

Abstract
Muscarinic m1 receptors are inhibited by local anaesthetics (LA) at nM concentrations. To elucidate in more detail the site(s) of LA interaction, we compared these findings with LA effects on m3 muscarinic receptors.
We expressed receptors in Xenopus oocytes. Using two-electrode voltage clamp, we measured the effects of lidocaine, QX314 (permanently charged) and benzocaine (permanently uncharged) on Ca2+-activated Cl−-currents (ICl(Ca)), elicited by acetyl-β-methylcholine bromide (MCh). We also characterized the interaction of lidocaine with [3H]-quinuclydinyl benzylate ([3H]-QNB) binding to m3 receptors. Antisense-injection was used to determine the role of specific G-protein α subunits in mediating the inhibitory effects of LA. Using chimeric receptor constructs we investigated which domains of the muscarinic receptors contribute to the binding site for LA.
Lidocaine inhibited m3-signalling in a concentration-dependent, reversible, non-competitive manner with an IC50 of 370 nM, approximately 21 fold higher than the IC50 (18 nM) reported for m1 receptors. Intracellular inhibition of both signalling pathways by LA was similar, and dependent on the Gq- protein α subunit. In contrast to results reported for the m1 receptor, the m3 receptor lacks the major extracellular binding site for charged LA. The N-terminus and third extracellular loop of the m1 muscarinic receptor molecule were identified as requirements to obtain extracellular inhibition by charged LA.
Keywords: G proteins, muscarinic acetylcholine receptors, local anaesthetics, Xenopus oocytes, chimeras, antisense
Introduction
Muscarinic acetylcholine signalling plays important roles in several organ functions of great relevance to anaesthesiologists. It is involved in modulation of the level of consciousness in the brainstem. Inhibition of muscarinic signalling (by reducing acetylcholine levels, inhibiting its release, or administration of scopolamine) decreases the minimum alveolar concentration (MAC) of inhaled anaesthetics. In contrast, physostigmine administration increases MAC, and reverses the action of propofol on the CNS (Bonhomme et al., 1998). In addition, m1 and m3 muscarinic receptors are largely responsible for maintenance of airway tone.
m1 Muscarinic acetylcholine receptors have been shown to be highly sensitive to local anaesthetics (LA) (Hollmann et al., 2000). Half-maximal inhibitory concentration for lidocaine on recombinantly expressed m1 muscarinic receptors was 18 nM, approximately 500 fold less than that required to block neuronal sodium channels (Scholz et al., 1998). This sensitivity can be explained by a superadditive interaction between two sites, one located extracellularly on the receptor (separate from the ligand binding domain), and one located intracellularly, on receptor or coupled G protein.
In an effort to define more precisely these interaction sites, we determined effects of LA on a related receptor, the m3 muscarinic receptor. Since m1 and m3 receptors are very similar in sequence (Bonner, 1989), any differences in local anaesthetic effect can be used to determine the site of action within the molecule. Our findings indicate the presence of several, molecularly well defined interaction sites for LA on the m3 muscarinic receptor and coupled G protein.
Methods
Oocyte experiments
Oocyte expression
The study protocol was approved by the Animal Care and Use Committee at the University of Virginia. Our methodology for oocyte harvesting and expression has been described previously (Hollmann et al., 2000). Briefly, oocytes were obtained from Xenopus laevis frogs, defolliculated with collagenase, and injected with complementary RNA (cRNA). Ca2+-activated Cl−-currents, induced by IP3-mediated intracellular Ca2+-release, were measured using 2-electrode voltage clamp.
cRNA synthesis and injection
The rat m3 muscarinic acetylcholine receptor complementary DNA (cDNA) was obtained from Dr T.I. Bonner (National Institute of Mental Health, Bethesda, MD, U.S.A.). It consists of a 2.8-kilobasepair fragment in a commercial vector (pGEM1; Promega, Madison, WI, U.S.A.). The construct was linearized by digestion with the nuclease HindIII and complementary RNA (cRNA) was prepared by transcription in vitro using the bacteriophage RNA polymerase T7 (Ambion T7 mMessage mMachine Kit, Austin, TX, U.S.A.). A capping analogue (7m GpppG) was included in the reaction to generate capped transcripts, as these are translated more efficiently in the oocyte. The resulting cRNA was quantified by spectometry, and 5 ng cRNA in a 30 nl volume was injected into the oocyte, using an automated microinjector (Nanoject; Drummond Scientific, Broomall, PA, U.S.A.). The adequacy of injection was confirmed by a slight increase in cell size during injection. The cells were then cultured in modified Barth's (in mM) NaCl 88, KCl 1, NaHCO3 2.4, N-[2-hydroxyethyl]piperazine-N′-[2-ethanesulphonic acid] (HEPES) 15, CaNO3·4H20 0.3, CaCl2·6H20 0.41, MgSO4·7H20 0.82, 10 μg ml−1 gentamicin solution for 72 h at 18°C before study.
Drug administration
Acetyl-β-methylcholin bromide (MCh), used as agonist for the m3 muscarinic receptor, was diluted in Tyrode's (in mM) (NaCl 150, KCl 5, MgCl2·6H20 1, CaCl2·2H2O 2, Dextrose 10, HEPES 10) solution to the required concentration and was superfused (3 ml min−1) over the oocyte for 10 s. The oocyte was positioned close to the inflow tubing, so that complete exposure to test solutions was obtained in 4.8±0.4 s (n=20). Lidocaine also was diluted in Tyrode's solution to various concentrations and superfused (3 ml min−1) for 10 min. Benzocaine and QX314 were diluted and administered extracellulary in the same manner as lidocaine.
