in BAL fluid. IL-1β was detected with a delayed kinetic profile compared to TNF-α and coincided with the sustained initial neutrophilic phase and the second peak of neutrophilia observed 8 – 12 h post-challenge. This result suggests that IL-1β may be involved in the neutrophilia observed after 2 h in this model but again this assumption may not be valid given the role of other neutrophil chemoattractants has not been examined. IL-1β has many of the same effects as TNF-α, including stimulation of chemokine release and upregulation of adhesion molecules (Dinarello, 1996). It has previously been shown that intratracheal injection of LPS induces IL-1β expression in vivo in Lewis rat lung with expression peaking 4 – 6 h after instillation (Ulich et al., 1991a). Furthermore, intratracheal injection of IL-1β replicates the kinetics and relative magnitudes of the acute neutrophilic inflammatory response (Ulich et al., 1991b). Furthermore, the IL-1 receptor antagonist coinjected intratracheally with LPS or IL-1β in rats significantly inhibits neutrophilic exudation into bronchoalveolar lavage (Ulich et al., 1991a; 1994). Furthermore, an anti-IL-1 antibody has been shown to reduce LPS-induced lung ICAM-1 mRNA expression which is necessary for neutrophil recruitment (Beck-Schimmer et al., 1997).
Studies in monocytes (Lee et al., 1994; 1996) and alveolar macrophages (Carter et al., 1999) have shown that the p38 MAPK pathway is critical for LPS-induced cytokine release. In these studies, specific inhibition of the p38 kinase pathway by SB 203580 resulted in reduced cytokine release secondary to a defect in translation (Lee et al., 1994). In alveolar macrophages, the regulatory effect of p38 kinase on LPS-induced TNF-α and IL-6 gene expression was mediated partly through changes in gene transcription (Carter et al., 1999). In our LPS inhalational challenge model, SB 203580 even at the top dose of 100 mg kg−1 was ineffective in inhibiting LPS-induced BAL TNF-α levels and the accompanied neutrophilia. However the PDE 4 inhibitor RP 73401 (Souness et al., 1996) completely abrogated the induced neutrophilia and significantly inhibited BAL TNF-α levels in agreement with data obtained with other PDE 4 inhibitors in rats (Turner et al., 1993) and mice (Pettipher et al., 1996; Griswold et al., 1998; Gonçalves de Moraes et al., 1998). The inability of SB 203580 to inhibit BAL TNF-α cannot be attributed to the bioavailability and tissue distribution of the compound as BAL fluid from SB 203580-treated animals was able to inhibit TNF-α release from cultured human monocytes. In murine monocytes cell lines, SB 203580 was shown to inhibit LPS-induced IL-1β transcription (Baldassare et al., 1999). In our model, there was a dose-related inhibition of BAL IL-1β and the associated BAL, but not lung tissue, neutrophilia by SB 203580. The lack of a significant effect of SB 203580 on lung tissue compared to BAL neutrophilia is not surprising given it is generally more difficult to impact upon given the basal granuloma burden in rat lungs. Furthermore, in this study a supramaximal dose of dexamethasone produced a greater inhibition of BAL, compared with lung tissue, neutrophilia following LPS challenge. It would be interesting to investigate the activity of a more potent p38 inhibitor (ED50 >15 mg kg−1 on LPS-induced TNF-α release in plasma) as it may be expected to have a greater impact on lung tissue neutrophilia induced by LPS.
The doses at which SB 203580 inhibited neutrophilia (between 10 and 100 mg kg−1) are similar to those necessary to inhibit collagen-induced arthritis in DBA1/LACJ mice (50 mg kg−1, Badger et al., 1996) and adjuvant-induced arthritis in Lewis rats (30 and 60 mg kg−1, Badger et al., 1996) consistent with an inhibitory action of the compound on p38 kinase. The fact that SB 203580 inhibited, in a similar fashion, both IL-1β release and neutrophilia in the BAL 4 h following LPS could suggest a causal relationship. However, a non-specific action of this compound, unrelated to its inhibitory action on p38 kinase, cannot be ruled out. In fact this study would benefit from the inclusion of data with other p38 inhibitors administered i.v. to account for any differences in the oral bioavailability of the compounds. However, for this data to be meaningful one would have to use inhibitors from different chemical series that were structurally distinct, which had varying inhibitory potencies on the enzyme. If the inhibitory activity of the compounds was related to inhibition of p38 then their activity in vivo should follow the same rank order of potency.
