

Abstract
We have studied the effects of four different phenol derivatives, with methyl and halogen substituents, on heterologously expressed human skeletal muscle sodium channels, in order to find structural determinants of blocking potency.
All compounds blocked skeletal muscle sodium channels in a concentration-dependent manner. The methylated phenol 3-methylphenol and the halogenated phenol 4-chlorophenol blocked sodium currents on depolarization from −100 mV to 0 mV with IC50 values of 2161 and 666 μM respectively. Methylation of the halogenated compound further increased potency, reducing the IC50 to 268 μM in 2-methyl-4-chlorophenol and to 150 μM in 3,5-dimethyl-4-chlorophenol.
Membrane depolarization before the test depolarization increased sodium channel blockade. When depolarizations were started from −70 mV or when a 2.5 s prepulse was introduced before the test pulse inducing slow inactivation, the IC50 was reduced more than 3 fold in all compounds. The values of KD for the fast-inactivated state derived from drug-induced shifts in steady-state availability curves were 14 μM for 3,5-dimethyl-4-chlorophenol, 19 μM for 2-methyl-4-chlorophenol, 26 μM for 4-chlorophenol and 115 μM for 3-methylphenol.
All compounds accelerated the current decay during depolarization and slowed recovery from fast inactivation. No relevant frequency-dependent block after depolarizing pulses applied at 10, 50 and 100 Hz was detected for any of the compounds.
All the phenol derivatives that we examined are effective blockers of skeletal muscle sodium channels, especially in conditions that are associated with membrane depolarization. Blocking potency is increased by halogenation and by methylation with increasing numbers of methyl groups.
Keywords: phenol derivatives, voltage-gated sodium channels, skeletal muscle
Introduction
The structural units that are characteristic of class I antiarrhythmic drugs and local anaesthetics are a hydrophobic aromatic group connected via an intermediate chain to a hydrophilic amine group (Ehring et al., 1988). In most of these drugs, the hydrophobic aromatic group is represented by a substituted phenol. Several studies have addressed the structural requirements for pharmacological effects (Ehring et al., 1988; Sheldon et al., 1991), but have not provided clues about which parts of the lidocaine molecule are responsible for its state-dependent block of voltage-operated sodium channels. The approach of dissecting the lidocaine molecule into phenol and diethylamide (Zamponi & French, 1993) did not take into account the fact that the aromatic group of the parent compound is a methylated phenol derivative. Although phenol block mimicked slow block of cardiac sodium channels seen with lidocaine, blocking potency was an order of magnitude lower and skeletal muscle sodium channels were only minimally affected.
In contrast, the halogenated and methylated phenol derivative 4-chloro-m-cresol (3-methyl-4-chlorophenol) turned out to be as effective as lidocaine in blocking skeletal muscle sodium channels (Haeseler et al., 1999). Analogous to lidocaine (Bean et al., 1983), 4-chloro-m-cresol showed higher affinity to inactivated channel states, but differed from lidocaine in that it accelerated the current decay during depolarization and that recovery from inactivation in the presence of drug was too fast to accumulated frequency-dependent block, even at high stimulating frequencies (Haeseler et al., 1999). We studied the effects of four structurally closely related compounds on voltage-gated skeletal muscle sodium channels. The aim was to elucidate the contribution of halogenation and/or methylation at the benzene ring to their blocking potency and their kinetics of binding and unbinding.
Methods
Molecular biology
Wild-type α-subunits of human skeletal muscle sodium channels were heterologously expressed in human embryonic kidney (HEK 293) cells, a stable cell line since 1962 (American Tissue Culture Collection CRL 1573). Transfection was performed using calcium phosphate precipitation (Graham & Van der Eb, 1973). Permanent expression was achieved by selection for resistance to the aminoglycoside antibiotic geneticin G418 (Life Technology, Eggenstein, Germany) (Mitrovic et al., 1994). Transfected cells were a gift from Prof Lehmann-Horn, Ulm, Germany. Successful channel expression was verified electrophysiologically. The clone has been used in several investigations (Haeseler et al., 1999; 2000; Mitrovic et al., 1994).
Solutions
The structures of all drugs examined are depicted in Figure 1. 3,5-dimethyl-4-chlorophenol and 3-methylphenol were purchased from Sigma Chemicals, Deisenhofen, Germany; 2-methyl-4-chlorophenol and 4-chlorophenol were from FLUKA, Deisenhofen, Germany. 3,5-Dimethyl-4-chlorophenol was prepared as a 1 M stock solution in methanol; 2-methyl-4-chlorophenol, 4-chlorophenol, and 3-methylphenol were dissolved directly in the bath solution immediately before the experiments. Concentrations were calculated from the amount injected into the glass vials. Drug-containing solutions were protected from light and were vigorously vortexed for 60 min. The solution was applied via a glass polytetrafluoroethylene perfusion system and a stainless steel superfusion pipette. The bath solution contained (mM) NaCl 140, MgCl2 1.0, KCl 4.0, CaCl2, HEPES 5.0, dextrose 5.0. Patch electrodes contained (mM) CsCl2 130, MgCl2 2.0, EGTA 5.0, HEPES 10. All solutions were adjusted to 290 mosm/l by the addition of mannitol and to pH 7.4 by the addition of CsOH2.