For intracellular administration of QX314 or lidocaine a third micropipette was inserted into the voltage-clamped oocyte. The micropipette was connected to an automated microinjector (Nanoject; Drummond Scientific, Broomall, PA, U.S.A.). Under voltage clamp, 50 nl (approximately 10% of total oocyte volume) of a 150 mM KCl solution was injected for determination of the control response; in the treatment group we injected 50 nl of KCl solution containing various concentrations of QX314 or lidocaine. Injection was followed by superfusion with Tyrode's solution for 10 min, preventing an extracellular effect of any QX314 or lidocaine leaked from the puncture site or through the membrane. ICl(Ca) was then induced by superfusion of MCh, as described previously.
Control, treatment and at times recovery responses were obtained from different oocytes to prevent the effects of receptor desensitization from obscuring the results.
Oligonucleotide injection
Phosphorothioate oligonucleotides were synthesized by the University of Virginia Biomolecular Research Facility. The antisense sequences are complementary to specific 20-base segments showing less than 50% homology with other types of Xenopus laevis Gα proteins (Shapira et al., 1999). Sense oligonucleotides were used as control. Oocytes injected 24 h prior with cRNA encoding the m3 receptor were injected with 50 nl sterile water containing 50 ng cell−1 antisense or sense oligonucleotides. Control cells were injected with the same amount of sterile water. Twenty-four and 48 h after oligonucleotide injection the cells were tested as described below.
Binding experiments
Membrane preparation
Chinese Hamster Ovary (CHO) cells stably transfected with the muscarinic m3 receptor were homogenized in 10 vol of ice-cold homogenization buffer (2-amino-2-hydroxymethylpropan-1,3-diol (Tris) 50 mM, MgCl2 5 mM, ethylene diamine tetraacetic acid (EDTA) 5 mM, ethylene glycol-bis (b-amino ethyl ether) tetraacetic acid (EGTA) 1 mM, aprotinin 2 μg ml−1, pH adjusted to 7.5) with an Overhead stirrer (Wheaton Instruments, Millville, NJ, U.S.A.) three times for 15 s at medium speed. The homogenate was centrifuged for 30 min at 4°C and 500×g. The supernatant was adjusted to 107 mM KCl and 20 mM 3-[N-morpholino]propanesulphonic acid (MOPS) (pH 7.4), mixed, incubated for 10 min on ice, and centrifuged for 60 min at 160,000×g at 4°. The pellet was resuspended in 160 mM KCl and 20 mM Tris (pH 7.4) with a short burst of an overhead stirrer at medium speed, and centrifuged at 160,000×g for 45 min at 4°C. The final pellet was resuspended in homogenization buffer and stored in aliquots at −20°C. Protein concentration was determined by the Lowry method using bovine serum albumin (BSA) for standards.
Ligand binding
Muscarinic m3 receptor density and equilibrium dissociation constants in CHO cell membranes were determined by specific binding of a muscarinic m1 receptor antagonist, [3H]-quinuclydinyl benzylate ([3H]-QNB) (0.1 – 16 nM). One hundred-μl aliquots of membrane (15 μg of protein) were incubated with [3H]-QNB in assay buffer (20 mM Tris, 100 mM NaCl, 0.5 mM EDTA, pH 7.4) for 90 min at 21°C. Membranes were collected onto Whatman GF/C glass fibre filters, which were washed three times for 10 s with ice-cold buffer (Tris 10 mM, MgCl2 5 mM, pH 7.4). Radioactivity trapped on filters was counted using a scintillation counter. All reactions were performed in triplicate. Nonspecific binding was determined by adding 5 μM atropine to displace specific binding of [3H]-QNB.
Specific binding was fit to a single site binding model using nonlinear least square curve fitting of the untransformed data to calculate receptor density (Bmax) and dissociation constants (Kd). To determine interaction of lidocaine with specific binding of [3H]-QNB, 100 μl aliquots of membrane (15 μg of protein) were incubated with various concentrations of lidocaine (10−2 to 10−10 M) and [3H]-QNB (at Kd) in assay buffer (in mM) Tris 20, NaCl 100, EDTA 0.5, pH 7.4) for 90 min at 21°C and ligand binding was determined as previously described.
In order to determine whether lidocaine acts as a competitive or non-competitive antagonist, we performed binding in the presence and absence of lidocaine. One hundred μl aliquots of membrane (15 μg of protein) and various concentrations of [3H]-QNB were incubated with 100 μl of assay buffer either with or without 10−3 M lidocaine (final concentration of 5×10−4 M). This concentration corresponds approximately to an IC20 (concentration of antagonist that reduces the response to a submaximal concentration of agonist by 80%) for lidocaine in the binding assay. After an incubation period of 90 min at 21°C (to obtain equilibrium between the membrane protein, [3H]-QNB and lidocaine) membranes were collected, radioactivity was counted and the data were analysed as described previously. Again all reactions were done in triplicate (n=3). Absence of ligand depletion in these studies was assured by determining the ratio of bound to total counts at high ligand concentration. This ratio was 37%, well below the 50% usually accepted as a cutoff. Also, the binding curve did not show an upward angle at higher [3H]-QNB concentrations, again indicating no significant depletion took place.
Chimera constructs
The chimeras between muscarinic m1 and m3 receptors were constructed using routine molecular biology techniques. Briefly, all cDNA fragments necessary for the construction of chimeras were obtained from PCR reactions using m1 and m3 receptor cDNA as templates. These PCR products carrying restriction enzyme sites at their 5′ and 3′ ends were gel purified and digested with the respective restriction enzymes. The matching fragments of cDNA for the respective chimeras were subcloned in pcDNA 3.1 (Invitrogen, Carlsbad, CA, U.S.A.) by transformation in DH5α cells (Gibco BRL, Gaithersburg, MD, U.S.A.) employing the protocols recommended by the suppliers. The transformants with the proper cDNA insert were chosen, sequenced (Biomolecular Research Facility, University of Virginia, VA, U.S.A.) and their homology with the parent cDNA was confirmed. These plasmid DNA were linearized and cRNA synthesized in vitro as described above.
