

Abstract
The endogenous cannabinoid receptor agonist anandamide (AEA) and the related compound palmitoylethanolamide (PEA) are inactivated by transport into cells followed by metabolism by fatty acid amide hydrolase (FAAH). The cellular uptake of AEA has been characterized in detail, whereas less is known about the properties of the PEA uptake, in particular in neuronal cells. In the present study, the pharmacological and functional properties of PEA and AEA uptake have been investigated in mouse Neuro-2a neuroblastoma and, for comparison, in rat RBL-2H3 basophilic leukaemia cells.
Saturable uptake of PEA and AEA into both cell lines were demonstrated with apparent KM values of 28 μM (PEA) and 10 μM (AEA) in Neuro-2a cells, and 30 μM (PEA) and 9.3 μM (AEA) in RBL-2H3 cells. Both PEA and AEA uptake showed temperature-dependence but only the AEA uptake was sensitive to treatment with Pronase and phenylmethylsulfonyl fluoride.
The AEA uptake was inhibited by AM404, 2-arachidonoylglycerol (2-AG), R1- and S1-methanandamide, arachidonic acid and olvanil with similar potencies for the two cell types. PEA, up to a concentration of 100 μM, did not affect AEA uptake in either cell line. AEA, 2-AG, arachidonic acid, R1-methanandamide, Δ9-THC, and cannabidiol inhibited PEA transport in both cell lines. The non-steroidal anti-inflammatory drug indomethacin inhibited the AEA uptake but had very weak effects on the uptake of PEA.
From these data, it can be concluded that PEA is transported in to cells both by passive diffusion and by a facilitated transport that is pharmacologically distinguishable from AEA uptake.
Keywords: Palmitoylethanolamide, anandamide, transport, Neuro-2a cells, RBL-2H3 cells, fatty acid amide hydrolase
Introduction
Arachidonoylethanolamide (anandamide, AEA), 2-arachidonoylglycerol (2-AG), and palmitoylethanolamide (PEA) belong to an emerging class of endogenous lipids including amides and esters of long chain polyunsaturated fatty acids, termed collectively cannabimimetic fatty acid derivatives (CFADs). AEA and 2-AG mimic the pharmacological effects of Δ9-tetrahydrocannabinol (Δ9-THC), the main active constituent of hashish and marijuana (Gaoni & Mechoulam, 1964), by binding to cannabinoid receptors (Di Marzo & Deutsch, 1998). To date, two cannabinoid receptor subtypes have been characterized, designated CB1 and CB2 (Matsuda et al., 1990; Munro et al., 1993), and both AEA and 2-AG bind to both receptor subypes (for a review, see Mechoulam et al., 1998).
With regard to PEA, Facci et al. (1995) proposed that it acts as an endogenous physiological ligand for the CB2 receptor. However, more recent data have indicated that PEA does not bind with high affinity to either of the two cannabinoid receptor subtypes (Showalter et al., 1996; Sheskin et al., 1997; Lambert et al., 1999). Nevertheless, PEA is found in neural and non-neural tissues, and has an anti-inflammatory action in mast cells and basophils (Facci et al., 1995) as well as a purported neuroprotective effect in cerebellar granule cells (Skaper et al., 1996) (although this is not seen in chick neurons, Andersson et al., 2000). The PEA level increases several-fold in inflamed tissue (Natarajan et al., 1982; Kondo et al., 1998), and PEA has been hypothesized to act as an autacoid capable of locally antagonizing inflammatory processes (Facci et al., 1995; Mazzari et al., 1996). Furthermore, peripheral administration of PEA produces an analgesic effect in formalin-induced inflammatory pain (Jaggar et al., 1998; Calignano et al., 1998), and the PEA-induced anti-nociceptive effects are believed to be mediated by a peripheral CB2-like receptor. This is of particular pharmacological interest since PEA, in contrast to AEA, has no action on central CB1 receptors, and so is unlikely to produce unwanted psychotropic effects.
The possible therapeutic potential of PEA (and AEA), however, is hampered by their rapid cellular inactivation. This process includes cellular uptake and intracellular degradation by the enzyme fatty acid amide hydrolase (FAAH) (Deutsch & Chin, 1993; Di Marzo et al., 1994). Cellular uptake of AEA has been demonstrated and characterized in neural-derived cell lines (Deutsch & Chin, 1993; Piomelli et al., 1999), primary cultures of cerebellar granule cells (Hillard et al., 1997), striatal neurons and astrocytes (Di Marzo et al., 1994; Beltramo et al., 1997), as well as in the immortalized rat basophilic leukaemia cell line RBL-2H3 (Bisogno et al., 1997; Rakhshan et al., 2000). The uptake mechanism has been shown to be mediated by a saturable, selective, temperature-dependent and Na+-independent transporter (Di Marzo et al., 1994; Beltramo et al., 1997; Hillard et al., 1997), supporting the hypothesis that the AEA uptake is carrier-mediated.
To this date, only one report gives experimental evidence for a PEA ‘carrier' (Bisogno et al., 1997), and the study was performed using RBL-2H3 cells. By better understanding of how PEA is transported and inactivated by cells, new pharmacological agents can be developed that selectively interfere with the PEA inactivation, increase the life span of the molecule, and hence, potentiate its anti-inflammatory and anti-nociceptive effects. This prompted us to examine the cellular uptake of PEA and thus, the aims with the present study are to: (a) study if PEA (and AEA for comparison) is transported by mouse Neuro-2a neuroblastoma cells; (b) investigate if the neuronal PEA (and AEA) transport is similar to the transport mechanism found in cells with immune origin, represented by the cognate mast cell line RBL-2H3; (c) examine the pharmacological properties of the PEA uptake and directly compare it with the more characterized AEA uptake, and finally; (d) determine if the cellular PEA accumulation is mediated by a membrane transport-protein as opposed to a process of passive diffusion. A preliminary report of this study was presented at the 30th Annual Meeting of the Society for Neuroscience, New Orleans, 2000 (Jacobsson et al., 2000).
Methods
Chemicals
Arachidonoylethanolamide (anandamide, AEA) and (R)-(+)-arachidonyl-1′-hydroxy-2′-propylamide (R1-methanandamide) were purchased from Cayman Chemical Company (Ann Arbor, MI, U.S.A.) and Tocris Cookson (Bristol, U.K.). Palmitoylethanolamide (PEA) was purchased from Cayman Chemical Company and ICN Biomedical, Inc. (Aurora, OH, U.S.A.). Anandamide [etanolamine-1-3H] ([3H]-AEA; specific activity 20 Ci mmol−1) and palmitoylethanolamide [palmitoyl-9,10-3H] (([3H]-PEA; specific activity 60 Ci mmol−1) were synthesized by American Radiolabeled Chemicals, Inc. (St. Louis, MO, U.S.A.). [3H]-WIN 55,212-2 [5,7-3H] (specific activity 50.8 Ci mmol−1) was obtained from NEN, Life Sciences Products, Inc. (Boston, MA, U.S.A.). N-piperidin-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-3-pyrazole-carboxamide (SR 141716A) and N-[(1S)-endo-1,3,3-trimethyl bicyclo [2.2.1] heptan-2-yl]-5-(4-chloro-3-methylphenyl)-1-(4-methylbenzyl)-pyrazole-3-carboxamide (SR 144528) were obtained from Sanofi Recherche (Montpellier, France). 2-Arachidonoylglycerol (2-AG), N-(4-hydroxyphenyl)arachidonylamide (AM404), N-[(4-hydroxy-3-methoxyphenyl)methyl]-9Z-octadecenamide (Olvanil), and (S)-(−)-arachidonyl-1′-hydroxy-2′-propylamide (S1-methanandamide) were purchased from Cayman Chemical Company. Arachidonic acid, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), ethylenediaminetetracetic acid (EDTA), ibuprofen, indomethacin, phenylmethylsulfonyl fluoride (PMSF) and protease (Pronase; EC 3.4.24.31) from Streptomyces griseus (specific activity 4.7 units mg−1) were purchased from the Sigma Chemical Co. (St. Louis, MO, U.S.A.). Δ9-Tetrahydrocannabinol (Δ9-THC) and cannabidiol were obtained from RBI/Sigma (Natick, MA, U.S.A.). Cytotoxicity Detection Kit (LDH) was purchased from Roche Molecular Biochemicals (Mannheim, Germany). All cell culture media, sera and supplements were from Gibco/Life Technologies (Sweden).