Figure 1.
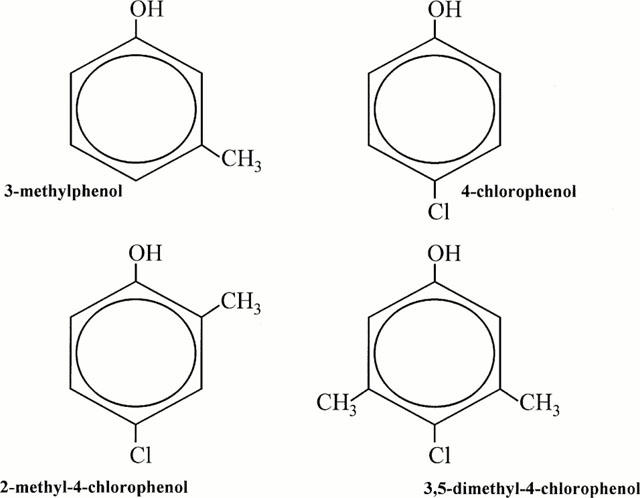
Structures of the phenol derivatives 3,5-dimethyl-4-chlorophenol, 2-methyl-4-chlorophenol, 4-chlorophenol and 3-methylphenol.
Experimental set-up
Standard whole-cell voltage-clamp experiments (Hamill et al., 1981) were performed at 20°C. Each experiment consisted of test recordings with the drug present at only one concentration, and of drug-free control recordings before and after the test. Each cell was exposed to one test concentration only. At least four experiments were performed at each concentration. The amount of the diluent methanol corresponding to the test concentration of 3,5-dimethyl-4-chlorophenol was added to the control solution. Patched cells were lifted into the visible stream of either bath solution or test solution, applied via a two-channel superfusion pipette close to the cell. To ensure adequate adjustment of the application device, one test experiment in distilled water reducing inward sodium current to zero was performed every 6 – 10 experiments.
Current recordings and analysis
For data acquisition and further analysis we used the EPC9 digitally-controlled amplifier in combination with Pulse and Pulse Fit software (HEKA Electronics, Lambrecht, Germany). The EPC9 provides automatic subtraction of capacitive and leakage currents by means of a prepulse protocol. The data were filtered at 10 kHz and digitized at 20 μs per point. Input resistance of the patch pipettes was at 1.8 – 2.5 MΩ. We used only small cells with capacitances of 9 – 15 pF; residual series resistance (after 50% compensation) was 1.2 – 2.5 MΩ; experiments with a rise in series resistance were rejected. The time constant of the voltage settling within the membrane (residual series resistance×cell capacitance) was less than 35 μs. To minimize a possible contribution of endogenous Na+ channels in HEK cells that conduct with amplitudes ranging from 50 to 350 pA (Mitrovic et al., 1994), we only analysed currents ranging between 1 and 7 nA. To minimize time-dependent shifts in the voltage-dependence of steady-state inactivation (Wang et al., 1996), all test experiments were performed within 5 min of patch rupture. Under these experimental conditions, time-dependent hyperpolarizing shifts in control conditions were less than −2 mV (Haeseler et al., 2000). Voltage activated currents were studied by applying different voltage-clamp protocols described in the results section or in the appropriated figure legends. Either exponential functions [I(t)=a0+a1exp(−t/τh1)+a2exp(−t/τh2)] or Boltzmann functions [I/Imax=(1+exp(−zF(Vtest−V0.5)/RT))−1] were fitted to the data, using a non-linear least-squares Marquardt – Levenberg algorithm, yielding the time constant τ of inactivation and recovery from inactivation, the membrane potential at half-maximum channel availability (V0.5), and the slope factor z of the steady-state availability curve. F is Faraday's constant (9.6487×104 C mol−1), R is the gas constant (8.315 J K−1 mol−1), and T is the temperature in degrees Kelvin. Drug effects on the peak current amplitude were assessed at different holding potentials (−70, −100 and −150 mV), or when a 2.5 s prepulse to −35 mV was introduced before the test pulse at −100 mV in order to induce slow inactivation. The residual sodium current (INa+) in the presence of drug (with respect to the current amplitude in control solution) was plotted against the applied concentration of each drug [C]. Fits of the Hill equation [INa+=(1+([C]/IC50)nH)−1] to the data yielded the concentration for half-maximum channel blockade (IC50) and the Hill coefficient nH. All data are presented as mean±s.d.
Results
Successful Na+ channel expression was verified electrophysiologically in almost all of the established whole-cell recordings. A total of 83 cells were included in the study. Average currents in the control experiments after depolarization from −100 mV to 0 mV were −4.9±2.1 nA.