We have also investigated the modulatory effect of SB 203580 on plasma TNF-α levels after intraperitoneal injection of LPS. LPS evoked a significant increase in plasma TNF-α levels which was inhibited in a dose-related manner by SB 203580. The derived potency (ED50 of 15.8 mg kg−1) agrees with data obtained in mice and rats with SB 203580 and the recently described p38 kinase inhibitor SB 220025 (Badger et al., 1996; Jackson et al., 1998). The PDE 4 inhibitor RP 73401, which was used as a positive control, also significantly reduced TNF-α levels in rat plasma following intraperitoneal administration of LPS. Thus, SB 203580 seems to differentially affect BAL and plasma TNF-α levels. Initially, this appeared to be a strange result however we have recently confirmed this observation by demonstrating the lack of inhibitory effect on BAL TNF-α levels of SB 203580 in a model of Sephadex-induced airway inflammation (Birrell et al., 2000). However, this phenomenon appears to be stimulus specific since SB 203580 has been shown to inhibit the increase in BAL TNF-α levels following allergen challenge in sensitized Brown Norway rats (Escott et al., 2000). Alternatively, the lack of effect of SB 203580 on BAL TNF-α levels in this model could indicate that the inhibitory effect on BAL IL-1β and neutrophilia at later time points is not due to inhibition of p38 MAPK inhibition.
The potential reasons for this differential effect are unclear but may be related to the differential expression and the sensitivity of the different p38 kinase isoforms to inhibition by SB 203580 and the possibility that TNF-α and IL-1β production is dependent on different isoforms or cell types. Indeed, four genes encode the known members of the p38 family, p38α (Lee et al., 1994), p38β (Jiang et al., 1996; Stein et al., 1997), p38γ (Lechner et al., 1996; Li et al., 1996), and p38δ (Wang et al., 1997; Jiang et al., 1997). Hale et al. (1999) have shown that the expression of p38 family members is not ubiquitous, but is controlled during cell differentiation and in a lineage-specific fashion. Differential expression was most striking in monocytes that strongly expressed p38α, but did not express p38β or p38γ. As blood monocytes differentiated into macrophages, a striking induction of p38Δ mRNA and protein occurred so that p38Δ protein became at least as abundant as p38α (Hale et al., 1999). In neutrophils, only p38α and p38Δ were detected (Nick et al., 1999). Although p38γ mRNA was present in endothelial cells, p38γ protein was not detected in any cell type (Hale et al., 1999). This is consistent with previous reports that p38γ is a muscle-specific protein (Lechner et al., 1996; Li et al., 1996). Furthermore, in terms of regulation it has been shown that p38α is preferentially activated by MKK-3 in PC-12 cells, whereas p38α is predominantly activated by MKK-6 in monocytes and KB cells suggesting that p38 is activated by different MKKs in a cell type-dependent manner (Hu et al., 1999). Unlike p38α, p38Δ was activated by MKK-3, -4, -6, and -7 in an approximately equal manner in 293 T cells, suggesting that the regulation of p38Δ may be distinct from p38α. However, it is still not clear whether the regulation of p38δ depends on cell type (Hu et al., 1999).
The role for p38Δ in airway structural and inflammatory cell function is not known. A role for p38Δ in inflammation was suggested recently by Jiang et al. (1997), who reported the activation of renal p38Δ during glomerulonephritis in rats. The widely used pyridinyl imidazole inhibitors of p38, SB 203580 and SB 202190, are nearly equipotent against p38α and p38β, but do not inhibit p38γ or p38Δ (Jiang et al., 1996, 1997; Stein et al., 1997; Wang et al., 1997; Kumar et al., 1997; Foey et al., 1998). Thus, if p38γ or p38Δ are the predominant isoforms involved in the release of TNF-α in BAL fluid following LPS inhalational challenge this could explain the apparent ineffectiveness of SB 203580. However, the development of inhibitors with greater selectivity for the different isoforms of p38 kinase will undoubtedly allow a more precise definition of the relative contribution of p38 kinases to airway inflammation and airway inflammatory diseases. Alternatively, this data may indicate that the release of TNF-α may not be solely controlled via the p38 kinase signal transduction pathway and could be due to the activation of other signalling pathways. In fact, both p38 and ERK are involved in LPS-induced TNF-α production from macrophages in vitro (Ajizian et al., 1999).