Cell cultures
Rat basophilic leukaemia (RBL-2H3) cells (passage range 7 – 29) and mouse neuroblastoma (Neuro-2a) cells (passage range 181 – 209), obtained from the American Type Culture Collection (MD, U.S.A.), were grown in 75 cm2 culturing flasks at 37°C in a humidified atmosphere of 5% CO2 in air. RBL-2H3 cells were maintained in Eagle's minimum essential medium (MEM) with 15% foetal bovine serum (FBS), 2 mM L-glutamine, and 100 units ml−1 penicillin+100 μg ml−1 streptomycin (PEST). Neuro-2a cells were maintained in MEM supplemented with 10% FBS, 0.1 mM non-essential amino acids (NEAA) and PEST. The culture media were changed three times a week.
Determination of cellular [3H]-AEA and [3H]-PEA uptake
The method used is essentially that of Rakhshan et al. (2000). Briefly, Neuro-2a cells (cultured in MEM with 10% FBS, 1% PEST and 1% NEAA) or RBL-2H3 cells (cultured in MEM with 15% FBS and 1% PEST) were added to 24-well culture plates at a plating density of 2 – 3×105 cells/well. After an incubation period of 18 h, the wells were washed once with 0.5 ml warm assay buffer (mM): NaCl 120, KCl 4.7, CaCl2 2.2, HEPES 10, KH2PO4 1.2, MgSO4 1.2, pH 7.4; gassed with 95% O2, 5% CO2 and pre-incubated in 300 μl buffer at 37°C for 10 min. [3H]-AEA or [3H]-PEA were added to the wells (assay volume 400 μl), and uptake at 37°C was allowed for 15 min. Saturation kinetics were determined using increasing concentrations of [3H]-AEA or [3H]-PEA diluted with unlabelled AEA and PEA, respectively. For inhibition assays, the cells were pre-incubated with the test compounds at appropriate concentrations followed by the addition of 10 μM [3H]-AEA, or 20 μM [3H]-PEA. After the incubation phase, the plates were placed on ice, the buffer aspirated and the transport was terminated by washing the cells three times with ice-cold buffer containing 1% BSA. An aliquot (0.5 ml) of 0.2 M NaOH was added and the plates were incubated for 15 min at 75°C to solubilize the cells. Aliquots (300 μl) were taken and assayed for tritium content by liquid scintillation spectroscopy with quench correction. Blanks, defined as the radioactivity recovered in the absence of cells, were determined in wells on the same culture plates. Nonspecific cellular association of [3H]-AEA or [3H]-PEA was determined in the presence of either 100 μM unlabelled AEA of PEA, or in parallel incubations performed at 4°C. All compounds were serial diluted in ethanol, acetonitrile or buffer, depending on the recommendation of the supplier, and added to the cultures from stock solutions to yield a constant solvent concentration in each well.
To study the uptake during protein (and/or FAAH) inactivating conditions, Neuro-2a cells were pre-incubated with various concentrations of Pronase and PMSF, before initiating the [3H]-AEA and [3H]-PEA uptake assays.
Membrane preparation
Neuro-2a and RBL-2H3 cells grown to confluence in 75 cm2 culture flasks were incubated 10 min at 37°C in pre-warmed phosphate-buffered saline (PBS), pH 7.4, with 1 mM EDTA. The cells were dislodged and collected in 15 ml Falcon conical tubes. After centrifugation (10 min at 200×g), the pellet was resuspended in ice-cold 50 mM Tris-HCl buffer, pH 7.4, with 0.32 M sucrose and homogenized using a glass homogenizer. The homogenate was centrifuged at 36,000×g for 20 min at 4°C and the pellet sonicated using a Branson Sonifier (output 5, pulsed 50% duty cycle, 30 s) in distilled water and re-centrifuged as above. Frozen rat brain (minus cerebellum) samples were thawed and homogenized at 4°C in 20 mM HEPES buffer, pH 7.0, with 1 mM MgCl2 using a glass homogenizer and the homogenates centrifuged twice at 36,000×g for 20 min at 4°C. The cell and tissue pellets were re-suspended in HEPES buffer and incubated at 37°C for 15 min to degrade any endogenous ligands that might interfere with the binding assay. After centrifugation at 36,000×g for 20 min at 4°C, membranes were re-suspended in 50 mM Tris-HCl buffer, pH 7.4, containing 1 mM EDTA and 3 mM MgCl2. The membrane preparations were determined for protein content according to the method of Harrington (1990), using bovine serum albumin as standard, and stored at −70°C until used for assay.
CB receptor binding assay
The protocol of Thomas et al. (1998) was used, with minor modifications. Flat bottomed 96-well microtitre plates (Nalge Nunc International) were used in all assays, with a total volume of 250 μl in each well. Membranes (50 μg) of either rat brain or Neuro-2a or RBL-2H3 cells were incubated at 30°C for 60 min with 50 μl test compounds, 50 μl [3H]-WIN 55,212-2 (∼0.8 nM assay concentration) in 50 mM Tris-HCl buffer, pH 7.4, containing 1 mM EDTA, 3 mM MgCl2 and 0.5% bovine serum albumin (w/v). The test compounds and radioligand were diluted to the appropriate concentrations with assay buffer. The labelled membranes were harvested and rinsed by vacuum filtration (Brandel Cell Harvester) using pre-soaked (0.3% polyethylenimine) filters and ice-cold 50 mM Tris-HCl, pH 7.4, containing 0.1% (w/v) BSA as wash buffer. Each filter was placed in a 5 ml scintillation vial, and the tritium contents were measured by liquid scintillation spectroscopy with quench correction.
Cytotoxicity assays
Cell viability after the various treatments was assessed using either the LDH activity assay or the MTT reduction assay (Mosmann, 1983). The cellular release of LDH was measured by transferring 100 μl aliquots of the culture medium to an optically clear 96-well flat bottom microtiter plate. To each well, 100 μl of a Cytotoxicity Detection Kit assay mixture was added and the samples were measured spectrophotometrically at 490 nm (reference wavelength 650 nm) in a ThermoMax microplate reader (Molecular Devices). To determine the total amount of LDH in the cultures, the cells were lyzed in 0.1% Triton X-100 solution. LDH release was expressed as medium LDH activity/total LDH activity. The MTT assay is based on the conversion of the yellow tetrazolium salt MTT by mitochondrial dehydrogenase of live cells to the purple MTT formazan, which can be measured spectrophotometrically at 570 nm (Mosmann, 1983). To each well, 50 μl MTT dissolved in PBS was added (0.5 mg/ml final concentration), and the plate was incubated for 2 – 3 h. After the incubation, 500 μl MTT solubilization solution (0.1 M HCl and 10% Triton X-100 in isopropanol) was added, and the plates were mixed thoroughly on a SMI Vortexer for 20 – 30 min. Aliquots of 200 μl from each well were transferred to an optically clear 96-well flat bottom microtiter plate and read spectrophotometrically at 570 nm.
Data analyses
KM and Vmax values for the accumulation of AEA and PEA (Figure 1) were determined by fitting the data to the Michaelis – Menten equation using nonlinear regression (GraphPad Prism, GraphPad Software, San Diego, CA, U.S.A.). IC50 and pI50 values were determined by non-linear least square fitting of the data (one site competition), by the GraphPad Prism software. Residual association of [3H]-AEA (33% of total [3H]-AEA accumulation in Neuro-2a cells; 28%, RBL-2H3) and [3H]-PEA (63%, Neuro-2a; 54%, RBL-2H3), determined as the labelled AEA or PEA accumulation in the presence of 100 μM unlabelled AEA or PEA, respectively, was not subtracted. Instead, the computer-generated curve (Figures 4, 5, 6 and 7) was adjusted by setting the bottom plateau uptake (maximal inhibition) to the corresponding values above. For the inhibition of [3H]-WIN 55,212-2 binding by SR 141716A and SR 144528 (Figure 2), the total binding at each inhibitor concentration (but not in the absence of inhibitor) were analysed using the built-in equation ‘one site competition' of the GraphPad Prism software. Temperature- or compound-induced changes in AEA and PEA uptake and cell viability were expressed as the percentage of control levels and the data were analysed for statistical significant differences using one-way ANOVA across each treatment with Bonferroni (or Dunnett's) multiple comparison test (GraphPad Prism software).
Figure 1.