Suppression of peak sodium currents – differences in potency related to halogenation and methylation of the phenol ring
Maximum inward currents elicited by 10 ms pulses going from either −150 mV, −100 mV, or −70 mV to 0 mV were reversibly suppressed by all substances in a concentration-dependent manner. A steady-state of block, tested with infrequent pulses, was reached within 60 s after the start of perfusion with the drug-containing solution. The currents in the presence of drug were normalized to the respective current elicited in control conditions. Normalized currents derived from at least four different experiments for each drug concentration were averaged to establish concentration-response plots at three different holding potentials (see Figure 2). The degree of suppression at all holding potentials increased with halogenation and with the number of methyl groups at the phenol ring. The phenol derivative 3-methylphenol, containing only one methyl group in the meta position with respect to the hydroxyl group, blocked inward sodium current at a holding potential of −100 mV, with an IC50 value of 2161 μM. The halogenated compound 4-chlorophenol was more potent than the methylated compound, and reduced the IC50 to 666 μM. Methylation in addition to halogenation further increased potency, reducing the IC50 about 2 fold for each methyl group inserted into the halogenated compound (268 μM for 2-methyl-4-chlorophenol and 156 μM for 3,5-dimethyl-4-chlorophenol).
Figure 2.
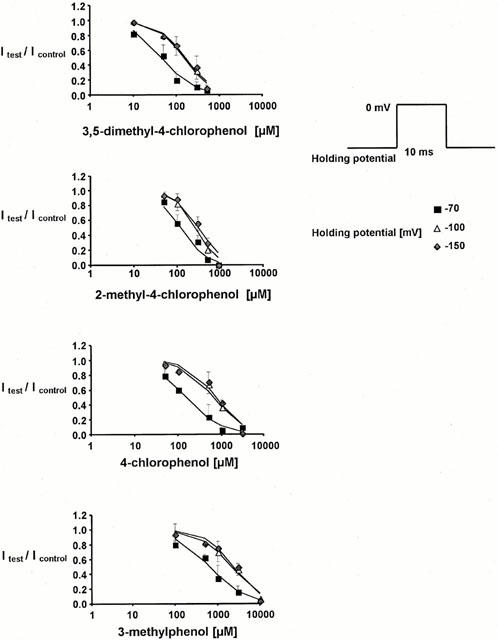
Concentration-dependent reduction in test pulse current with respect to control (Itest/Icontrol, mean±s.d.) induced by the different compounds. Drug concentrations on the x-axes were plotted on a logarithmic scale. The data were derived from at least four different experiments for each concentration tested. Depolarizing pulses to 0 mV (10 ms duration) were started from −150, −100, or −70 mV. Solid lines are Hill fits (INa+=[1+([C]/IC50)nH]−1) to the data. Concentration-response plots at −100 and −150 mV were nearly superimposable for all compounds, while the potency of the drugs was markedly increased at −70 mV. At the highest drug concentration examined in the respective compound, all the data at −100 and −150 mV are below the expected one from the Hill fit. It cannot be excluded that very high concentrations of the compounds have toxic effects on the membranes leading to an additional block due to irreversible damage.
Differences in potency at depolarized vs hyperpolarized holding potentials, and in the presence of slow inactivation
Concentration-response plots were nearly superimposable when depolarizations were started from −100 or −150 mV, respectively. The IC50 values derived from those experiments represent half-maximum concentrations required for block of resting channels. Upon depolarization, voltage-gated sodium channels assume non-conducting states that are kinetically distinct and may be characterized as ‘fast' or ‘slow' depending on the repolarization period needed for recovery. When depolarizations were started from −70 mV, where 20 – 30% of channels are in the fast-inactivated state, the IC50 values were reduced 3 – 4 fold in all compounds, to 661 μM (3-methylphenol), 154 μM (4-chlorophenol), 119 μM (2-methyl-4-chlorophenol), and 46 μM (3,5-dimethyl-4-chlorophenol).
Prolonged depolarization induces a slow-inactivated state that requires much longer periods for recovery (>1 s), and thus, assumes particular importance in pathological conditions, such as ischaemia, in which tissues are depolarized for longer periods (Balser et al., 1996b). To examine drug effects in the presence of a definite fraction of slow-inactivated channels, we stepped up the holding potential from −100 mV to −35 mV for 2.5 s. The membrane potential was then returned to −100 mV for 10 ms, allowing recovery from fast inactivation. The availability of unblocked resting channels was assessed at −100 mV by a 40 ms test pulse to 0 mV. In control solution, the inactivating prepulse caused a reduction of the current elicited by the test pulse by 32±13%, owing to slow inactivation during the long conditioning prepulse, from which channels did not recover during the 10 ms repolarization. All compounds blocked the peak current amplitude at −100 mV in the presence of 32% slow inactivation (with respect to the current amplitude obtained with the same protocol in control solution) almost completely at concentrations slightly exceeding the IC50 for rest block (see Figure 3). The Hill plots (Figure 4) showed a significant increase in potency when 2.5 s inactivating pulses were before the test pulse. The IC50 values in the presence of slow inactivation were reduced about 3 fold to 40 μM (3,5-dimethyl-4-chlorophenol), 116 μM (2-methyl-4-chlorophenol), 184 μM (4-chlorophenol), and 667 μM (3-methylphenol).
Figure 3.