In conclusion, p38 kinase appears to be involved in the release of IL-1β and the associated sustained phase of neutrophilia following aerosolized LPS. This may suggest a role for p38 inhibitors in the treatment of airway inflammatory diseases (e.g. septic shock, chronic obstructive pulmonary disease and acute respiratory distress syndrome) associated with neutrophilia of the airways.
Acknowledgments
Many thanks to Ian McLay and John Souness for their technical and intellectual contribution.
Abbreviations
- BAL
bronchoalveolar lavage
- CMC
carboxymethyl cellulose
- CSBP
cytokine suppressive binding protein
- ELISA
enzyme-linked immunosorbant assay
- ERK
extracellular-regulated kinases
- FCS
foetal calf serum
- ICAM-1
intercellular adhesion molecule-1
- IL-1β
interleukin 1β
- JNK
c-Jun N-terminal kinase
- LPS
lipopolysaccharide
- MAPK
mitogen-activated kinase
- PBMC
peripheral blood mononuclear cells
- PBS
phosphate buffered saline
- PMSF
phenylmethyl sulphonyl fluoride
- RPMI
Roswell Park Memorial Institute
- SAPK
stress-activated protein kinases
- SDS – PAGE
SDS-polyacrylamide gel
- TNF-α
tumour necrosis factorα
References
- AJIZIAN S.J., ENGLISH B.K., MEALS E.A. Specific inhibitors of p38 and extracellular signal-regulated kinase and mitogen-activated protein kinase pathways block inducible nitric oxide synthase and tumor necrosis factor accumulation in murine macrophages stimulated with lipopolysaccharide and interferon-gamma. J. Infect. Dis. 1999;179:939–944. doi: 10.1086/314659. [DOI] [PubMed] [Google Scholar]
- BADGER A.M., BRADBEER J.N., VOTTA B., LEE J.C., ADAMS J.L., GRISWOLD D.E. Pharmacological profile of SB 203580, a selective inhibitor of cytokine suppressive binding protein/p38 kinase, in animal models of arthritis, bone resorption, endotoxin shock and immune function. J. Pharmacol. Exp. Ther. 1996;279:1453–1461. [PubMed] [Google Scholar]
- BALDASSARE J.J., BI Y., BELLONE C.J. The Role of p38 Mitogen-Activated Protein Kinase in IL-1β Transcription. J. Immunol. 1999;162:5367–5373. [PubMed] [Google Scholar]
- BECK-SCHIMMER B., SCHIMMER R.C., WARNER R.L., SCHMAL H., NORDBLOM G., FLORY C.M., LESCH M.E., FRIEDL H.P., SCHRIER D.J., WARD P.A. Expression of Lung Vascular and Airway ICAM-1 after Exposure to Bacterial Lipopolysaccharide. Am. J. Respir. Cell Mol. Biol. 1997;17:344–352. doi: 10.1165/ajrcmb.17.3.2861. [DOI] [PubMed] [Google Scholar]
- BIRRELL M.A., BATTRAM C.H., MCCLUSKIE K., PECORARO M., WEBBER S.E., FOSTER M.L., BELVISI M.G. Effect of the p38 kinase inhibitor, SB 203580, on sephadex induced airway inflammation in the rat. Br. J. Pharmacol. 2000;129:224P. doi: 10.1183/09031936.00.16594700. [DOI] [PubMed] [Google Scholar]
- BLASKI C.A., CLAPP W.D., THORNE P.S., QUINN T.J., WATT J.L., FRESS K.L., YAGLA S.J., SCHWARTZ P.A. The role of atopy in grain dust-induced airway disease. Am. J. Respir. Crit. Care Med. 1996;154:334–340. doi: 10.1164/ajrccm.154.2.8756803. [DOI] [PubMed] [Google Scholar]
- CARTER A.B., MONICK M.M., HUNNINGHAKE G.W. Both Erk and p38 Kinases Are Necessary for Cytokine Gene Transcription. Am. J. Respir. Cell Mol. Biol. 1999;20:751–758. doi: 10.1165/ajrcmb.20.4.3420. [DOI] [PubMed] [Google Scholar]