Analysis
Results are reported as mean±s.d.. Measurements of at least 12 oocytes were averaged to generate each data point. As variability between batches of oocytes is common, responses were at times normalized to control response. Statistical tests employed are indicated in the Results section. P<0.05 was considered significant. Concentration-response curves were fit to the following logistic function, derived from the Hill equation: y=ymin+(ymax−ymin) {1−xn/(x50n+xn)} where ymax and ymin are the maximum and minimum response obtained, n is the Hill coefficient, and X50 is the half-maximal effect concentration (EC50 for agonist) or the half-maximal inhibitory effect concentration (IC50 for antagonist).
Materials
Molecular biology reagents were obtained from Promega (Madison, WI, U.S.A.) and other chemicals were obtained from Sigma (St. Louis, MO, U.S.A.). CHO cells (CRL-1982), stably transfected with the rat muscarinic m3 receptor, were purchased from ATCC (Manassas, VA, U.S.A.). QX314 was a gift from Astra Pharmaceuticals, L.P. (Westborough, MA, U.S.A.).
Results
Functional expression of m3 muscarinic receptors in Xenopus oocytes
Whereas uninjected oocytes were unresponsive to MCh, oocytes injected with m3 muscarinic receptor cRNA responded to application of MCh (10−4 – 10−9 M) with a transient ICl(Ca) (e.g. Figure 1A). We have shown previously that this response is mediated by m3 muscarinic receptors, as it is inhibited by atropine and the selective m3 antagonist 4-Diphenylacetoxy-N-methylpiperidine (4-DAMP) (Nietgen et al., 1998). As shown in Figure 1B, the response was concentration-dependent. EC50, calculated from the Hill equation, was 4.2±0.4×10−7 M (nH=0.6). Maximal responses of 1.9±0.2 μA were obtained at a MCh concentration of 0.1 mM. Calculated maximal response size (Emax)was 2.0±0.4 μA. These findings compare closely with data reported in our previous study (Nietgen et al., 1998).
Figure 1.
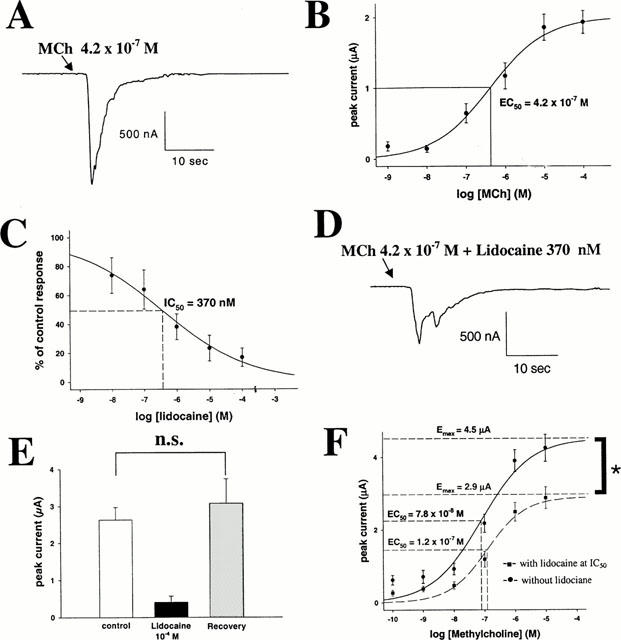
(A) Sample trace of a Ca-activated Cl current (ICl(Ca)) induced by 10 s administration of MCh at approximately EC50 (4.2×10−7 M) in an oocyte expressing the muscarinic m3 receptor. Peak current is 1.12 μA. (B) MCh evokes ICl(Ca) in a concentration-dependent manner. EC50 is 4.2±0.4×10−7 M, Emax is 2.0±0.4 μA. (C) Lidocaine inhibits MCh (at EC50)-induced ICl(Ca) in a concentration-dependent manner. IC50 is 3.7±0.8×10−7 M. (D) Sample trace of a m3 response elicited by stimulation with MCh (at EC50) in the presence of lidocaine at IC50. Peak current is 0.48 μA (A). (E) Mean±s.d. of m3 responses elicited with MCh (at EC50). Left bar indicates the control response, middle bar represents the response after 10 min incubation in 10−4 M lidocaine, and right bar represents the response after a 10 min incubation in 10−4 M lidocaine and another 10 min wash with Tyrode's solution. The third response is not significantly different in size from the first one, indicating reversibility of lidocaine inhibition. (F) Concentration-response curve for MCh on m3 receptors in the presence or absence of lidocaine (at IC50). Lidocaine inhibition cannot be overcome with maximal agonist concentrations (Emax remains inhibited by 36%) suggesting a primarily non-competitive antagonism.
Lidocaine inhibits m3 signalling
Administration for 10 min of various concentrations of lidocaine resulted in a concentration-dependent inhibition (Figure 1C) of muscarinic responses, evoked by stimulation with MCh at EC50 (0.42 μM). Half-maximal inhibitory effect concentration (IC50) for lidocaine was 3.7±0.8×10−7 M (Figure 1D and A) (nH=0.5). Although much lower than that required for blockade of sodium channels (Scholz et al., 1998), this concentration is approximately 21 fold greater than that reported for inhibition of muscarinic m1 signalling (Hollmann et al., 2000). Maximal inhibition was obtained with lidocaine 100 μM; at this concentration muscarinic responses were inhibited by 84%. As shown in Figure 1E, the lidocaine effect was reversible. Per cent inhibition by lidocaine (10−4 M) was similar to that shown in Figure 1C. Control, treatment and recovery responses to MCh 0.42 μM were 2.6±0.3, 0.4±0.1, and 3.1±0.6 μA, respectively (n=16). The holding current of the oocytes did not change significantly during these experiments.