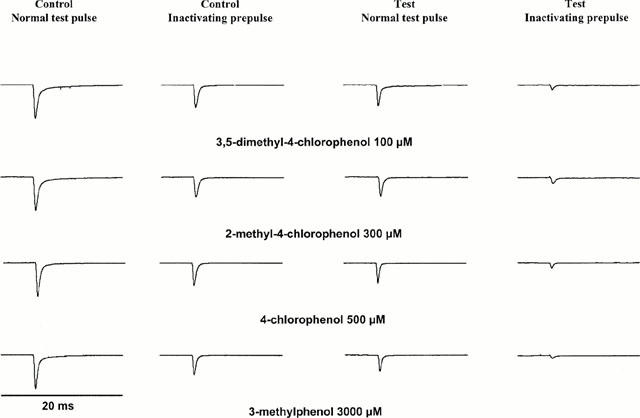
Representative current traces (in control conditions and in one representative concentration for each compound) evoked by 40 ms test pulses from −100 mV to 0 mV in the absence (traces 1 and 3) and presence (traces 2 and 4) of a 2.5 s inactivating prepulse introduced before the test pulse. All currents were normalized to the peak current in control conditions without an inactivating prepulse (trace 1). In control conditions, the inactivating prepulse caused a reduction in the current elicited by the test pulse, owing to slow inactivation, from which channels did not recover during the 10 ms repolarization (compare traces 1 and 2). Note that the channel blockade achieved by drug concentrations in the range of the IC50 for rest block was more than 90% with slow inactivation.
Figure 4.
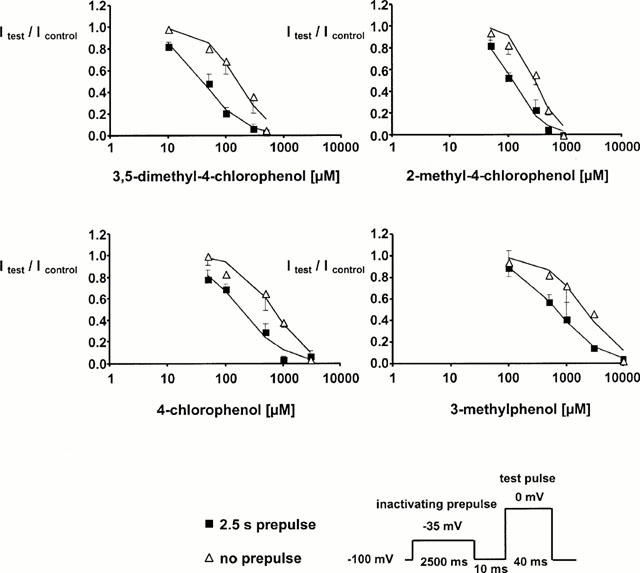
Concentration-dependent reduction in test pulse current with (filled squares) or without (empty triangles) a 2.5 s inactivating prepulse introduced before the test pulse (n>4, mean±s.d.). Drug concentrations on the x-axes were plotted on a logarithmic scale. Currents were normalized to the current elicited with the same protocol in control conditions. The solid lines are Hill fits to the data. The 2.5 s prepulse uniformly enhanced sensitivity to all compounds examined.
Calculated values for the Hill coefficients nH ranged from 1.1 to 1.5 with all protocols. Currents reached 0.72±0.1% of control values after 2 min of wash-out for all substances and drug concentrations, except the highest one.
Affinity for the fast-inactivated state estimated by steady-state availability shifts
After brief depolarizations, Na+ channels enter a fast-inactivated state, from which they cannot readily reopen. Steady-state availability curves assessed by a double-pulse protocol reflect the availability of Na+ channels to open on depolarization to 0 mV as a function of membrane potential. In control conditions, currents elicited by test pulses (Itest), starting from varying prepulse potentials Vtest (from −150 mV to −5 mV), normalized to the current elicited at the most hyperpolarized prepotential −150 mV (Imax), represent the relative fraction of channels that have not been inactivated during the 50 ms inactivating prepulse. In the presence of a channel blocker, the position of the steady-state availability curve relative to control conditions reflects drug association with channels in resting as well as fast-inactivated states. Boltzmann fits to the resulting current – voltage plots yield the membrane potential at half-maximum channel availability (V0.5) and the slope factor z: I/Imax=(1+exp(−zF(Vtest−V0.5)/RT))−1.
Summarized control data (n=65) showed that half of the channels were unavailable at −59.5±4.7 mV, because of fast inactivation. The slope factor z was −3.5±0.4. With exposure to all phenol derivatives, V0.5 was shifted considerably in the direction of more negative prepulse potentials; the degree of alteration showed concentration dependence. These drug-induced hyperpolarizing shifts (illustrated in Figure 5) reflect an additional reduction of channel availability induced by all compounds in the voltage range of channel inactivation compared with −150 mV. The voltage-dependence of inactivation, reflected by the slope factor z, remained unchanged in the presence of drug (see Figure 5). Assuming that the inactivated state has a higher binding affinity for a blocking drug than the resting state does, the higher amount of channel block achieved with consecutive membrane depolarization, revealed by the drug-induced hyperpolarizing shift, reflects the apportionment of channels between resting and fast-inactivated states, as well as the different binding affinities for the two channel states. For the example of the effect of lidocaine on Purkinje fibres, Bean et al. (1983) developed a model that predicted the shift in the midpoint of the steady-state inactivation curve as a function of lidocaine concentration, using estimates for the affinity of lidocaine for the resting and inactivated states, respectively derived from experiments at depolarized or hyperpolarized membrane potentials. We used the model developed by Bean et al. (1983) to estimate the dissociation constant (Kd) of the different drugs for the fast-inactivated state of the channel, fitting the concentration-dependence of V0.5 according to: ΔV0.5=k·ln[(1+[C]/ECR50)×(1+[C]/Kd)−1] where ΔV0.5 is the shift in the midpoint at each concentration of drug (mean, n>4), k the slope factor for the availability curve (k=−RT/Fz), [C] the applied drug concentration, ECR50 the concentration for half-maximum effect derived from the concentration – response plots at −150 mV membrane potential, and Kd the dissociation constant from the inactivated state (see Figure 5e, lower left). The estimated values of Kd were 14 μM (3,5-dimethyl-4-chlorophenol), 19 μM (2-methyl-4-chlorophenol), 26 μM (4-chlorophenol) and 115 μM (3-methylphenol).