- CRAWLEY J.B., RAWLINSON L., LALI F.V., PAGE T.H., SAKLATVALA J., FOXWELL B.M.J. T cell proliferation in response to interleukins 2 and 7 requires p38 MAP kinase activation. J. Immunol. 1997;272:15023–15027. doi: 10.1074/jbc.272.23.15023. [DOI] [PubMed] [Google Scholar]
- CUENDA A., ROUSE J., DOZA Y.N., MEIER R., COHEN P., GALLAGHER T.F., YOUNG P.R., LEE J.C. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 1995;364:229–233. doi: 10.1016/0014-5793(95)00357-f. [DOI] [PubMed] [Google Scholar]
- DAVIS R.J. Transcriptional regulation by MAP kinases. Mol. Reprod. Dev. 1995;42:459–467. doi: 10.1002/mrd.1080420414. [DOI] [PubMed] [Google Scholar]
- DINARELLO C.A. Biologic basis for interleukin-1 in disease. Blood. 1996;87:2095–2147. [PubMed] [Google Scholar]
- ESCOTT K.J., BIRRELL M.A., BELVISI M.G., WEBBER S.E., FOSTER M.L., SARGENT C.A. Effect Of The P38 Kinase Inhibitor, SB 203580, On Allergic Airway Inflammation In The Rat. Br. J. Pharmacol. 2000;13:173–176. doi: 10.1038/sj.bjp.0703605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FOEY A.D., PARRY S.L., WILLIAMS L.M., FELDMANN M., FOXWELL B.M.J., BRENNAN D.F.M. Regulation of monocyte IL-10 synthesis by endogenous IL-1 and TNF-α: role of the p38 and p42/44 mitogen-activated protein kinases. J. Immunol. 1998;160:920–928. [PubMed] [Google Scholar]
- FUCHS H.J., DEBS R., PATTON J.S., LIGGITT H.D. The pattern of lung injury induced after exposure to tumor necrosis factor-α depends on the route of administration. Diagn. Microbiol. Infect. Dis. 1990;13:397–404. doi: 10.1016/0732-8893(90)90010-s. [DOI] [PubMed] [Google Scholar]
- GONÇALVES DE MORAES V.L., SINGER M., VARGAFTIG B.B., CHIGNARD M. Effects of rolipram on cyclic AMP levels in alveolar macrophages and lipopolysaccharide-induced inflammation in mouse lung. Br. J. Pharmacol. 1998;123:631–636. doi: 10.1038/sj.bjp.0701649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GONÇALVES DE MORAES V.L., VARGAFTIG B.B., LEFORT J., MEAGER A., CHIGNARD M. Effect of cyclo-oxygenase inhibitors and modulators of cyclic AMP formation on lipopolysaccharide-induced neutrophil infiltration in mouse lung. Br. J. Pharmacol. 1996;117:1792–1796. doi: 10.1111/j.1476-5381.1996.tb15356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GORDON T., BALMES J., FINE J., SHEPPARD D.D. Airway oedema and obstruction in guinea pigs exposed to inhaled endotoxin. Br. J. Ind. Med. 1991;48:629–635. doi: 10.1136/oem.48.9.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRISWOLD D.E., WEBB E.F., BADGER A.M., GORYCKI P.D., LEVANDOSKI P.A., BARNETTE M.A., GROUS M., CHRISTENSEN S., TORPHY T.J. SB 207499 (Ariflo), a second generation phosphodiesterase 4 inhibitor, reduces tumor necrosis factor alpha and interleukin-4 production in vivo. J. Pharmacol. Exp. Ther. 1998;287:705–711. [PubMed] [Google Scholar]
- HALE K.K., TROLLINGER D., RIHANEK M., MANTHEY C.L. Differential expression and activation of p38 Mitogen-Activated Protein Kinase α, β, γ, and Δ in inflammatory cell lineages. J. Immunol. 1999;162:4246–4252. [PubMed] [Google Scholar]