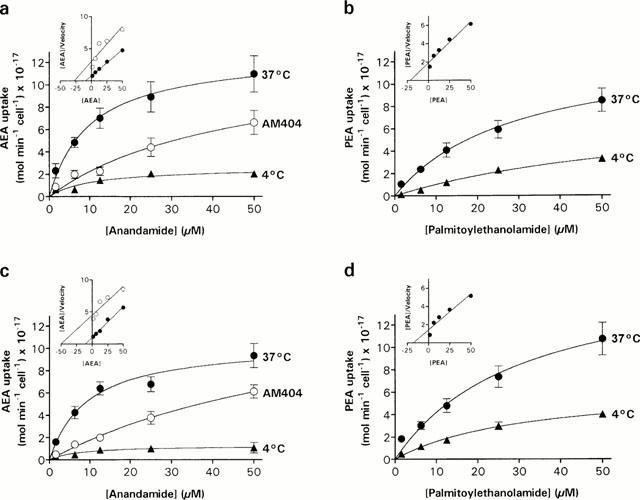
Saturation kinetics of AEA (a,c) and PEA (b,c) uptake in Neuro-2a cells (a,b) and RBL-2H3 cells (c,d). The cells were incubated with increasing concentrations of [3H]-AEA or [3H]-PEA for 15 min at 37°C or 4°C. The effect of 100 μM AM404 upon AEA uptake was determined by pre-incubating the cells for 15 min at 37°C with AM404 followed by the addition of [3H]-AEA. Each value represents the mean±s.e.mean (n=3 – 6). Insets, Hanes-Woolf plots of the respective saturation curves obtained at 37°C.
Figure 4.
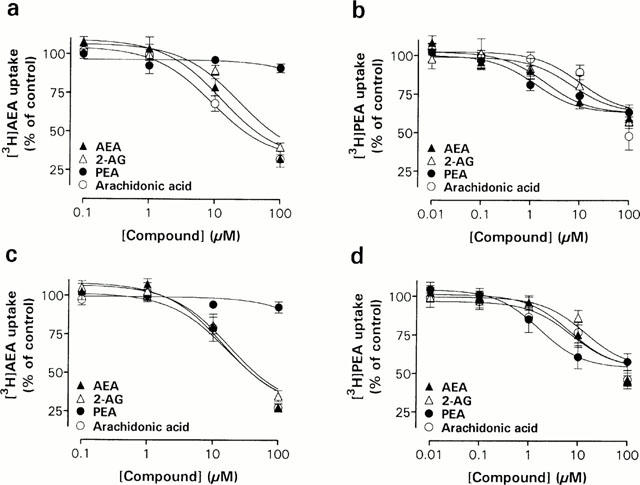
Concentration-dependent effects of putative endocannabinoids and arachidonic acid on [3H]-AEA (a,c) and [3H]-PEA (b,d) uptake in Neuro-2a cells (a,b) and RBL-2H3 cells (c,d). The cells were incubated 15 min with the indicated concentration of the test compound, followed by the addition of 10 μM [3H]-AEA or 20 μM [3H]-PEA. After 15 min at 37°C, the cellular uptake of AEA and PEA was determined. Data are plotted as percentage of total [3H]-AEA or [3H]-PEA uptake and represent means±s.e.mean (n=3 – 5).
Figure 5.
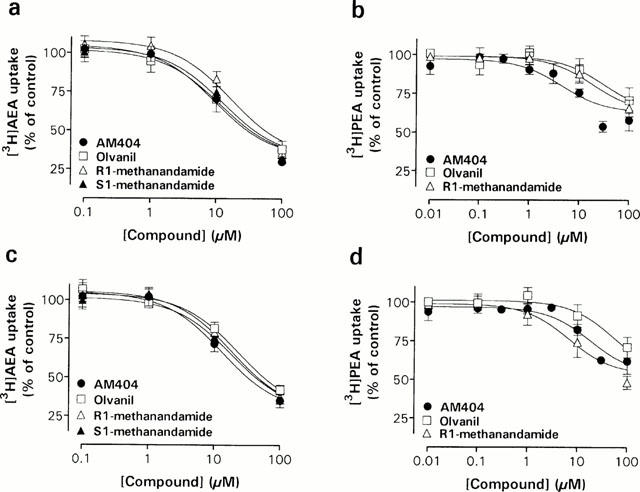
Concentration-dependent effects of AEA derivatives and the synthetic vanilloid agonist olvanil on [3H]-AEA (a,c) and [3H]-PEA (b,d) uptake in Neuro-2a cells (a,b) and RBL-2H3 cells (c,d). The experiments were undertaken as described in the legend to Figure 4. Data are plotted as percentage of total [3H]-AEA or [3H]-PEA uptake and represent means±s.e.mean (n=3 – 5).
Figure 6.
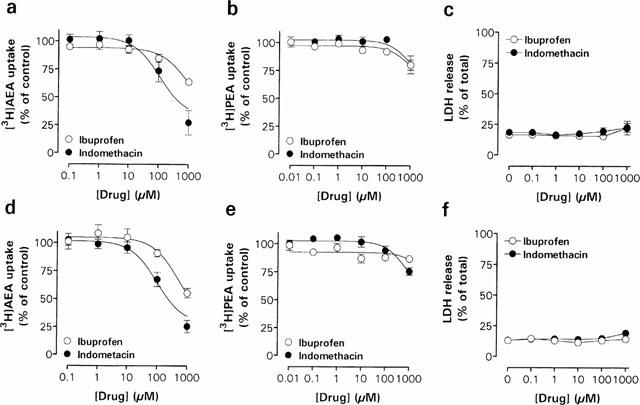
Concentration-dependent effects of the nonsteroidal anti-inflammatory drugs ibuprofen and indomethacin on [3H]-AEA (a,d) and [3H]-PEA (b,e) uptake and LDH release (c,f) in Neuro-2a cells (a,b,c) and RBL-2H3 cells (d,e,f). The experiments were undertaken as described in the legend to Figure 4. Data are plotted as percentage of total [3H]-AEA or [3H]-PEA uptake and represent means±s.e.mean (n=3 – 5).
Figure 7.
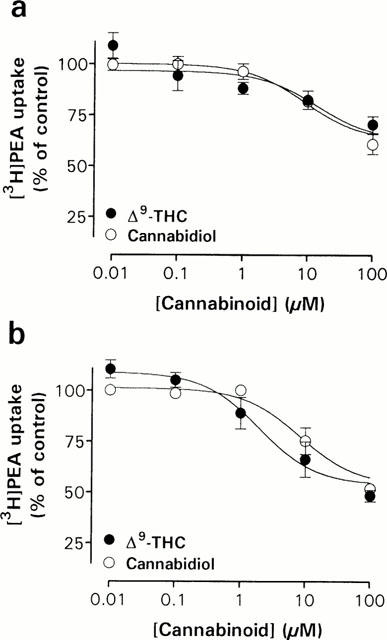
Concentration-dependent effects of cannabinoids on [3H]-PEA uptake in Neuro-2a cells (a) and RBL-2H3 cells (b). The experiments were undertaken as described in the legend to Figure 4. Data are plotted as percentage of total [3H]-PEA uptake and represent means±s.e.mean (n=3 – 5).
Figure 2.
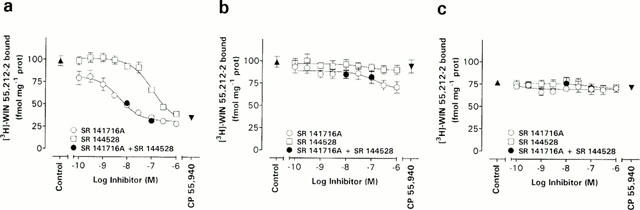
Competition of [3H]-WIN 55,212-2 binding to rat brain (a), Neuro-2a cell (b), and RBL-2H3 cell (c) membranes by the cannabinoid receptor antagonists SR 141716A and SR 144528. The control values were obtained in the absence of inhibitors and CP 55,940 values were obtained in the presence of 1 μM of this compound. Data represent the mean±s.e.mean (n=3). The radiolabelled ligand concentration used was in the range of 0.6 – 0.8 nM.
Results
AEA and PEA uptake in Neuro-2a and RBL-2H3 cells
Neuro-2a and RBL-2H3 cells rapidly accumulate [3H]-AEA and [3H]-PEA (Figure 1). Both cell types accumulated [3H]-AEA in a saturable manner with apparent KM values of 10±3.8 μM (Neuro-2a) and 9.3±3.0 μM (RBL-2H3), and Vmax values of 13±1.7×10−17 mol min−1 cell−1 (n=3) and 11±1.1×10−17 mol min−1 cell−1 (n=5), respectively. Given that each well initially contained 2×105 cells, these Vmax values correspond to 26±3.4 pmol min−1 well−1 and 22±2.2 pmol min−1 well−1. The [3H]-AEA uptake was greatly reduced at 4°C (Figure 1a,c), and the Vmax values decreased to 2.6±1.3 and 1.3±0.3×10−17 mol min−1 cell−1, respectively. In addition, the [3H]-AEA uptake decreased in both cell lines by pre-incubating the cultures with 100 μM of AM404. [3H]-PEA was also transported in the two cell types by a saturable mechanism that showed temperature-sensitivity (Figure 1b,d). The apparent KM and Vmax values of this transport process were in Neuro-2a cells 28±11 μM and 13±2.5×10−17 mol min−1 cell−1 (n=4), respectively, and in RBL-2H3 cells 30±13 μM and 17±3.5×10−17 mol min−1 cell−1 (n=6), respectively. At 4°C, the [3H]-PEA uptake was decreased as reflected by the decrease in Vmax values to 7.8±2.3×10−17 mol min−1 cell−1 in Neuro-2a cells, and 6.5±1.0×10−17 mol min−1 cell−1 in RBL-2H3 cells.