The effect of lidocaine was non-competitive. Responses to various concentrations of MCh (10−10 – 10−5 M) were measured under control conditions, or after a 10-min superfusion with lidocaine at IC50 (370 nM, n=20 at each MCh concentration, Figure 1F). The calculated control EC50 for MCh was 7.8±2.3×10−8 M (nH=0.8); Emax was 4.5±0.4 μA. In the presence of lidocaine the EC50 did not change significantly (1.2±0.3×10−7 M; nH=0.7; P=0.293, t-test), but Emax was reduced significantly to 2.9±0.2 μA (36% inhibition, P=0.005, t-test). This non-competitive antagonism suggests that lidocaine does not interact primarily with the ligand binding site. To confirm this, we studied the effect of lidocaine on [3H]-QNB binding to m3 muscarinic receptors. We first characterized [3H]-QNB binding to membranes prepared from CHO cells, stably transfected with the rat muscarinic m3 receptor. Over a range of 0.1 to 16 nM free drug specific binding was saturable and reached a maximum at 2.8 – 4.2 nM. The saturation curve and Scatchard analysis (Figure 2A) conform closely to a single site model with a Kd of 0.15±0.03 nM (n=5) and Bmax of 3517±154 fmol mg−1 protein (n=5). The action of lidocaine was tested in concentrations ranging between 10−10 and 10−2 M (Figure 2B). Lidocaine, at concentrations that inhibit m3 muscarinic signalling, did not affect specific binding of [3H]-QNB to the muscarinic m3 receptor. Lidocaine concentrations of 10−3 and 10−2 M modestly reduced binding, but in this concentration range nonspecific effects could not be excluded. Calculated IC50 using the Hill equation was 78±9 mM (n=5). We then performed [3H]-QNB binding in the presence and absence of lidocaine at IC20 (5×10−4 M, Figure 2C). The binding curve was not shifted significantly by lidocaine (P=0.39, unpaired t-test): Kd was 0.57±0.01 nM in the absence and 0.48±0.04 nM in the presence of lidocaine. Lidocaine inhibition on [3H]-QNB-binding could not be overcome with higher concentrations of [3H]-QNB. Bmax for [3H]-QNB-binding to muscarine m3 receptors decreased in the presence of lidocaine by 33±4% of control binding, from 1482.1±88 to 984.3±45.5 fmol mg−1 protein (P<0.001, t-test). Those results confirm that lidocaine acts as a non-competitive antagonist on muscarinic m3 receptors.
Figure 2.
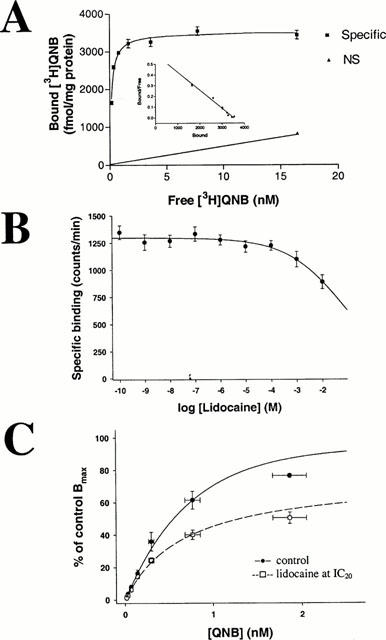
(A) Characterization of [3H]-QNB binding to membranes prepared from CHO cells, stably transfected with the rat muscarinic m3 receptor. The saturation curve and Scatchard analysis conform to a single site model with a Kd of 0.15±0.03 nM (n=5) and Bmax of 3517±154 fmol mg−1 protein (n=5). (B) Effects of lidocaine on specific binding of [3H]-QNB to muscarinic m3 receptors. IC50 is 78±9 mM (n=5). Lidocaine at functionally determined IC50 (3.7×10−7 M) does not affect agonist binding. (C) Binding curves for [3H]-QNB to m3 receptors in the absence (control, – • – ) and presence (–□amp;–) of lidocaine at IC20 for binding effect (5×10−4 M). Whereas Kd is not significantly shifted (0.57±0.01 nM in the absence and 0.48±0.04 nM in the presence of lidocaine), Bmax decreases in the presence of lidocaine by 33.5±3.8% of control, from 1482.1±88 to 984.3±45.5 fmol mg−1 protein.
Muscarinic m3 signalling is not inhibited by extracellular administered QX314
We then proceeded to determine the cause for the 21 fold difference in lidocaine sensitivity between m1 and m3 receptors. At the m1 receptor, a major part of the inhibition is due to an extracellularly charged interaction site (Hollmann et al., 2000). To determine if the same is true for the m3 receptor, we used extracellularly administered QX314, a permanently charged and therefore membrane impermeant lidocaine analogue. In contrast to our results obtained using m1 signalling (IC50 2.4×10−6 M) (Hollmann et al., 2000), QX314 in relevant concentrations was without effect on m3 signalling (Figure 3A). Only 10−3 M or 10−2 M QX314 inhibited partially (to 78±9 and 55±5% of control response, respectively), but at these concentrations nonspecific effects are likely. Thus, the absence on the m3 receptor of an extracellular interaction site for charged LA may explain in part its lower sensitivity to these compounds as compared with the m1 receptor.
Figure 3.