- HARMSEN A.G. Role of alveolar macrophages in lipopolysaccharide-induced neutrophil accumulation. Infect. Immun. 1988;56:1858–1863. doi: 10.1128/iai.56.8.1858-1863.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HU M.C., WANG Y.P., MIKHAIL A., QIU W.R., TAN T.H. Murine p38-delta mitogen-activated protein kinase, a developmentally regulated protein kinase that is activated by stress and proinflammatory cytokines. J. Biol. Chem. 1999;274:7095–7102. doi: 10.1074/jbc.274.11.7095. [DOI] [PubMed] [Google Scholar]
- JACKSON J.R., BOLOGNESE B., HILLEGASS L., KASSIS S., ADAMS J., GRISWOLD D.E., WINKLER J.D. Pharmacological effects of SB 220025, a selective inhibitor of p38 mitogen-activated protein kinase, in angiogenesis and chronic inflammatory disease models. J. Pharmacol. Exp. Ther. 1998;284:687–692. [PubMed] [Google Scholar]
- JIANG Y., CHEN C., LI Z., GUO W., GENGER J., LIN S., HANN J. Characterization of the structure and function of a new mitogen-activated protein kinase (p38β) J. Biol. Chem. 1996;271:17920–17926. doi: 10.1074/jbc.271.30.17920. [DOI] [PubMed] [Google Scholar]
- JIANG Y., GRAM H., ZHAO M., NEW L., GU J., FENG L., DI PADOVA F., ULEVITCH R., HAN J. Characterization of the structure and function of the fourth member of p38 group mitogen-activated protein kinases, p38Δ. J. Biol. Chem. 1997;272:30122–30128. doi: 10.1074/jbc.272.48.30122. [DOI] [PubMed] [Google Scholar]
- KARIN M. Mitogen-activated protein kinase cascades as regulators of stress responses. Ann. NY Acad. Sci. 1998;851:139–146. doi: 10.1111/j.1749-6632.1998.tb08987.x. [DOI] [PubMed] [Google Scholar]
- KOH Y., HYBERTSON B.M., JEPSON E.K., REPINE J.E. Tumor necrosis factor induced acute lung leak in rats: less than with interleukin-1. Inflammation. 1996;20:461–469. doi: 10.1007/BF01487039. [DOI] [PubMed] [Google Scholar]
- KOLLS J.K., LEI D., NELSON S., SUMMER W.R., GREENBERG S., BEUTLER B. Adenovirus-mediated blockade of tumor necrosis factor in mice protects against endotoxic shock yet impairs pulmonary host defense. J. Infect. Dis. 1995;171:570–575. doi: 10.1093/infdis/171.3.570. [DOI] [PubMed] [Google Scholar]
- KUMAR S., MCDONNELL P.C., GUM R.J., HAND A.T., LEE J.C., YOUNG P.R. Novel homologues of CSBP/p38 MAP kinase: activation, substrate specificity and sensitivity to inhibition by pyridinyl imidazoles. Biochem. Biophys. Res. Commun. 1997;235:533–538. doi: 10.1006/bbrc.1997.6849. [DOI] [PubMed] [Google Scholar]
- LECHNER C., ZAHYALKA M., GIOT J., MOLLER N., ULLRICH A. ERK6, a mitogen-activated protein kinase involved in C2C12 myoblast differentiation. Proc. Natl. Acad. Sci. U.S.A. 1996;93:4355–4359. doi: 10.1073/pnas.93.9.4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE J.C., ADAMS J.L. Inhibitors of serine/threonine kinases. Curr. Opin. Biotechnol. 1995;6:657–661. doi: 10.1016/0958-1669(95)80108-1. [DOI] [PubMed] [Google Scholar]
- LEE J.C., BADGER A.M., GRISWOLD D.E., DUNNINGTON D., TRUNEH A., VOTTA B., WHITE J.R., YOUNG P.R., BENDER P.E. Bicyclic imidazoles as a novel class of cytokine biosynthesis inhibitors. Ann. NY Acad. Sci. 1993;696:149–170. doi: 10.1111/j.1749-6632.1993.tb17149.x. [DOI] [PubMed] [Google Scholar]