CB receptor binding in Neuro-2a and RBL-2H3 cells
It is possible that some of the [3H]-AEA and [3H]-PEA measured in fact are not taken up by the cells, but rather bound to surface CB receptors. This question was addressed in a radioligand binding study using [3H]-WIN 55,212-2, a synthetic cannabinoid which binds to both CB1 and CB2 receptors, albeit with a slight preference for the CB2 receptor subtype (Showalter et al., 1996). In rat brain membrane, used as positive control, [3H]-WIN 55,212-2 binding was concentration-dependently inhibited by SR 141716A, a CB receptor antagonist with marked selectivity for CB1 receptors (Rinaldi-Carmona et al., 1994), with a greater potency than SR 144528 (Figure 2a), a CB receptor antagonist with a 700 fold higher affinity for the CB2 receptor than the CB1 receptor (Rinaldi-Carmona et al., 1998). This is in line with previous results showing that the brain primarily express CB1 receptor (Matsuda et al., 1990; Westlake et al., 1994). In Neuro-2a membranes, SR 144528 was not able to block the [3H]-WIN 55,212-2 binding, whereas SR 141617A produced a weak inhibition at concentrations above 10 nM (Figure 2b). Neither the SR compounds nor 1 μM CP 55,940, a CB receptor agonist used to determine non-specific binding (Pertwee, 1997), were able to inhibit the [3H]-WIN 55,212-2 binding to RBL-2H3 cells (Figure 2c). The results suggest a very low CB receptor density in the two cell lines, as measured by the ability of selective CB receptor antagonists to inhibit [3H]-WIN 55,212-2 binding. Other authors have reported the presence of CB receptor-like binding sites in RBL-2H3 cells (Ross et al., 1999; but see Lambert et al., 1999), although their expression is dependent upon the passage number of the cells (Lambert & Di Marzo, 1999). Be that as it may, the lack of CB receptor binding sites in the RBL-2H3 (and Neuro-2a) cells used in the present study is an advantage, since it means that [3H]-AEA and possibly [3H]-PEA binding to CB receptors will not be an interfering factor in the uptake experiments.
Effect of ethanol upon the accumulation of AEA in RBL-2H3 cells
Before embarking upon a pharmacological characterization of the cellular cannabinoid uptake, it is important to show that the solvent used to dissolve the very lipophilic compounds does not, at the concentrations used, affect per se the uptake. As can be seen from the data shown in Figure 3, the cellular uptake of [3H]-AEA was unaffected by ethanol concentrations up to 5%, and the final solvent concentration in our experiments never exceeded 4%. The fact that the cells tolerate such high ethanol concentrations presumably reflects the short exposure time to this solvent.
Figure 3.
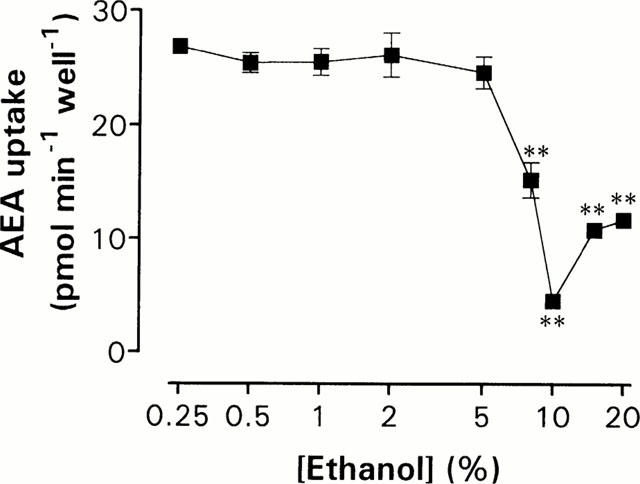
Uptake of 10 μM [3H]-AEA in various concentrations of ethanol. Stock solutions of AEA in ethanol was added to the RBL-2H3 cells and the cultures were incubated 15 min at 37°C. Data represent means±s.e.mean (n=4). Statistically significant differences (one-way ANOVA with Dunnett's multiple comparison test) are indicated as **P<0.01 when compared with corresponding cellular uptake of AEA in 0.07% ethanol (26±1.3 pmol min−1 well−1).
Pharmacological characterization of PEA uptake in Neuro-2a and RBL-2H3 cells
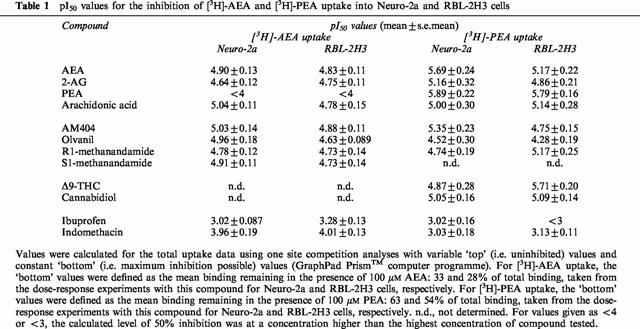
A series of compounds known to interfere with the transport and metabolism of AEA were tested for their ability to inhibit PEA uptake in Neuro-2a and RBL-2H3 cells. A major difference between the transport of AEA and that of PEA, is the level of residual accumulation of the compounds, determined as the percentage recovered radioactivity of controls in the presence of 100 μM of the unlabelled counterpart. Whereas 100 μM AEA inhibited the [3H]-AEA uptake to 33% of total in Neuro-2a cells (n=3) and 28% in RBL-2H3 cells (n=3), 100 μM PEA only inhibited the [3H]-PEA uptake to 63% of total in Neuro-2a cells (n=5) and 58% in RBL-2H3 cells (n=4). In order to compare pI50 values for the two uptake systems, the ‘specific uptake' was defined for [3H]-AEA as the binding sensitive to inhibition by 100 μM non-radioactive AEA, and for [3H]-PEA as the binding sensitive to inhibition by 100 μM non-radioactive PEA. These reference values were then used to calculate pI50 values (summarized in Table 1).
Table 1.
pI50 values for the inhibition of [3H]-AEA and [3H]-PEA uptake into Neuro-2a and RBL-2H3 cells
In the first series of experiments, the effects of the CFADs and arachidonic acid, the structural precursor to AEA, on PEA (Figure 4b,d) and, for comparison, AEA (Figure 4a,b) transport in the two cell lines were studied. No obvious difference in pharmacological profiles between Neuro-2a cell and RBL-2H3 cell uptake of [3H]-AEA could be observed. The [3H]-AEA uptake of both the cell lines was concentration-dependently inhibited by the endocannabinoid 2-AG and the long-chain fatty acid arachidonic acid, whereas PEA had no effect on the [3H]-AEA uptake in either cell line. [3H]-PEA uptake, on the other hand, was concentration-dependently blocked not only by 2-AG and arachidonic acid but also by AEA in both cell lines (Figure 4b,d).
The AEA transport inhibitor AM404, potently inhibited [3H]-AEA transport in both Neuro-2a and RBL-2H3 cells (Figure 5a,c), with an apparent pI50 of 5.03±0.14 (n=3) and 4.88±0.11 (n=3), respectively. Similar potencies of AM404 towards [3H]-PEA uptake were also seen (Table 1). The long-chain analogue of capsaicin, olvanil, concentration-dependently inhibited [3H]-AEA uptake in both cell types, supporting previous results obtained in RBL-2H3 cells (Di Marzo et al., 1998b) and in CCF-STTG1 human astrocytoma cells (Beltramo & Piomelli, 1999), but had a weaker effect upon [3H]-PEA uptake (Figure 5b,d). Furthermore, two derivatives of AEA, R1-methanandamide and S1-methanandamide, demonstrated [3H]-AEA uptake inhibiting activities (Figure 5a,c) with essentially equal potency in the two cell lines (apparent pI50 values ranging from 4.72 – 4.91; n=3). R1-methanandamide also blocked the [3H]-PEA uptake in RBL-2H3 cells (Figure 5d), but had little effect on the Neuro-2a transport of [3H]-PEA. The effect of S1-methanandamide on [3H]-PEA uptake was not investigated.