Figure 5.
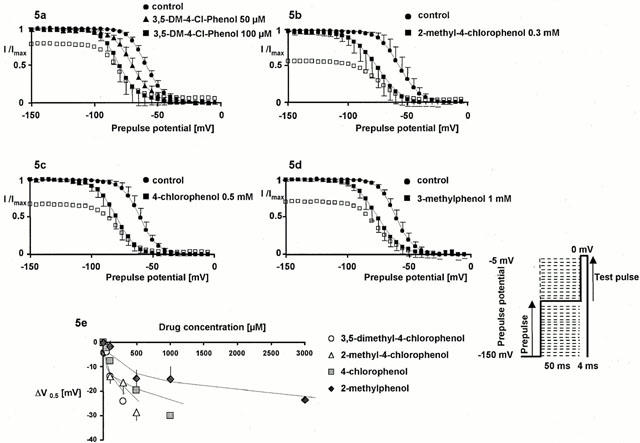
Interference of phenol derivatives with fast inactivated channels assessed by shifts in the steady-state availability curve. (a – d) Steady-state availability curves assessed by a two-pulse protocol in control conditions (circles) and in the presence of representative concentrations of each compound (triangles and squares). Each symbol represents the mean fractional current (n>4 for each concentration and each clone) elicited by a 4 ms test pulse to 0 mV, following a 50 ms inactivating prepulse from −150 mV to the indicated prepulse potential. Currents were normalized to maximum value (in each series at −150 mV prepotential); solid lines represent the best Boltzmann fit (I/Imax=(1+exp(−zF(Vtest−V0.5)/RT))−1) to the data. Error bars are standard deviations. In the presence of drug, currents were normalized either to the maximum value in the presence of drug (filled symbols) or to the maximum value in control conditions (empty symbols). All compounds shifted the midpoints of the curves in the direction of more negative prepulse potentials, reflecting an additional reduction in channel availability at depolarized prepotentials (compared with the blockade achieved at −150 mV). For 3,5-dimethyl-4-chlorophenol two representative concentrations were depicted to show the concentration dependence of this effect. (e) Concentration dependence of negative voltage shifts in the midpoints (ΔV0.5 [mV], mean±s.d.) of the steady-state availability plots relative to the starting values for all drugs examined. Solid lines are least-squares fits to the equation ΔV0.5=k·ln[(1+[C]/ECR50)×(1+[C]/Kd)−1], yielding the dissociation constant Kd from the inactivated state for each compound. Obviously, the fit rather tends to underestimate the shift in higher drug concentrations.
Acceleration of the Na+ current decay phase by phenol derivatives
To examine the time course of Na+ channel inactivation during a depolarization, 40 ms voltage steps from a holding potential of −100 mV to 0 mV were performed. The time constant of channel inactivation τh was obtained by fitting a single exponential to the decay of current during depolarizations: I(t)=a0+a1exp(−t/τh). In control conditions, τh was 0.43±0.08 ms (n=72). All phenol derivatives accelerated the decay of whole-cell currents. For all compounds, however, this effect was apparent only at concentrations that exceeded the IC50 values at a holding potential of −70 mV. Values obtained for τh in the presence of drug were: 0.27±0.02 ms in 50 μM 3,5-dimethyl-4-chlorophenol, 0.27±0.05 ms in 100 μM 2-methyl-4-chlorophenol, 0.23±0.03 ms in 500 μM 4-chlorophenol, and 0.30±0.08 ms in 1000 μM 3-methylphenol.
Effects of phenol derivatives on recovery from fast inactivation
After inactivation, channel reopenings are impossible until the channels recover from inactivation, a process that requires several ms after membrane repolarization. Further information about drug effects on the stability of the fast-inactivated state or the kinetics of drug dissociation from the fast-inactivated state can be derived from the rate at which the channels recover from inactivation in the presence of the drug. The time of membrane repolarization required to remove fast inactivation was assessed at −100 mV by a two-pulse protocol with varying time intervals (up to 100 ms) between the inactivating prepulse and the test pulse (see Figure 6). The time constants of recovery, τrec, were derived from monoexponential or biexponential fits to the fractional current after recovery from inactivation, plotted against the time interval between the inactivating prepulse and the test pulse: I(t)=a0+a1exp(−t/τrec1)+a2exp(−t/τrec2). Without drug, the data fitted well to a monoexponential, yielding a time constant, τrec1, of 2.3±0.7 ms (n=34). In the presence of drug, the fit contained a second, slow component of recovery, τrec2 of 94±8 ms (3,5-dimethyl-4-chlorophenol), 36±5 ms (2-methyl-4-chlorophenol), and 30±0.1 ms (4-chlorophenol and 3-methylphenol). For all drugs, however, the slow component made up less than 10% of the current amplitude at concentrations close to the IC50 for rest block. The fast component, τrec1, was prolonged to 3.8±0.8 ms in 100 μM 3,5-dimethyl-4-chlorophenol, to 4.3±1.6 ms in 300 μM 2-methyl-4-chlorophenol, to 3.0±0.7 ms in 500 μM 4-chlorophenol, and to 2.8±0.6 ms in 1000 μM 3-methylphenol. Figure 6 shows the time-course of recovery from fast inactivation with and without 100 μM 3,5-dimethyl-4-chlorophenol.