- LEE J.C., LAYDON J.T., MCDONNELL P.C., GALLAGHER T.F., KUMAR S., GREEN D., MCNULTY D., BLUMENTHAL M.J., HEYS J.R., LANDVATTER S.W., STRICKLER J.E., MCLAUGHLIN M.M., SIEMENS L.R., FISHER S.M., LIVI G.P., WHITE J.R., ADAMS J.L., YOUNG P.R. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- LEE J.C., YOUNG P.R. Role of CSBP/p38/RK stress response kinase in LPS and cytokine signaling mechanisms. J. Leukocyte Biol. 1996;59:152–157. doi: 10.1002/jlb.59.2.152. [DOI] [PubMed] [Google Scholar]
- LI Z., JIANG Y., ULEVITCH R.J., HAN J. The primary structure of p38γ: a new member of the p38 group of MAP kinases. Biochem. Biophys. Res. Commun. 1996;228:334–340. doi: 10.1006/bbrc.1996.1662. [DOI] [PubMed] [Google Scholar]
- MANTHEY C.L., WANG S.-W., KINNEY S.D., YAO Z. p38 mitogen-activated protein kinase plays a major role in LPS-induced mRNA expression in monocytes. J. Leukocyte Biol. 1998;64:409–417. doi: 10.1002/jlb.64.3.409. [DOI] [PubMed] [Google Scholar]
- MICHEL O., NAGY A.M., SCHROEVEN M., DUCHATEAU J., NEVE J., FONDU P., SERGYSELS R. Dose-response relationship to inhaled endotoxin in normal subjects. Am. J. Respir. Crit. Care Med. 1997;156:1157–1164. doi: 10.1164/ajrccm.156.4.97-02002. [DOI] [PubMed] [Google Scholar]
- MILLER-LARSSON A., RUNSTRÖM A., BRATTSAND R. Adrenalectomy permits a late, local TNF-a release in LPS-challenged rat airways. Eur. Respir. J. 1999;13:1310–1317. [PubMed] [Google Scholar]
- MULLIGAN M.S., VAPORCIYAN A.A., MIYASAKA M., TAMATANI T., WARD P.A. Tumor necrosis factor-alpha regulates in vivo intrapulmonary expression of ICAM-1. Am. J. Pathol. 1993;142:1739–1749. [PMC free article] [PubMed] [Google Scholar]
- NICK J.A., AVDI N.J., YOUNG S.K., LEHMAN L.A., MCDONALD P.P., FRASCH S.C., BILLSTROM M.A., HENSON P.M., JOHNSON G.L., WORTHEN G.S. Selective activation and functional significance of p38α mitogen-activated protein kinase in lipopolysaccharide-stimulated neutrophils. J. Clin. Invest. 1999;103:851–858. doi: 10.1172/JCI5257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIGHTENGALE J.A., ROGERS D.F., HART L.A., KHAIRITONOV S.A., CHUNG K.F., BARNES P.J. Effect of inhaled endotoxin on induced sputum in normal, atopic and atopic asthmatic subjects. Thorax. 1998;53:563–571. doi: 10.1136/thx.53.7.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'LEARY E.C., MARDER P., ZUCKERMAN S.H. Glucocorticoid effects in an endotoxin-induced rat pulmonary inflammation model: differential effects on neutrophil influx, integrin expression, and inflammatory mediators. Am. J. Respir. Cell Mol. Biol. 1996;15:97–106. doi: 10.1165/ajrcmb.15.1.8679228. [DOI] [PubMed] [Google Scholar]
- PAUWELS R.A., KIPS J.C., PELEMAN R.A., VAN DER STRAETEN M.E. The effect of endotoxin inhalation on airway responsiveness and cellular influx in rats. Am. Rev. Respir. Dis. 1990;141:540–545. doi: 10.1164/ajrccm/141.3.540. [DOI] [PubMed] [Google Scholar]
- PETTIPHER E.R., LABASI J.M., SALTER E.D., STAM E.J., CHENG J.B., GRIFFITHS R.J. Regulation of tumour necrosis factor production by adrenal hormones in vivo: insights into the anti-inflammatory activity of rolipram. Br. J. Pharmacol. 1996;117:1530–1534. doi: 10.1111/j.1476-5381.1996.tb15317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRICHETT W., HAND A., SHEILDS J., DUNNINGTON D.D. Mechanism of action of bicyclic imidazoles defines a translational regulatory pathway for TNF-α. J. Inflamm. 1995;45:97–105. [PubMed] [Google Scholar]