As previously reported by Fowler et al. (1997; 1999; 2000), the non-steroidal anti-inflammatory drugs (NSAIDs) ibuprofen, ketorolac, flurbiprofen and indomethacin inhibit AEA hydrolysis in rat brain. This motivated us to investigate the effects of ibuprofen and indomethacin, on the transport mechanism(s) of AEA and PEA (Figure 6). Indomethacin produced a concentration-dependent inhibition of [3H]-AEA, with an apparent pI50 of 3.96±0.19 in Neuro-2a cells (n=3) and 4.01±0.13 in RBL-2H3 cells (n=3). The effect of indomethacin upon [3H]-AEA uptake was not associated with cytotoxic effects, as assessed by the cellular release of LDH (Figure 6c,f). The uptake of [3H]-PEA was less sensitive to indomethacin than the uptake of [3H]-AEA, pI50 values of 3.03±0.18 and 3.13±0.11 being found for Neuro-2a (n=4) and RBL-2H3 cells (n=3), respectively (Figure 6b,e). Ibuprofen inhibited [3H]-AEA transport, albeit with a low potency, but had modest effects upon [3H]-PEA uptake (Figure 6a,b,d and e, Table 1).
Finally, we investigated the effects of the plant-derived cannabinoids Δ9-THC and the non-psychoactive precursor/metabolite of Δ9-THC, cannabidiol on PEA transport, given that these compounds are able to inhibit the uptake of [3H]-AEA in RBL-2H3 cells with reported mean Ki values of 16.5 and 11.4 μM (Rakhshan et al., 2000). Both cannabinoids concentration-dependently blocked [3H]-PEA uptake in Neuro-2a cells (Figure 7a) and in RBL-2H3 cells (Figure 7b). Cannabidiol showed about the same potency in Neuro-2a cells (apparent pI50 5.05±0.16; n=4) as in RBL-2H3 cells (5.09±0.14; n=3), whereas Δ9-THC proved more potent in blocking [3H]-PEA uptake in RBL-2H3 cells (apparent pI50 5.71±0.20; n=4) than in Neuro-2a cells (4.87±0.28; n=5). Furthermore, Δ9-THC was more efficacious in RBL-2H3 cells, producing 41±5% [3H]-PEA accumulation of total at a concentration of 100 μM, as compared to 71±4% in Neuro-2a cells.
Effects of Pronase and PMSF upon PEA transport in Neuro-2a cells
Proteolytic digestion of cells with Pronase did not produce any changes in [3H]-PEA or [3H]-AEA uptake that could be explained by anything else than a general cytotoxic effect (Figure 8). Neuronal accumulation of AEA was greatly reduced by the alkylating agent/fatty acid amide hydrolase inhibitor PMSF at 100 μM, a concentration that did not produce any significant cytotoxicity (Figure 8). The PMSF-treatment decreased the [3H]-AEA transport to 47±3% of control accumulation (n=4), and this decrease was significantly greater than the observed decrease in [3H]-PEA uptake (91±5% of control levels).
Figure 8.
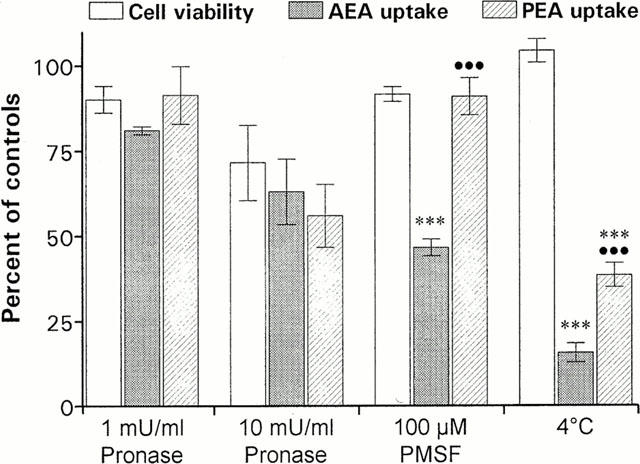
The effects of treatment with Pronase, PMSF and 4°C upon cell viability, AEA and PEA transport in Neuro-2a cells. The cells were incubated with the indicated concentration of Pronase or PMSF (b), followed by the addition of 10 μM [3H]-AEA or 20 μM [3H]-PEA. After 15 min at 37°C or 4°C, the cellular uptake of AEA and PEA and the cellular release of LDH or cellular MTT reduction were determined. Data are plotted as percentage of untreated control cultures and represent means±s.e.mean (n=3 – 8). Statistically significant differences (one-way ANOVA with Bonferroni multiple comparison test) are indicated: ***P<0.001 when cellular AEA or PEA uptake is compared with corresponding cell viability values, and •••P<0.001 between AEA and PEA uptake at respective treatment.
Discussion
The present study was undertaken in order to improve the understanding of the mechanisms behind the cellular uptake of PEA. In particular, we were interested in determining whether the uptake is similar in neuronal and non-neuronal cells, and to determine the degree to which PEA utilized a facilitated transport mechanism. Although the lipophilicity of PEA (and AEA) allows these compounds passively to diffuse through the cell membrane, facilitated transport mechanisms have been described for AEA in rat cortical neurons (Di Marzo et al., 1994; Beltramo et al., 1997), RBL-2H3 cells (Bisogno et al., 1997; Rakhshan et al., 2000), cerebellar granules (Hillard et al., 1997) and astrocytes (Beltramo et al., 1997). The mechanism appears to be mediated by a specific Na+-independent carrier protein in as much as it is saturable, temperature-dependent, selective, and apparently inhibited by various AEA analogues such as AM404 (for review, see Hillard & Jarrahian, 2000). The presence of a defined transport mechanism for PEA would provide an interesting pharmacological target for the treatment of inflammatory diseases, since a blockade of PEA transport would result in elevated extracellular concentrations of PEA, and hence, augmentation of the anti-inflammatory and anti-nociceptive effects of this compound (Facci et al., 1995; Mazzari et al., 1996; Jaggar et al., 1998; Calignano et al., 1998). This would be analogous to the situation for AEA, where both the duration and intensity of the hypotensive effects of i.v. administered AEA are potentiated by AM404 (Calignano et al., 1997, see also Giuffrida et al., 2000).
In the present study, saturable uptake of both PEA and AEA, that was reduced by reduction of the assay temperature to 4°C, was found for both Neuro-2a and RBL-2H3 cells. This would at first sight suggest a facilitated transport process for PEA as well as for AEA. Consistent with the notion of a facilitated transport process, Bisogno et al. (1997) reported that the uptake of both [3H]-AEA and [3H]-PEA into RBL-2H3 cells was sensitive to the alkylating agents N-ethyl-maleimide and PMSF, a finding also seen for [3H]-AEA uptake into human platelets (Maccarrone et al., 1999). In our hands, however, only AEA uptake was sensitive to PMSF. One explanation for this discrepancy may lie in differences in expression levels of the enzyme fatty acid amide hydrolase (FAAH, the enzyme responsible for the metabolism of AEA and PEA) in ostensibly the same cell line cultured in different laboratories. It is well established that upon uptake of AEA by the transporter, a substantial proportion (68% for RBL-2H3 cells under the conditions used here) is metabolized by FAAH (Rakhshan et al., 2000; see also Bisogno et al., 1997). This metabolism will function to reduce the AEA gradient across the cell membrane. PMSF (or indeed N-ethylmaleimide), by acting as an inhibitor of FAAH (Deutsch & Chin, 1993; Bisogno et al., 1997), will increase the level of unmetabolized AEA and hence the gradient, making uptake against the gradient more difficult. Indeed, Rakhshan et al. (2000) found that two other FAAH inhibitors (arachidonoyl trifluoromethylketone, methyl arachidonoyl fluorophosphonate) reduced the uptake of [3H]-AEA into RBL-2H3 cells more effectively than PMSF. In addition, PMSF has been shown to reduce the uptake of AEA in HeLa cells transfected with FAAH but to be without effect on the uptake in untransfected cells, which have a very low endogenous level of FAAH (E.L. Barker, personal communication). A similar result has recently been reported by Deutsch et al. (2001): in their study, it was found that PMSF and methylarachidonylfluorophosphonate reduced the uptake of AEA into FAAH-expressing N18 neuroblastoma and C6 glioma cells, but not into laryngeal carcinoma (Hep2) cells, which have very low levels of endogenous FAAH. In addition, transfection of the Hep2 cells with FAAH resulted in an increased rate of AEA uptake (Deutsch et al., 2001).