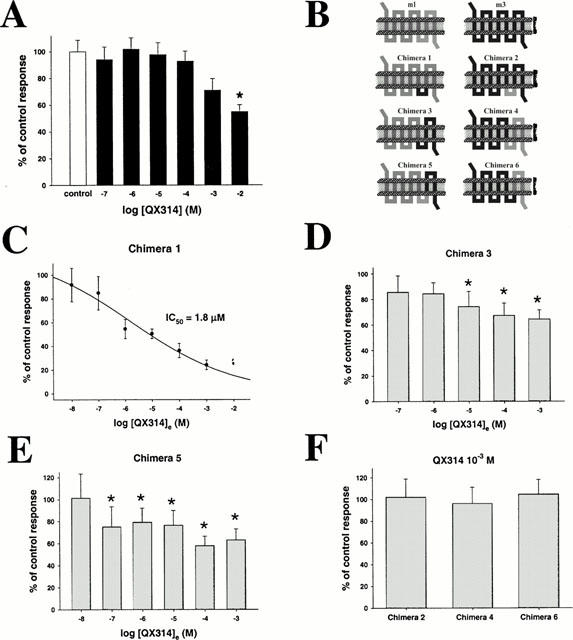
(A) m3 muscarinic signalling induced by MCh (4.2×10−7 M) is not affected by relevant concentrations of QX314. Only concentrations of 10−3 M (78±8.7% of control response) and 10−2 M (55±5.1% of control response) inhibit partially. (B) Schematic illustration of the chimeras employed. (C) Concentration-response curve for effect of extracellularly applied QX314 on chimera 1. IC50 is 1.8±0.6 μM. Mean control response was 4.9±0.4 μA. (D – E) Effects of various concentrations of extracellularly administered QX314 on chimera 3 (D) and chimera 5 (E). Modest inhibition was observed in both cases (* indicates significant differences vs control). Maximal inhibition is obtained using 10−3 M QX314 on chimera 3 (64.2±7.5% of control response (1.9±0.2 μA)) and using 10−4 M QX314 on chimera 5 (58.2±8.5% of control response (1.5±0.2 μA)). (F) Extracellularly applied QX314 (10−3 M) does not inhibit chimera 2, 4 or 6. Mean response sizes are 102.1±8.5, 96.2±15 and 104.8±13.5% of control response for chimera 2, 4 and 6, respectively.
Extracellular inhibition of m1 receptors by QX314 and lidocaine depends on N-terminus and third extracellular loop
The m1 and m3 receptors are highly similar structurally, with the major differences located in the N-terminus and the third intracellular loop. To localize further the extracellular site of local anaesthetic interaction on the m1 receptor, we used chimeric m1/m3 receptors (Figure 3B). EC50 for MCh on each chimera was between 10−7 M and 10−6 M, similar therefore to that of the parent m1 and m3 receptors. We then tested the effect of 10 min incubation in QX314 (10−3 M) on each of the six chimeras. Chimeras were stimulated with their corresponding EC50 of MCh. Only chimera 1, containing both the N-terminus and the third extracellular loop of the m1 receptor, was inhibited by QX314 in a manner similar to that obtained with the m1 receptor (IC50 1.8±0.6×10−6 M (nH=0.5), as compared with 2.4×10−6 M for the m1 receptor (Figure 3C) (Hollmann et al., 2000). Chimeras 3 (Figure 3D) and 5 (Figure 3E), containing the N-terminus, but not the third extracellular loop of m1 were also significantly (P<0.05, one-way ANOVA, Dunnett correction) inhibited at QX314 concentrations higher than 10−6 M (for chimera 3) or 10−8 M (for chimera 5), but only partially. Calculated IC50s were beyond the greatest concentration tested (10−3 M). Chimeras 2, 4 and 6 (Figure 3F), containing the N-terminus of the m3 receptor, were not inhibited. Thus, effective inhibition by extracellularly applied QX314 requires the N-terminus and the third extracellular loop of the m1 receptor. The m1 N-terminal domain is necessary, but not fully sufficient for inhibition to take place.
We repeated these experiments with lidocaine itself, at pH 5, when it is almost completely charged, and obtained similar results. Only chimera 1 was inhibited, with a calculated IC50 of 4.0±0.8×10−6 M (data not shown). Partial but significant inhibition was observed for chimeras 3 and 5, whereas chimeras 2, 4 and 6 were not inhibited (data not shown).
Benzocaine, intracellular QX314 and intracellular lidocaine inhibit m3 muscarinic signalling
We next investigated the effects of intracellularly injected QX314 on m3 signalling. QX314 inhibited concentration-dependently (Figure 4A). IC50 was 445±226×10−6 M (nH=0.6), approximately three orders of magnitude less potent than lidocaine, and comparable to the IC50 of 962±204×10−6 M obtained on the m1 receptor (Hollmann et al., 2000). Thus, the more potent inhibitory effect of lidocaine as compared with intracellular QX314 cannot be explained solely by an intracellular charged site of action.
Figure 4.
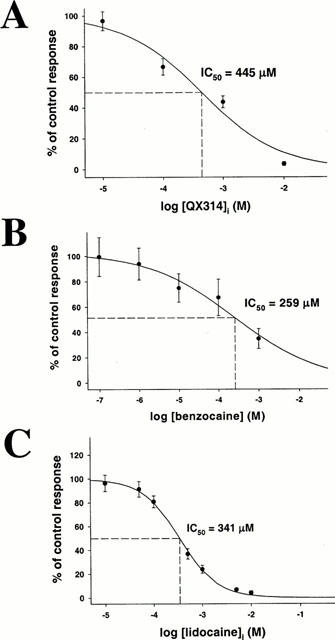
Concentration-dependent inhibition of MCh (4.2×10−7 M)-induced ICl(Ca) in oocytes expressing m3 muscarinic receptors by (A) intracellularly injected QX314 (IC50 445±226 μM), (B) extracellularly administered benzocaine (IC50 258±182 μM) and (C) intracellularly applied lidocaine (IC50 341±33 μM).
To determine if an additional uncharged site of action may play a role, we studied the effect of the permanently uncharged and therefore membrane permeant local anaesthetic benzocaine. Benzocaine, extracellularly applied, inhibited with an IC50 of 259±182×10−6 M (nH=0.4; Figure 4B). That benzocaine alone is also not able to mimick m3 receptor inhibition with lidocaine suggests that two separate binding sites contribute to lidocaine inhibition: a charged intracellular site, and an uncharged site which could be located either intra- or extracellularly.