- RIDLEY S.H., SARSFIELD S.J., LEE J.C., BIGG H.F., CAWSTON T.E., TAYLOR D.J., DEWITT D.L., SAKLATVALA J. Actions of IL-1 are selectively controlled by p38 mitogen-activated protein kinase: regulation of prostaglandin H synthase-2, metalloproteinases, and IL-6 at different levels. J. Immunol. 1997;158:3165–3173. [PubMed] [Google Scholar]
- SANDSTROM T., BJERMER L., RYLANDER R. Lipopolysaccharide (LPS) inhalation in healthy subjects causes bronchoalveolar neutrophilia, lymphocytosis, and fibronectin increase. Am. J. Ind. Med. 1994;25:103–104. doi: 10.1002/ajim.4700250127. [DOI] [PubMed] [Google Scholar]
- SKERRETT S.J., MARTIN T.R., CHI E.Y., PESCHON J.J., MOHLER K.M., WILSON C.B. Role of the type 1 TNF receptor in lung inflammation after inhalation of endotoxin or Pseudomonas aeruginosa. Am. J. Physiol. Lung Cell Mol. Physiol. 1999;276:L715–L727. doi: 10.1152/ajplung.1999.276.5.L715. [DOI] [PubMed] [Google Scholar]
- SOUNESS J.E., GRIFFIN M., MASLEN C., EBSWORTH K., SCOTT L.C., POLLOCK K., PALFREYMAN M.N., KARLSSON J-A. Evidence that cyclic AMP phophodiesterase inhibitors suppress TNFa generation from human monocytes by interacting with a ‘low-affinity' phosphodiesterase 4 conformer. Br. J. Pharmacol. 1996;118:649–658. doi: 10.1111/j.1476-5381.1996.tb15450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEIN B., YANG M.X., YOUNG D.B., JANKNECHT R., HUNTER T., MURRAY B.W., BARBOSA M.S. p38-2, a novel mitogen-activated protein kinase with distinct properties. J. Biol. Chem. 1997;272:19509–19517. doi: 10.1074/jbc.272.31.19509. [DOI] [PubMed] [Google Scholar]
- STRIETER R.M., KUNKEL S.L. Acute lung injury: the role of cytokines in the elicitation of neutrophils. J. Investig. Med. 1994;42:640–651. [PubMed] [Google Scholar]
- TANG G., WHITE J.E., LUMB P.D., LAWRENCE D.A., TSAN M.-F. Role of endogenous cytokines in endotoxin- and interleukin-1-induced pulmonary inflammatory response and oxygen tolerance. Am. J. Respir. Cell Mol. Biol. 1995a;12:339–344. doi: 10.1165/ajrcmb.12.3.7873200. [DOI] [PubMed] [Google Scholar]
- TANG W., YI E., REMICK D., WITTWER A. , YIN S., QI M., ULICH T. Intratracheal injection of endotoxin and cytokines: IX. Contribution of CD11a/ICAM-1 to neutrophil emigration. Am. J. Physiol. 1995b;269:L653–L659. doi: 10.1152/ajplung.1995.269.5.L653. [DOI] [PubMed] [Google Scholar]
- TONG L., PAV S., WHITE D.M., ROGERS S., CRANE K.M., CYWIN C.L., BROWN M.L., PARGELLIS C.A. A highly specific inhibitor of human p38 MAP kinase binds in the ATP pocket. Nat. Struc. Biol. 1997;4:311–316. doi: 10.1038/nsb0497-311. [DOI] [PubMed] [Google Scholar]
- TURNER C., ESSER K.M., WHEELDON E.B. Therapeutic intervention in a rat model of ARDS: IV. Phosphodiesterase IV inhibition. Cir. Shock. 1993;39:237–245. [PubMed] [Google Scholar]
- ULICH T.R., SONGMEI Y., GUO K., DEL CASTILLO J., EISENBERG S.P., THOMPSON R.C. The intratracheal administration of endotoxin and cytokines. III. The interleukin-1 (IL-1) receptor antagonist inhibits endotoxin- and IL-1-induced acute inflammation. Am. J. Pathol. 1991a;138:521–524. [PMC free article] [PubMed] [Google Scholar]