In a recent survey of AEA uptake into human neuroblastoma cells, it was reported that ‘only in those cell lines that rapidly metabolized labelled AEA, was uptake inhibited by competing AEA and by the putative transport inhibitor, AM404' (Verity et al., 2000). The lack of effect of PMSF upon [3H]-PEA uptake found in the present study may therefore reflect the fact that this substrate is not metabolized by FAAH as well as AEA (see Bisogno et al., 1997), although competition for FAAH by PEA might have expected to reduce the rate of [3H]-AEA metabolism and hence its uptake. Given that the expression of FAAH varies from cell to cell, it is thus likely that the contribution of FAAH to the rate of AEA uptake will also vary from cell to cell.
The KM values estimated for the facilitated transport component of AEA are rather similar to those found previously (Hillard et al., 1997; Bisogno et al., 1997; Rakhshan et al., 2000), although other authors have reported KM values near or below 1 μM (Beltramo et al., 1997; Maccarrone et al., 1998; 1999; 2000b). One explanation for this discrepancy may be differences in curve fitting techniques (see Rakhshan et al., 2000), and it can be noted that in a recent study using human CCF-STTG1 astrocytoma cells, the IC50 of non-radioactive AEA as an inhibitor of the uptake of 30 nM AEA (15.1±3.0 μM) was considerably higher than its KM value (0.6±0.1 μM) (Piomelli et al., 1999). A more exciting possibility, however, is the presence of heterogeneity in AEA uptake mechanisms (for discussion, see Hillard & Jarrahian, 2000). In this respect, Maccarrone et al. (1998; 2000a,2000b) found that the cellular uptake rate of AEA in various cells of human origin was greatly enhanced by nitric oxide donors, such as sodium nitroprusside (SNP). In contrast, AEA (and PEA) uptake in Neuro-2a cells and RBL-2H3 cells was not altered by exposure to 5 mM SNP (S.O.P. Jacobsson & C.J. Fowler, unpublished data).
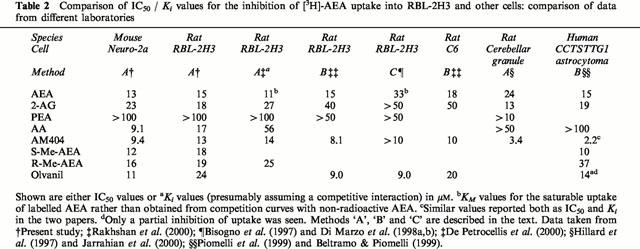
Evidence against heterogeneity of AEA uptake comes from the pharmacological data presented in the present and previous studies. Initially, it should be pointed out that different research groups employ different methods for calculation of inhibitory potencies. We have essentially adopted the method described by Jarrahian et al. (2000) and Rakhshan et al. (2000), whereby the total tritium content accumulated within the cell is defined as the total uptake, and the fraction of this sensitive to 100 μM non-radioactive AEA as the uptake due to facilitated transport (termed ‘Method A' in Table 2). Unsurprisingly, the present study shows similar pI50 values of the tested compounds towards [3H]-AEA uptake as those reported for RBL-2H3 cells by Rakhshan et al. (2000). For ease of comparison, the mean pI50 values have been converted to IC50 values and compared with the literature data in Table 2. An essentially similar approach has been used by Piomelli et al. (1999) who measured the rate of disappearance of label from the medium rather than accumulation in the cell (termed ‘Method B' in Table 2). In contrast, Di Marzo and colleagues, in some of their studies using RBL-2H3 and other cells have separated the accumulated radioactivity as unchanged and as metabolized (i.e. [14C]-ethanolamine) AEA (see e.g. Bisogno et al., 1997), thereafter to calculate inhibitory potencies upon the effect of unchanged AEA (termed ‘Method C' in Table 2). Whilst the latter method undoubtedly gives a clearer picture as to the dynamics of the total inactivation process, inhibitory potencies calculated using the two methods may vary depending upon the degree to which FAAH is also affected by the test compound (either by direct inhibition, or by an ‘entourage effect'). This, together with differences in FAAH expression, might explain some of the discrepancies seen between groups. For example, most studies indicate an inhibitory effect of 2-AG upon AEA uptake, whereas this was not seen by Di Marzo et al. (1998b) (see Table 2). However, they found that ‘when intact RBL-2H3 cells were co-incubated with 50 μM 2-AG and 4 μM [14C]-AEA, the hydrolysis, but not the uptake, of the latter compound was significantly inhibited (60.0 and 94.2% respectively of control)', indicating a reduction in the total cellular accumulation of label. A similar argument can be made concerning AM404 (compare values in Table 2), given its potent effects on FAAH. Indeed, Rakhshan et al. (2000) found that AM404 not only increased the KM value of [3H]-AEA uptake into RBL-2H3 cells but also increased the Vmax, the latter presumably a reflection of the more potent effects of this compound upon FAAH than upon AEA transport (Jarrahian et al., 2000). Consistent with this conclusion, 10 μM AM404 only produced 15% inhibition of AEA uptake into Hep2 cells, which have very low levels of endogenous FAAH activity (Deutsch et al., 2001). Once again, this would suggest that the contribution of FAAH to the uptake process is of pharmacological importance. This may also explain the differences in sensitivity to arachidonic acid found in the different laboratories, given that it is an FAAH inhibitor (Maccarrone et al., 1998), and that the expression of FAAH was low in the cells used by Piomelli et al. (1999).
Table 2.
Comparison of IC50 / Ki values for the inhibition of [3H]-AEA uptake into RBL-2H3 and other cells: comparison of data from different laboratories
In contrast to the situation for [3H]-AEA uptake, the uptake of [3H]-PEA was only partially inhibited by an excess of non-radioactive PEA. In addition, a residual [3H]-PEA uptake (considerably larger than that for [3H]-AEA uptake) was seen when the cells were incubated at 4°C. This would suggest that passive diffusion contributes to the PEA uptake. Such diffusion means that the apparent KM and Vmax values found for PEA are overestimates of the true values for the facilitated uptake. By assuming: (a) a linear relationship between the [3H]-PEA concentration and the rate of passive diffusion; and (b) a contribution to the uptake of 50% by passive diffusion at 25 μM [3H]-PEA, KM and Vmax values can be estimated for the component of the uptake due to facilitated transport. These values, calculated from the raw data for the four concentrations ⩽25 μM (the degree of passive diffusion was deemed to be too large at 50 μM PEA to allow accurate estimation of the facilitated transport), were for Neuro-2a cells, KM 6.6±3.8 μM, Vmax 3.8±0.76×10−17 mol min−1 cell−1; RBL-2H3 cells, KM 3.4±1.6 μM, Vmax 3.9±0.49×10−17 mol min−1 cell−1 (means±s.e.mean for analyses of 4 – 6 experiments).
The facilitated transport component of PEA appears to be pharmacologically similar in the RBL-2H3 cells and the Neuro-2a cells. In contrast, this transport process is pharmacologically distinguishable from that of AEA, a finding in line with the study of Bisogno et al. (1997). Thus, AEA uptake is sensitive to inhibition by indomethacin and not PEA, whereas PEA uptake is sensitive to AEA, but less so to indomethacin, except at the highest concentration tested. Bisogno et al. (1997) also found that 50 μM AEA reduced the accumulation of PEA in RBL-2H3 cells, a 15 min incubation resulting in a reduction to midway between the samples incubated either with [14C]-PEA alone and those incubated at 4°C. In contrast, Di Marzo et al. (1998a) reported as ‘data not shown' that olvanil did not affect the uptake of [14C]-PEA. The compound is, however, not potent in our hands (IC50 value ∼50 μM), and, given the relatively small component of PEA uptake that is due to facilitated transport, might not have been observable in the study of Di Marzo et al. (1998a) who used 50 μM as maximum concentration (at least in the data shown for AEA uptake).
In conclusion, our results demonstrate that the cellular transport of PEA is produced by similar processes in both neurons and in cells of the immune system. The PEA uptake is mediated partly by a facilitated transport mechanism separate to that for AEA, and partly by a passive diffusion through the cellular membrane.
Acknowledgments
This work was supported by generous grants from the Swedish Medical Research Foundation (Grant no. 12158), Borgerskapets i Umeå Research Foundation, and the Research Fund of the Medical Faculty, Umeå University. The excellent technical assistance of Britt Jacobsson is gratefully acknowledged.