To determine the location of this uncharged LA site we injected lidocaine into the oocyte and prevented any extracellular effect by continuous copious superfusion of the cell with Tyrode's solution. If the uncharged site of action is located intracellularly we would expect intracellular lidocaine to show an IC50 similar to that obtained when administered extracellularly (since it would have access to both charged and uncharged sites). Conversely, if the uncharged binding site is located on the extracellular domain of the m3 receptor, we would expect lidocaine, intracellularly injected, to inhibit at similar concentrations as did intracellularly injected QX314 (since it would only have access to the charged site). As shown in Figure 4C, lidocaine inhibited m3 responses to a similar degree as did QX314. Calculated IC50 was 341±33×10−6 M (nH=0.8). We therefore hypothesize that m3 receptor inhibition by lidocaine is due to interactions at two sites, where the uncharged form acts on the extracellular receptor domain and the charged one acts intracellularly.
Inhibition of m3 signalling by intracellular QX314 depends on Gαq
We studied in more detail the location of the intracellular site of action of QX314. The concentration-response curve of intracellular QX314 on m3 receptor signalling is highly similar to that obtained on lysophosphatidic acid (LPA) receptors and m1 receptors (Hollmann et al., 2000). Since LPA and muscarinic receptors show very little sequence similarity, this suggests that the local anaesthetic acts at a common coupled G protein, rather than at the receptors (effects downstream of the G protein have been ruled out (Sullivan et al., 1999). We have shown previously that m1 muscarinic receptors couple selectively to Gαq and Gα11, whereas LPA receptors couple to Gαq and Gαo, (Hollmann et al., 2001) suggesting Gq as the site of action. Using antisense constructs directed against G protein α subunits, we investigated the G protein coupling of m3 muscarinic receptors. Experiments were performed 48 h after antisense injection. Injection of anti-Gαq (49±6% of control response) or anti-Gα11 (65.4±6.8% of control response) affected MCh-induced responses significantly (P<0.05, one-way ANOVA, Dunnett correction), whereas anti-Gαo- (111±14% of control response) or anti-Gα14- (113±13% of control response) injected oocytes showed responses not significantly different from those observed in control cells (P>0.05, one-way ANOVA, Dunnett correction) (Figure 5A). Similar to our findings for muscarinic m1 signalling (Hollmann et al., 2001), m3 signalling is mainly mediated by Gαq and Gα11.
Figure 5.
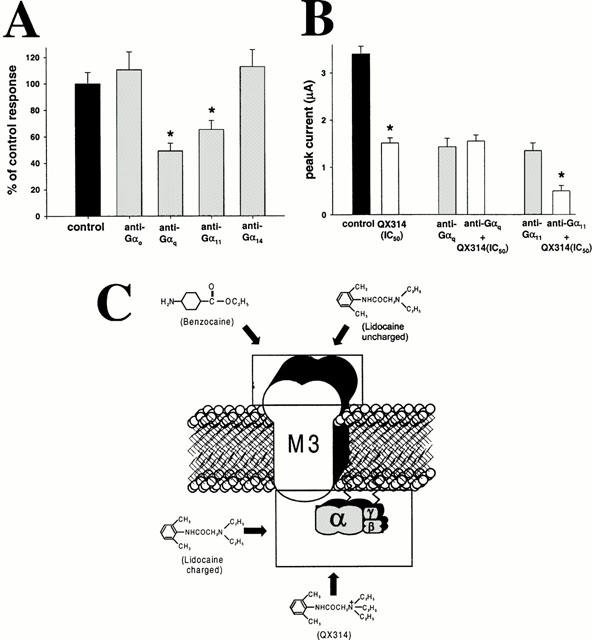
(A) Responses to MCh (4.2×10−7 M) after selective G-protein α-subunit knockdown using oligonucleotides directed against Gαo, Gαq, Gα11 or Gα14, as compared with control responses. Data were collected 48 h after antisense injection. Knockdown of Gαq (49.3±5.7% of control) or Gα11 (65.4±6.8% of control) significantly affected MCh-induced responses (P<0.05), whereas anti-Gαo (110.7±13.6% of control) or anti-Gα14 (112.9±12.9% of control) injected cells showed responses similar to those observed in control cells (P<0.05). Therefore, m3 signalling is mediated primarily by Gq and G11. (B) Mean±s.d. of ICl(Ca), elicited by MCh (10−7 M) in oocytes expressing m3 muscarinic receptors, injected with 50 nl of 150 mM KCl (control, 3.4±0.16 μA, black bar); QX314 (445×10−6 M) reduced m3 responses to 44±3% of the control response (1.51±0.11 μA, white bar) 150 mM KCl 48 h after injection of either anti-Gαq (1.43±0.18 μA) or anti-Gα11 (1.35±0.24 μA, grey bars); or QX314 (445×10−6 M) 48 h after injection of either anti-Gαq (1.55±0.13 μA) or anti-Gα11 (0.5±0.11 μA, white bars). Lack of effect of the local anaesthetic after Gq knockdown indicates that inhibition requires this G proteins subunit. (C) Schematic summary of hypothesized sites of action on m3 muscarinic signalling for the local anaesthetics studied.
These findings suggest that Gαq may be the target for intracellular charged LA. We therefore determined the ability of intracellular QX314 to inhibit m3 muscarinic signalling in cells in which Gαq had been depleted by prior injection of antisense constructs. We chose an MCh concentration somewhat less than EC50, because injection of 150 nM KCl (50 nl) used in control cells and in treatment cells as buffer for QX314 caused substantially increased response sizes as a result of the increased intracellular Cl−-load. The inhibitory effect of intracellularly injected QX314 at IC50 (445×10−6 M) was studied in oocytes injected 48 h prior with solvent, anti-Gαq or anti-Gα11, which, as shown above, mediate the m3 response. Oocytes were stimulated with 0.1 μM MCh. As shown in Figure 5B, intracellularly injected QX314 at IC50 (445×10−6 M) reduced the responses to 0.1 μM MCh to 44.4±3.2% (1.51±0.11 μA) of control response. In addition, significant inhibition of m3 responses by intracellularly injected QX314 was obtained only in anti-Gα11 injected cells (0.5±0.1 μA vs 1.4±0.2 μA, P<0.001, t-test). In contrast, in anti-Gαq injected cells QX314 had no significant inhibitory effect (1.4±0.2 μA vs 1.6±0.1 μA, P=0.724, t-test). These findings indicate that QX314 inhibits only when functional Gαq is present, suggesting that it mediates its inhibitory effect by acting on this G protein subunit.