Figure 6.
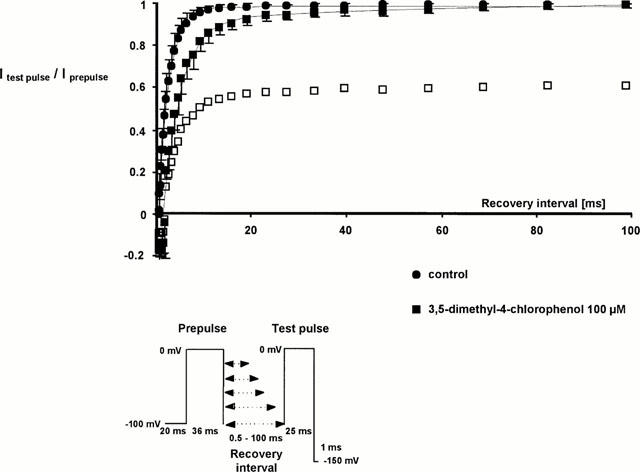
Recovery from fast inactivation assessed by a two-pulse protocol in control conditions (circles) and in the presence of 100 μM 3,5-dimethyl-4-chlorophenol (squares). The abscissa represents the recovery time interval between prepulse and test pulse (up to 100 ms), the ordinate represents the fractional current (mean±s.d., n=5) after recovery from fast inactivation, induced by the prepulse in the same series. In the presence of drug, currents were normalized either to the prepulse in the presence of drug (filled symbols) or in the corresponding control conditions (empty symbols). Solid lines are exponential fits I(t)=a0+a1exp(−t/τh1)+a2exp(−t/τh2) to the fractional currents after recovery from inactivation or inactivated channel block. Without drug, the data fitted to a monoexponential. In the presence of drug, recovery was delayed and contained a second slow component, which made up 11% of the current amplitude.
Frequency-dependent block
The accumulation of block during trains of depolarizing pulses indicates that the interval between pulses is too short to allow recovery of Na+ channel availability. To derive an estimate of the kinetics of drug binding and unbinding during the interpulse interval, we applied series of 1 – 10 ms depolarizing pulses from −100 mV to 0 mV at high frequencies (10, 50, and 100 Hz). Frequency-dependent block was defined as the additional reduction in INa+ for the last pulse relative to the first pulse in a test train in the presence of drug. In control conditions, the amplitude of the last pulse relative to the first pulse in a test train was 99±1% at 10 Hz and 96±3% at 50 and 100 Hz. Neither compound induced frequency-dependent block over 10% at 10 Hz. At 50 and 100 Hz, only concentrations exceeding the IC50 for rest block produced a small amount of frequency-dependent block. During a 100 Hz train, the additional fall relative to the first pulse was 17±5% in 300 μM 3,5-dimethyl-4-chlorophenol, 16±5% in 500 μM 2-methyl-4-chlorophenol, 13±2% in 1000 μM 4-chlorophenol, and 8±2% in 3000 μM 3-methylphenol.
Discussion
The principal new finding in this work is that the addition of one to three substituents in the phenol molecule is sufficient to form compounds with equal or higher potency in blocking voltage-operated muscle sodium channels than lidocaine. For example, the IC50 value for lidocaine block of heterologously expressed muscle sodium channels at −100 mV was 500 μM (Fan et al., 1996), compared with 150 μM for 3,5-dimethyl-4-chlorophenol. The blocking potency of phenol derivatives is increased up to 30 fold by halogenation and by increasing the numbers of methyl groups on the benzene ring. In addition, we have shown that, despite the differences in potency, voltage-dependent block by all compounds retains a characteristic set of features that describes local anaesthetic block (Balser et al., 1996a, 1996b; Bean et al., 1983; Fan et al., 1996; Scheuer, 1999).
Comparison with previous work shows that compounds with a methyl group attached directly to the benzene ring (e.g. 3-methylphenol) are more potent that compounds with a methyl group inserted between the ring and the phenolic hydroxyl group (benzylalcohol, (Haeseler et al., 2000)). The halogenated compound 4-chlorophenol is even more potent than the methylated compound (3-methylphenol). Insertion of methyl groups into the halogenated compound increased potency about 2 fold with each methyl group; 2-methyl-4-chlorophenol, in which the methyl group is attached at position 2 of the benzene ring, is slightly more potent than 3-methyl-4-chlorophenol, in which the methyl group is attached at position 3 (Haeseler et al., 1999).