Abbreviations
- 2-AG
2- arachidonoylglycerol
- AEA
arachidonoylethanolamide (anandamide)
- BSA
bovine serum albumin
- CB
cannabinoid
- CFAD
cannabimimetic fatty acid derivative
- Δ9-THC
Δ9-tetrahydrocannabinol
- EDTA
ethylenediaminetetracetic acid
- FBS
foetal bovine serum
- HEPES
4-(2-hydroxyethyl)-1-piperazineethane sulphonic acid
- LDH
lactate dehydrogenase
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NEAA
non-essential amino acids
- NO
nitric oxide
- NSAID
non-steroidal anti-inflammatory drug
- PEA
palmitoylethanolamide
- PEST
100 units ml−1 penicillin+100 μg ml−1 streptomycin
- PMSF
phenylmethylsulfonyl fluoride
- Pronase
protease from Streptomyces griseus
- SNP
sodium nitroprusside
- Tris
Tris-(hydroxymethyl)-amino-methane
- WIN 55,212-2
(R)-(+)-[2,3-dihydro-5-methyl-3-[(4-morpholino)methyl]pyrrolo-[1,2,3-de]-1,4-benzoxazin-6-yl](1-naphthyl)methone
References
- ANDERSSON M., JACOBSSON S.O.P., JONSSON K.-O., TIGER G., FOWLER C.J. Neurotoxicity of glutamate in chick telencephalon neurons. Reduction of toxicity by preincubation with carbachol, but not by the endogenous fatty acid amides anandamide and palmitoylethanolamide. Arch. Toxicol. 2000;74:161–164. doi: 10.1007/s002040050669. [DOI] [PubMed] [Google Scholar]
- BELTRAMO M., PIOMELLI D. Anandamide transport inhibition by the vanilloid agonist olvanil. Eur. J. Pharmacol. 1999;364:75–78. doi: 10.1016/s0014-2999(98)00821-8. [DOI] [PubMed] [Google Scholar]
- BELTRAMO M., STELLA N., CALIGNANO A., LIN S.Y., MAKRIYANNIS A., PIOMELLI D. Functional role of high-affinity anandamide transport, as revealed by selective inhibition. Science. 1997;277:1094–1097. doi: 10.1126/science.277.5329.1094. [DOI] [PubMed] [Google Scholar]
- BISOGNO T., MAURELLI S., MELCK D., DE PETROCELLIS L., DI MARZO V. Biosynthesis, uptake, and degradation of anandamide and palmitoylethanolamide in leukocytes. J. Biol. Chem. 1997;272:3315–3323. doi: 10.1074/jbc.272.6.3315. [DOI] [PubMed] [Google Scholar]
- CALIGNANO A., LA RANA G., BELTRAMO M., MAKRIYANNIS A., PIOMELLI D. Potentiation of anandamide hypotension by the transport inhibitor, AM404. Eur. J. Pharmacol. 1997;337:R1–R2. doi: 10.1016/s0014-2999(97)01297-1. [DOI] [PubMed] [Google Scholar]
- CALIGNANO A., LA RANA G., GIUFFRIDA A., PIOMELLI D. Control of pain initiation by endogenous cannabinoids. Nature. 1998;394:277–281. doi: 10.1038/28393. [DOI] [PubMed] [Google Scholar]
- DE PETROCELLIS L., BISOGNO T., DAVIS J.B., PERTWEE R.G., DI MARZO V. Overlap between the ligand recognition properties of the anandamide transporter and the VR1 vanilloid receptor: inhibitors of anandamide uptake with negligible capsaicin-like activity. FEBS Lett. 2000;483:52–56. doi: 10.1016/s0014-5793(00)02082-2. [DOI] [PubMed] [Google Scholar]
- DEUTSCH D.G., CHIN S.A. Enzymatic synthesis and degradation of anandamide, a cannabinoid receptor agonist. Biochem. Pharmacol. 1993;46:791–796. doi: 10.1016/0006-2952(93)90486-g. [DOI] [PubMed] [Google Scholar]
- DEUTSCH D.G., GLASER S.T., HOWELL J.M., KUNZ J.S., PUFFENBARGER R.A., HILLARD C.J., ABUMRAD N.The cellular uptake of anandamide is coupled to its breakdown by fatty acid amide hydrolase (FAAH) J. Biol. Chem. 2001. in press [DOI] [PubMed]
- DI MARZO V., BISOGNO T., SUGIURA T., MELCK D., DE PETROCELLIS L. The novel endogenous cannabinoid 2-arachidonoylglycerol is inactivated by neuronal- and basophil-like cells: connections with anandamide. Biochem. J. 1998a;331:15–19. doi: 10.1042/bj3310015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DI MARZO V., BISOGNO T., MELCK D., ROSS R., BROCKIE H., STEVENSON L., PERTWEE R., DE PETROCELLIS L. Interactions between synthetic vanilloids and the endogenous cannabinoid system. FEBS Lett. 1998b;436:449–454. doi: 10.1016/s0014-5793(98)01175-2. [DOI] [PubMed] [Google Scholar]
- DI MARZO V., DEUTSCH D.G. Biochemistry of the endogenous ligands of cannabinoid receptors. Neurobiol. Dis. 1998;5:386–404. doi: 10.1006/nbdi.1998.0214. [DOI] [PubMed] [Google Scholar]
- DI MARZO V., FONTANA A., CADAS H., SCHINELLI S., CIMINO G., SCHWARTZ J.C., PIOMELLI D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature. 1994;372:686–691. doi: 10.1038/372686a0. [DOI] [PubMed] [Google Scholar]
- FACCI L., DAL TOSO R., ROMANELLO S., BURIANI A., SKAPER S.D., LEON A. Mast cells express a peripheral cannabinoid receptor with differential sensitivity to anandamide and palmitoylethanolamide. Proc. Natl. Acad. Sci. U.S.A. 1995;92:3376–3380. doi: 10.1073/pnas.92.8.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FOWLER C.J., BÖRJESSON M., TIGER G. Differences in the pharmacological properties of rat and chicken brain fatty acid amidohydrolase. Br. J. Pharmacol. 2000;131:498–504. doi: 10.1038/sj.bjp.0703569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FOWLER C.J., JANSON U., JOHNSON R.M., WAHLSTRÖM G., STENSTRÖM A., NORSTRÖM Å., TIGER G. Inhibition of anandamide hydrolysis by the enantiomers of ibuprofen, ketorolac, and flurbiprofen. Arch. Biochem. Biophys. 1999;362:191–196. doi: 10.1006/abbi.1998.1025. [DOI] [PubMed] [Google Scholar]
- FOWLER C.J., TIGER G., STENSTRÖM A. Ibuprofen inhibits rat brain deamidation of anandamide at pharmacologically relevant concentrations. Mode of inhibition and structure-activity relationship. J. Pharmacol. Exp. Therapeut. 1997;283:729–734. [PubMed] [Google Scholar]
- GAONI Y., MECHOULAM R. Isolation, structure and partial synthesis of an active constituent of hashish. J. Am. Chem. Soc. 1964;86:1646–1648. [Google Scholar]
- GIUFFRIDA A., RODRIGUEZ DE FONSECA F., NAVA F., LOUBET-LESCOULIÉ P., PIOMELLI D. Elevated circulating levels of anandamide after administration of the transport inhibitor, AM404. Eur. J. Pharmacol. 2000;408:161–168. doi: 10.1016/s0014-2999(00)00786-x. [DOI] [PubMed] [Google Scholar]
- HARRINGTON C.R. Lowry protein assay containing sodium dodecyl sulfate in microtiter plates for protein determination on fractions from brain tissue. Anal. Biochem. 1990;186:285–287. doi: 10.1016/0003-2697(90)90081-j. [DOI] [PubMed] [Google Scholar]
- HILLARD C.J., EDGEMOND W.S., JARRAHIAN A., CAMPBELL W. B. Accumulation of N-arachidonoylethanolamine (anandamide) into cerebellar granule cells occurs via facilitated diffusion. J. Neurochem. 1997;69:631–638. doi: 10.1046/j.1471-4159.1997.69020631.x. [DOI] [PubMed] [Google Scholar]
- HILLARD C.J., JARRAHIAN A. The movement of N-arachidonoylethanolamine (anandamide) across cellular membranes. Chem. Phys. Lipids. 2000;108:123–134. doi: 10.1016/s0009-3084(00)00191-2. [DOI] [PubMed] [Google Scholar]
- JACOBSSON S., TIGER G., FOWLER C.J. Uptake of anandamide and palmitoylethanolamide into neuroblastoma cells: separate or common transport mechanisms. Soc. Neurosci. Abstr. 2000;26:2163. [Google Scholar]