Discussion
In the present study we investigated the effect of LA on the functioning of muscarinic m3 receptors. We demonstrated that lidocaine reversibly inhibits signalling of m3 receptors recombinantly expressed in Xenopus oocytes. The calculated IC50 (370 nM) is significantly less than that required for blocking sodium channels (60 – 200 μM, depending on the state of the Na+-channel) (Scholz et al., 1998). (For comparison, clinically relevant blood concentrations during i.v. infusion or epidural anaesthesia are in the range of 1 – 15 μM, corresponding to a plasma level of 0.3 to 4.5 μg ml−1). However, compared with inhibition of m1 muscarinic signalling (IC50 18 nM) (Hollmann et al., 2000), m3 signalling is approximately 21 fold less sensitive to lidocaine. This discrepancy is explained most parsimoniously by absence on the m3 receptor of the major extracellular binding site for charged LA that is present on the m1 receptor. The N-terminus and third extracellular loop of the m1 receptor molecule were identified as requirements for this extracellular binding site for charged LA. In contrast, intracellular inhibition of both receptors was quite similar. As determined for m1 signalling (Hollmann et al., 2001), m3 signalling is primarily mediated by Gq and G11; of these, Gq was shown to be a likely target for intracellular inhibition by LA.
It is of interest that, despite their sequence similarity except for the amino-terminus and the third intracellular loop (Bonner, 1989), the m3 receptor lacks the extracellular binding site for charged LA present on the m1 receptor (Hollmann et al., 2000). Since the greatest variety in amino acid sequence extracellularly between both receptors is attributable to their N-terminus, we built chimeric constructs to identify domains of the muscarinic receptor molecule required for this extracellular binding site for charged LA. Our results suggest that the N-terminal as well as the third extracellular loop of the m1 receptor molecule are required to form the binding pocket for charged LA. The amino terminus is quite divergent between m1 and m3 receptors, and significant additional work will be required to delineate the amino acids involved in the binding site. However, since in the third extracellular loop only five amino acids are different between the m1 and m3 receptors, further investigations might be able to exactly characterize the amino acid residues involved in this part of the binding site for LA on muscarinic receptors.
In contrast to the difference in extracellular actions, intracellular injection of lidocaine or QX314, a permanently charged and therefore membrane impermeant lidocaine analogue, inhibited m3 signalling quite similarly to their effect on lysophosphatidate (LPA) and m1 muscarinic signalling (Hollmann et al., 2000), suggesting a common intracellular site of action for LA. Similar to m1 signalling, we found m3 signalling primarily mediated by Gαq and Gα11. This is in agreement with findings by Stehno-Bittel et al. (1995). Employing the Xenopus oocyte expression system, they showed that antisense oligonucleotides and antibodies to Gαq and Gα11 blocked m3-mediated signal transduction by inhibiting interaction of the muscarinic receptor with the G protein. Interestingly, agents that specifically bound free Gβγ also inhibited acetylcholine-induced activation of phospholipase C-β (PLC-β). Conversely, direct injection of Gβγ subunits into oocytes induced release of intracellular Ca2+, suggesting that receptor coupling is determined by Gαq but that Gβg is the predominant signalling molecule activating oocyte PLC-β (Stehno-Bittel et al., 1995). These findings are not specific to the oocyte. Morel et al. (1997) studied coupling of G protein subunits involved in acetylcholine-induced Ca2+ release in mouse duodenal myocytes. Intracellular dialysis with a patch pipette solution containing anti-Gαq/11 antibodies inhibited acetylcholine-induced Ca2+ release, and antisense oligonucleotide studies showed that only Gαq was involved.
Further studies should elucidate the exact mechanism by which LA inhibit Gq functioning. Several possible explanations exist. First, LA might interfere selectively with the coupling of the Gq protein to the muscarinic receptor. Either decreasing the affinity of Gαq for the receptor (preventing activation of the G protein), but also stabilization of the receptor-G protein complex (preventing uncoupling of the G protein from the receptor), as suggested for volatile anaesthetics (Aronstam & Dennison, 1989), could be underlying mechanisms. Second, LA might stabilize the Gαq-βγ complex, thus reducing free Gβγ and preventing PLC activation. A maybe less likely possibility would be LA-enhanced guanosinetriphosphatase (GTPase) activity, due to more effective GTPase-activating protein (GAP) activity of a regulator of G protein signalling (RGS) protein. This would lead to faster reassembly of the heterotrimeric complex. Since the guanosindiphosphate (GDP)/guanosintriphosphate (GTP) binding site is highly conserved among all Gα-subunits, it seems unlikely that LA interact selectively with the nucleotide binding site on Gαq. Whatever the underlying mechanism, it will have to account for the ability of local anaesthetic to differentiate between Gq and G11, two very closely related G protein α subunits.
However, the profound sensitivity of m3 receptor signalling to extracellularly applied lidocaine cannot be solely explained by a single intracellular site of action. Our findings using benzocaine and intracellularly injected lidocaine suggest that inhibition of m3 signalling by lidocaine is most likely due to a combined effect on an intracellular charged site on Gq and an extracellular uncharged site on the m3 receptor molecule, as illustrated in Figure 5. Interaction of two separate binding sites explaining LA inhibition of G protein-coupled receptors (m1 muscarinic and LPA receptors) were demonstrated in previous studies from our group (Hollmann et al., 2000; Sullivan et al., 1999).