- ULICH T.R., WATSON L.R., YIN S.M., GUO K.Z., WANG P., THANG H., DEL CASTILLO J. The intratracheal administration of endotoxin and cytokines. I. Characterization of LPS-induced IL-1 and TNF mRNA expression and the LPS-, IL-1-, and TNF-induced inflammatory infiltrate. Am. J. Pathol. 1991b;138:1485–1496. [PMC free article] [PubMed] [Google Scholar]
- ULICH T.R., YI E.S., SMITH C., REMICK D. Intratracheal administration of endotoxin and cytokines VII. The soluble interleukin-1 receptor and the soluble tumor necrosis factor receptor II (p80) inhibit acute inflammation. Clin. Immunol. Immunopathol. 1994;72:137–140. doi: 10.1006/clin.1994.1117. [DOI] [PubMed] [Google Scholar]
- ULICH T.R., YIN S., REMICK D.G., RUSSELL D., EISENBERG S.P., KOHNO T. Intratracheal administration of endotoxin and cytokines IV. The soluble tumor necrosis factor receptor type 1 inhibits acute inflammation. Am. J. Pathol. 1993;142:1335–1338. [PMC free article] [PubMed] [Google Scholar]
- WANG X.S., DIENER K., MANTHEY C.L., WANG S.-W., ROSENSWEIG B., BRAY J., DELANEY J., COLE C.N., CHAN-HUI P.-Y., MANTLO N., LICHENSTEIN H.S., ZUKOWSKI M., YAO Z. Molecular cloning and characterization of a novel p38 mitogen-activated protein kinase. J. Biol. Chem. 1997;272:23668–23674. doi: 10.1074/jbc.272.38.23668. [DOI] [PubMed] [Google Scholar]
- WARREN J.S., YABROFF K.R., REMICK D.G., KUNKEL S.L., CHENSUE S.W., KUNKEL R.G., JOHNSON K.J., WARD P.A. Tumor necrosis factor participates in the pathogenesis of acute immune complex alveolitis in the rat. J. Clin. Invest. 1989;84:1873–1882. doi: 10.1172/JCI114374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WESSELIUS L.J., SMIRNOV I.M., O'BRIEN-LADNER A.R., NELSON M.E. Synergism of intratracheally administered tumor necrosis factor with interleukin-1 in the induction of lung edema in rats. J. Lab. Clin. Med. 1995;125:618–625. [PubMed] [Google Scholar]
- WOHLFORD-LENANE CL., DEETZ D.C., SCHWARTZD A. Cytokine gene expression after inhalation of corn dust. Am. J. Physiol. 1999;276:L736–L743. doi: 10.1152/ajplung.1999.276.5.L736. [DOI] [PubMed] [Google Scholar]
- YI E.S., REMICK D.G., LIM Y., TANG W., NADZIENKO A.B., YIN S., ULICH T.R. The intratracheal administration of endotoxin: X. Dexamethasone down-regulates neutrophil emigration and cytokine expression in vivo. Inflammation. 1996;20:165–175. doi: 10.1007/BF01487403. [DOI] [PubMed] [Google Scholar]
- YOUNG P., MCDONNELL P., DUNNINGTON D., HAND A., LAYDON J., LEE J. Pyridinyl imidazoles inhibit IL-1 and TNF production at the protein level. Agents Actions. 1993;39:C67–C69. doi: 10.1007/BF01972723. [DOI] [PubMed] [Google Scholar]
- YOUNG P.R., MCLAUGHLIN M.M., KUMAR S., KASSIS S., DOYLE M.L., MCNULTY D., GALLAGHER T.F., FISHER S., MCDONNELL P.C., CARR S.A., HUDDLESTON M.J., SEIBEL G., PORTER T.G., LIVI G.P., ADAMS J.L., LEE J.C. Pyridinyl imidazole inhibitors of p38 mitogen-activated protein kinase bind in the ATP site. J. Biol. Chem. 1997;272:12116–12121. doi: 10.1074/jbc.272.18.12116. [DOI] [PubMed] [Google Scholar]
- ZU Y.-L., QI J., GILCHRIST A., FERNANDEZ G.A., VAZQUEZ-ABOD D., KREUTZER D.L., HUANG C.-K., SHA'AFI R.I. p38 mitogen-activated protein kinase activation is required for human neutrophil function triggered by TNF-α or FMLP stimulation. J. Immunol. 1998;160:1982–1989. [PubMed] [Google Scholar]