Thus, one can postulate that there are two important structural groups that confer reactivity on the molecule. Methyl groups, which partition in a hydrophobic pocket, are electron donors and most likely render the chloride at position 4 of the aromatic ring more electronegative (Zorzato et al., 1993). Substitution of the methyl groups with a bulkier side-chain might produce a more potent sodium channel blocker. In addition, the position of the methyl group (attached directly to the benzene ring or separating the benzene ring and the hydroxyl group), as well as halogenation in position 4 of the phenol ring, has a major effect on the acidity of the hydroxyl hydrogen and on the hydrogen bond donor versus acceptor character of the molecule (Abraham et al., 1994; Elliott & Elliott, 1997).
The actions of all phenolic compounds show remarkable parallels with those of lidocaine. In analogy to local anaesthetics and antidysrhythmic drugs such as lidocaine (Balser et al., 1996b; Pugsley & Goldin, 1998; Scheuer, 1999), the blocking potency of all phenol derivatives strongly depends on the kinetic state of the channel, reducing the IC50 values more than 3 fold when membrane depolarization before the test pulse induces either fast or slow channel inactivation. This finding might be particularly important in pathological conditions, such as hypoxia or ischaemia, in which the muscle membrane is depolarized for longer periods (Lehmann-Horn & Rüdel, 1995). The estimated dissociation constants from the fast inactivated state were more than one order of magnitude below the respective concentration for half-maximum block of resting channels for all compounds. This means that preferential drug binding to inactivated channels is retained by all phenol derivatives, despite the structural differences that determine blocking potency.
However, all compounds differed from lidocaine in that equilibration of drug binding to inactivated with respect to resting channels was reached within several milliseconds. Recovery from inactivation in the presence of all phenol derivatives reached 90% within 15 ms. Rapid recovery from inactivation explains the lack of use-dependent block at stimulating frequencies below 50 Hz. Lack of use-dependent block has equally been described for the structurally related local anaesthetic benzocaine (Quan et al., 1996). In contrast, lidocaine induces substantial use-dependent block, even at lower stimulating frequencies than those used in our experiments (Fan et al., 1996). Structure – activity studies of different local anaesthetics and their derivatives have shown that the rate of dissociation from inactivated channels, which determines the accumulation of frequency-dependent block during repetitive stimulation, is related primarily to the size of the aliphatic side-chains (Quan et al., 1996), the molecular weight, and charge (Ehring et al., 1988). Thus, the lack of use-dependent block seen with all the phenol derivatives we have studied compared with lidocaine could be attributed to differences in molecular weight.
All compounds uniformly accelerated the current decay during depolarization at concentrations close to the respective IC50 at −70 mV. Although there seems to be a common underlying mechanism for this effect, the acceleration of the current decay in normally gating sodium channels induced by all phenol derivatives was only discernible in the case of lidocaine in mutated channels, in which fast inactivation was disabled and channel closure occurred on a time-scale of about 100 ms (Balser et al., 1996b). These results suggest that, in contrast to lidocaine, the kinetic of drug binding in the case of all phenol derivatives is fast enough to interfere with normal channel inactivation, which occurs on a time-scale of 1 – 2 ms. The underlying mechanism for this effect – allosteric stabilization of the inactivated state or open channel block – remains to be elucidated.
Our results show that phenol derivatives constitute a new group of sodium channel blockers. The potency of these compounds, even with only a single substituent in the benzene ring, may exceed the potency of lidocaine. All the phenol derivatives we have examined retain the principal features of local anaesthetic block described for lidocaine (Balser et al., 1996a, 1996b; Bean et al., 1983; Fan et al., 1996; Scheuer, 1999), although the kinetics of drug binding and unbinding are more than one order of magnitude faster.
Conclusions
In these studies of the effects of four different phenol derivatives, with methyl and halogen substituents, on heterologously expressed human skeletal muscle sodium channels, we have shown that substituted phenols block skeletal muscle sodium channels in a concentration-dependent manner. All the phenol derivatives that we examined were effective blockers of skeletal muscle sodium channels, especially in conditions that are associated with membrane depolarization. Blocking potency was increased by halogenation and by methylation with increasing numbers of methyl groups.
Acknowledgments
We are indebted to Prof Lehmann-Horn, Ulm, for providing us with transfected cells. We thank Birgitt Nentwig, Department of Anaesthesiology, Hannover, for taking care of the cell culture, Dr Horst Rückoldt and Dr Burkhard Vangerow, Department of Anaesthesiology, Hannover, for help with software problems, and Jobst Kilian, Department of Neurology, for technical support.