In summary, our present study adds to previous findings that LA at clinically relevant concentrations can inhibit G protein-coupled receptors. The interactions between the anaesthetics and the receptor pathways are complex, and involve multiple sites of action, on receptor as well as coupled G protein. In view of the important roles of m1 and m3 muscarinic signalling in the brain and peripheral tissues, LA inhibition of these receptors is likely to be of relevance. The clinical implications remain to be addressed.
Acknowledgments
We gratefully acknowledge Prof Dr med Eike Martin (Ruprecht-Karls-Universität Heidelberg, Germany) and Anne M. Byford (University of Virginia, U.S.A.) for their support. Sincere thanks to Astra Pharmaceuticals, LP (Westborough, MA, U.S.A.) for supplying QX314. Dr Hollmann is supported in part by the Department of Anesthesiology, University of Heidelberg, Heidelberg, Germany, by the 2000 BenCovino Research Award, sponsored by AstraZeneca Pain Control, Sweden and by a grant of the German Research Society (DFG HO 2199/1-1), Bonn, Germany. Supported in part by National Institutes of Health grant GMS 52387, Bethesda, MD, U.S.A. and an American Heart Association grant (Mid-Atlantic Affiliation) VHA 9920345U, Baltimore, MD, U.S.A.
Abbreviations
- Bmax
calculated receptor density
- 4-DAMP
4-diphenylacetoxy-N-methylpiperidine
- cDNA
complementary DNA
- CHO
Chinese hamster ovaries
- cRNA
complementary RNA
- Emax
maximal response size
- GAP
GTPase-activating protein
- GDP
guanosindiphosphate
- GTP
guanosintriphosphate
- [3H]-QNB
[3H] quinuclydinyl benzylate
- ICl(Ca)
Ca2+-activated Cl−-currents
- Kd
dissociation constant
- LA
local anaesthetics
- LPA
lysophosphatidic acid
- MCh
acetyl-β-methylcholine bromide
- MAC
minimal alveolar concentration
- MOPS
3-[N-morpholino]propanesulphonic acid
- RGS
regulator of G protein signalling
References
- ARONSTAM R.S., DENNISON R.L. Anesthetic effects on muscarinic signal tranduction. Int. Anesthesiol. Clin. 1989;27:265–272. doi: 10.1097/00004311-198902740-00005. [DOI] [PubMed] [Google Scholar]
- BONHOMME V., MEURET P., BACKMAN S.B., PLOURDE G., FISET P.Cholinergic mechanisms mediating propofol-induced loss of consciousness in man Can. J. Anaesth. 199845A52(Abstract) [Google Scholar]
- BONNER T.I. The molecular basis of muscarinic receptor diversity. TINS. 1989;12:148–151. doi: 10.1016/0166-2236(89)90054-4. [DOI] [PubMed] [Google Scholar]
- HOLLMANN M.W., WIECZOREK K., BERGER A., DURIEUX M.E. Local anesthetic inhibition of G protein-coupled receptor signaling by interference with Gαq protein function. Mol. Pharmacol. 2001;59:294–301. doi: 10.1124/mol.59.2.294. [DOI] [PubMed] [Google Scholar]
- HOLLMANN M.W., FISCHER L.G., BYFORD A.M., DURIEUX M.E. Local anesthetic inhibition of m1 muscarinic acetylcholine signaling. Anesthesiology. 2000;93:497–509. doi: 10.1097/00000542-200008000-00030. [DOI] [PubMed] [Google Scholar]
- MOREL J.L., MACREZ N., MIRONNEAU J. Specific Gq protein involvement in muscarinic M3 receptor-induced phosphatidylinositol hydrolysis and Ca2+ release in mouse duodenal myocytes. Br. J. Pharmacol. 1997;121:451–458. doi: 10.1038/sj.bjp.0701157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIETGEN G.W., HOENEMANN C.W., CHAN C.K., KAMATCHI G.L., DURIEUX M.E. Volatile anaesthetics have differential effects on recombinant m1 and m3 muscarinic acetylcholine receptor function. Br. J. Anaesth. 1998;81:569–577. doi: 10.1093/bja/81.4.569. [DOI] [PubMed] [Google Scholar]
- SCHOLZ A., KUBOYAMA N., HEMPELMANN G., VOGEL W. Complex blockade of TTX-resistant Na+ currents by lidocaine and bupivacaine reduce firing frequency in DRG neurons. J. Neurophysiol. 1998;79:1746–1754. doi: 10.1152/jn.1998.79.4.1746. [DOI] [PubMed] [Google Scholar]
- SHAPIRA H., AMIT I., REVACH M., ORON Y., BATTEY J.F. Gα14 and Gαq mediate the response to trypsin in Xenopus oocytes. J. Biol. Chem. 1999;273:19431–19436. doi: 10.1074/jbc.273.31.19431. [DOI] [PubMed] [Google Scholar]
- STEHNO-BITTEL L., KRAPIVINSKY G., KRAPIVINSKY L., PEREZ-TERZIC C., CLAPHAM D.E. The G protein beta gamma subunit transduces the muscarinic receptor signal for Ca2+ release in Xenopus oocytes. J. Biol. Chem. 1995;270:30068–30074. doi: 10.1074/jbc.270.50.30068. [DOI] [PubMed] [Google Scholar]
- SULLIVAN L.M., HOENEMANN C.W., ARLEDGE J.A.M., DURIEUX M.E. Synergistic inhibition of lysophosphatidic acid signaling by charged and uncharged local anesthetics. Anesth. Analg. 1999;88:1117–1124. doi: 10.1097/00000539-199905000-00029. [DOI] [PubMed] [Google Scholar]