- JAGGAR S.I., HASNIE F.S., SELLATURAY S., RICE A.S.C. The anti-hyperalgesic actions of the cannabinoid anandamide and the putative CB2 receptor agonist palmitoylethanolamide in visceral and somatic inflammatory pain. Pain. 1998;76:189–199. doi: 10.1016/s0304-3959(98)00041-4. [DOI] [PubMed] [Google Scholar]
- JARRAHIAN A., MANNA S., EDGEMOND W.S., CAMPBELL W.B., HILLARD C.J. Structure-activity relationships among N-arachidonoylethanolamine (anandamide) head group analogues for the anandamide transporter. J. Neurochem. 2000;74:2597–2606. doi: 10.1046/j.1471-4159.2000.0742597.x. [DOI] [PubMed] [Google Scholar]
- KONDO S., SUGIURA T., KODAKA T., KUDO N., WAKU K., TOKUMURA A. Accumulation of various N-acylethanolamines including N-arachidonoylethanolamine (anandamide) in cadmium chloride-administered rat testis. Arch. Biochem. Biophys. 1998;354:303–310. doi: 10.1006/abbi.1998.0688. [DOI] [PubMed] [Google Scholar]
- LAMBERT D.M., DI MARZO V. The palmitoylethanolamide and oleamide enigmas: are these two fatty acid amides cannabimimetic. Curr. Med. Chem. 1999;6:757–773. [PubMed] [Google Scholar]
- LAMBERT D.M., DIPAOLO F.G., SONVEAUX P., KANYONYO M., GOVAERTS S.J., HERMANS E., BUEB J., DELZENNE N.M., TSCHIRHART E.J. Analogues and homologues of N-palmitoylethanolamide, a putative endogenous CB2 cannabinoid, as potential ligands for the cannabinoid receptors. Biochim. Biophys. Acta. 1999;1440:266–274. doi: 10.1016/s1388-1981(99)00132-8. [DOI] [PubMed] [Google Scholar]
- MACCARRONE M., BARI M., LORENZON T., BISOGNO T., DI MARZO V., FINAZZI-AGRO A. Anandamide uptake by human endothelial cells and its regulation by nitric oxide. J. Biol. Chem. 2000a;275:13484–13492. doi: 10.1074/jbc.275.18.13484. [DOI] [PubMed] [Google Scholar]
- MACCARRONE M., BARI M., MENCHELLI A., DEL PRINCIPE D., FINAZZI-AGRO A. Anandamide activates human platelets through a pathway independent of the arachidonate cascade. FEBS Lett. 1999;447:277–282. doi: 10.1016/s0014-5793(99)00308-7. [DOI] [PubMed] [Google Scholar]
- MACCARRONE M., FIORUCCI L., ERBA F., BARI M., FINAZZI-AGRO A., ASCOLI F. Human mast cells take up and hydrolyze anandamide under the control of 5-lipoxygenase and do not express cannabinoid receptors. FEBS Lett. 2000b;468:176–180. doi: 10.1016/s0014-5793(00)01223-0. [DOI] [PubMed] [Google Scholar]
- MACCARRONE M., VAN DER STELT M., ROSSI A., VELDINK G.A., VLIEGENTHART J.F., FINAZZI-AGRO A. Anandamide hydrolysis by human cells in culture and brain. J. Biol. Chem. 1998;273:32332–32339. doi: 10.1074/jbc.273.48.32332. [DOI] [PubMed] [Google Scholar]
- MATSUDA L.A., LOLAIT S.J., BROWNSTEIN B.J., YOUNG A.C., BONNER T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- MAZZARI S., CANELLA R., PETRELLI L., MARCOLONGO G., LEON A. N-(2-hydroxyethyl)hexadecanamide is orally active in reducing edema formation and inflammatory hyperalgesia by down-modulating mast cell activation. Eur. J. Pharmacol. 1996;300:227–236. doi: 10.1016/0014-2999(96)00015-5. [DOI] [PubMed] [Google Scholar]
- MECHOULAM R., FRIDE E., DI MARZO V. Endocannabinoids. Eur. J. Pharmacol. 1998;359:1–18. doi: 10.1016/s0014-2999(98)00649-9. [DOI] [PubMed] [Google Scholar]
- MOSMANN T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Meth. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- MUNRO S., THOMAS K.L., ABU-SHAAR M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- NATARAJAN V., REDDY P.V., SCHMID P.C., SCHMID H.H. N-Acylation of ethanolamine phospholipids in canine myocardium. Biochim. Biophys. Acta. 1982;712:342–355. doi: 10.1016/0005-2760(82)90352-6. [DOI] [PubMed] [Google Scholar]
- PERTWEE R.G. Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol. Ther. 1997;74:130–178. doi: 10.1016/s0163-7258(97)82001-3. [DOI] [PubMed] [Google Scholar]
- PIOMELLI D., BELTRAMO M., GLASNAPP S., LIN S.Y., GOUTOPOULOS A., XIE X.Q., MAKRIYANNIS A. Structural determinants for recognition and translocation by the anandamide transporter. Proc. Natl. Acad. Sci. U.S.A. 1999;96:5802–5807. doi: 10.1073/pnas.96.10.5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAKHSHAN F., DAY T.A., BLAKELY R.D., BARKER E.L. Carrier-mediated uptake of the endogenous cannabinoid anandamide in RBL-2H3 cells. J. Pharmacol. Exp. Ther. 2000;292:960–967. [PubMed] [Google Scholar]
- RINALDI-CARMONA M., BARTH F., HEAULME M., SHIRE D., CALANDRA B., CONGY C., MARTINEZ S., MARUANI J., NELIAT G., CAPUT D., FERRARA P., SOUBRIÉ P., BRELIÈRE J.-C., LE FUR G. SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett. 1994;350:240–244. doi: 10.1016/0014-5793(94)00773-x. [DOI] [PubMed] [Google Scholar]
- RINALDI-CARMONA M., BARTH F., MILLAN J., DEROCQ J.M., CASELLAS P., CONGY C., OUSTRIC D., SARRAN M., BOUABOULA M., CALANDRA B., PORTIER M., SHIRE D., BRELIERE J.C., LE FUR G.L. SR 144528, the first potent and selective antagonist of the CB2 cannabinoid receptor. J. Pharmacol. Exp. Ther. 1998;284:644–650. [PubMed] [Google Scholar]
- ROSS R.A., MURPHY V.L., MCKAY N.G., ASHFORD M.L.J., PERTWEE R.G.Characterization of cannabinoid receptors in the spleen and in mast cells Essential Fatty Acids and Eicosanoids: Invited papers from the 4th International Congress 1999AOCS Press: Champaign, IL, U.S.A; 376–379.ed. Riemersma, R.A., Armstrong, R., Kelly, R.W. & Wilson, R. pp [Google Scholar]
- SHESKIN T., HANUS L., SLAGER J., VOGEL Z., MECHOULAM R. Structural requirements for binding of anandamide-type compounds to the brain cannabinoid receptor. J. Med. Chem. 1997;40:659–667. doi: 10.1021/jm960752x. [DOI] [PubMed] [Google Scholar]
- SHOWALTER V.M., COMPTON D.R., MARTIN B.R., ABOOD M.E. Evaluation of binding in a transfected cell line expressing a peripheral cannabinoid receptor (CB2): identification of cannabinoid receptor subtype selective ligands. J. Pharmacol. Exp. Ther. 1996;278:989–999. [PubMed] [Google Scholar]
- SKAPER S.D., BURIANI A., DAL TOSO R., PETRELLI L., ROMANELLO S., FACCI L., LEON A. The ALIAmide palmitoylethanolamide and cannabinoids, but not anandamide, are protective in a delayed postglutamate paradigm of excitotoxic death in cerebellar granule neurons. Proc. Natl. Acad. Sci. U.S.A. 1996;93:3984–3989. doi: 10.1073/pnas.93.9.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THOMAS B., GILLIAM A., BURCH D., ROCHE M., SETZMAN H. Comparative receptor binding analyses of cannabinoid agonists and antagonists. J. Pharmacol. Exp. Ther. 1998;285:285–292. [PubMed] [Google Scholar]
- VERITY A.N., TALI N., CHI T., DOWELL D., GABRIEL S., BAECKER P., DENAGEL D. AM404 and inhibitors of fatty acid amide hydrolase prevent anandamide uptake and metabolism in SK-N-AS cells. Soc. Neurosci. Abstr. 2000;26:2163. [Google Scholar]
- WESTLAKE T.M., HOWLETT A.C., BONNER T.I., MATSUDA L.A., HERKENHAM M. Cannabinoid receptor binding and messenger RNA expression in human brain. An in vitro receptor autoradiography and in situ hybridization histochemistry study of normal aged and Alzheimer's brains. Neuroscience. 1994;63:637–652. doi: 10.1016/0306-4522(94)90511-8. [DOI] [PubMed] [Google Scholar]