Abbreviations
- ECR50
drug concentration producing half-maximum effect at hyperpolarized membrane potentials
- F
Faraday's constant (9.6487×104 C mol−1)
- R
Gas constant (8.315 J K−1 mol−1)
References
- ABRAHAM M.H., CHADHA H.S., WHITING G.S., MITCHELL R.C. Hydrogen bonding. 32. An analysis of water-octanol and water-alkane partitioning and the log p parameter of Seiler. J. Pharm. Sci. 1994;83:1085–1100. doi: 10.1002/jps.2600830806. [DOI] [PubMed] [Google Scholar]
- BALSER J.R., BRADLEY NUSS H., ORIAS D.W., JOHNS D.C., MARBAN E., TOMASELLI G.F., LAWRENCE J.H. Local anesthetics as effectors of allosteric gating. J. Clin. Invest. 1996a;98:2874–2886. doi: 10.1172/JCI119116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BALSER J.R., BRADLEY NUSS H., ROMASHKO D.N., MARBAN E., TOMASELLI G.F. Functional consequences of lidocaine binding to slow-inactivated sodium channels. J. Gen. Physiol. 1996b;107:643–658. doi: 10.1085/jgp.107.5.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BEAN B.P., COHEN C.J., TSIEN R.W. Lidocaine block of cardiac sodium channels. J. Gen. Physiol. 1983;81:613–642. doi: 10.1085/jgp.81.5.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EHRING G.R., MOYER J.W., HONDEGHEM L.M. Quantitative structure activity studies of antiarrhythmic properties in a series of lidocaine and procainamide derivatives. J. Pharmacol. Exp. Therapeut. 1988;244:479–492. [PubMed] [Google Scholar]
- ELLIOTT A.A., ELLIOTT J.R. Voltage-dependent inhibition of RCK1 K+ channels by phenol, p-cresol, and benzyl alcohol. Mol. Pharmacol. 1997;51:475–483. [PubMed] [Google Scholar]
- FAN Z, , GEORGE A.L., KYLE J.W., MAKIELSKI J.C. Two human paramyotonia congenita mutations have opposite effects on lidocaine block of Na+ channels expressed in a mammalian cell line. J. Physiol. 1996;496:275–286. doi: 10.1113/jphysiol.1996.sp021684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRAHAM F.L., VAN DER EB A.J. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology. 1973;52:456–467. doi: 10.1016/0042-6822(73)90341-3. [DOI] [PubMed] [Google Scholar]
- HAESELER G., LEUWER M., KAVAN J., WÜRZ A., DENGLER R., PIEPENBROCK S. Voltage-dependent block of normal and mutant muscle sodium channels by 4-chloro-m-cresol. Br. J. Pharmacol. 1999;128:1259–1267. doi: 10.1038/sj.bjp.0702896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAESELER G., MAMARVAR M., BUFLER J., DENGLER R., HECKER H., ARONSON J.K., PIEPENBROCK S., LEUWER M. Voltage-dependent blockade of normal and mutant muscle sodium channels by benzylalcohol. Br. J. Pharmacol. 2000;130:1321–1330. doi: 10.1038/sj.bjp.0703447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAESELER G., PETZOLD J., HECKER H., WÜRZ A., DENGLER R., PIEPENBROCK S., LEUWER M. Succinylcholine metabolite succinic acid alters steady-state activation in muscle sodium channels. Anesthesiology. 2000;92:1385–1392. doi: 10.1097/00000542-200005000-00029. [DOI] [PubMed] [Google Scholar]
- HAMILL O.P., MARTY A., NEHER E., SAKMANN B., SIGWORTH F.J. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- LEHMANN-HORN F., RÜDEL R. Hereditary nondystrophic myotonias and periodic paralyses. Curr. Op. Neurol. 1995;8:402–410. doi: 10.1097/00019052-199510000-00014. [DOI] [PubMed] [Google Scholar]
- MITROVIC N., GEORGE A.L., HEINE R., WAGNER S., PIKA U., HARTLAUB U., ZHOU M., LERCHE H., FAHLKE C., LEHMANN-HORN F. K+-aggravated myotonia: destabilization of the inactivated state of the human muscle sodium channel by the V1589M mutation. J. Physiol. 1994;478:395–402. doi: 10.1113/jphysiol.1994.sp020260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PUGSLEY M.K., GOLDIN A.L. Effects of bisaramil, a novel class I antiarrhythmic agent, on heart, skeletal muscle and brain Na+ channels. Eur. J. Pharmacol. 1998;342:93–104. doi: 10.1016/s0014-2999(97)01420-9. [DOI] [PubMed] [Google Scholar]
- QUAN C., MOK W.M., WANG G.K. Use-dependent inhibition of Na+ currents by benzocaine homologs. Biophys. J. 1996;70:194–201. doi: 10.1016/S0006-3495(96)79563-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHEUER T. A revised view of local anesthetic action: What channel state is really stabilized. J. Gen. Physiol. 1999;113:3–6. doi: 10.1085/jgp.113.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHELDON R.S., HILL R.J., TAOUIS M., WILSON L.M. Aminoalkyl structural requirements for interactions of lidocaine with the class I antiarrhythmic drug receptor on rat cardiac myocytes. Mol. Pharmacol. 1991;39:609–614. [PubMed] [Google Scholar]
- WANG D.W., GEORGE A.L., BENNETT P.B. Comparison of heterologously expressed human cardiac and skeletal muscle sodium channels. Biophys. J. 1996;70:238–245. doi: 10.1016/S0006-3495(96)79566-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZAMPONI G.W., FRENCH R.J. Dissecting lidocaine action: Diethylamide and phenol mimic separate modes of lidocaine block of sodium channels from heart and rat skeletal muscle. Biophys. J. 1993;65:2335–2347. doi: 10.1016/S0006-3495(93)81292-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZORZATO F., SCUTARI E., TEGAZZIN V., CLEMENTI E., TREVES S. Chlorocresol: An activator of ryanodine receptor-mediated Ca2+-release. Mol. Pharmacol. 1993;44:1192–1201. [PubMed] [Google Scholar]