

Abstract
β-Amyloid peptide (Aβ), a 39 – 43 amino acid peptide, is believed to induce oxidative stress and inflammation in the brain, which are postulated to play important roles in the pathogenesis of Alzheimer's disease. Ferulic acid is an antioxidant and anti-inflammatory agent derived from plants; therefore, the potential protective activity of ferulic acid against Aβ toxicity in vivo was examined.
Mice were allowed free access to drinking water (control) or water containing ferulic acid (0.006%). After 4 weeks, Aβ1-42 (410 pmol) was administered via intracerebroventricular injection.
Injection of control mice with Aβ1-42 impaired performance on the passive avoidance test (35% decrease in step-through latency), the Y-maze test (19% decrease in alternation behaviour), and the water maze test (32% decrease in percentage time in platform-quadrant). In contrast, mice treated with ferulic acid prior to Aβ1-42 administration were protected from these changes (9% decrease in step-through latency; no decrease in alternation behaviour; 14% decrease in percentage time in platform-quadrant). Aβ1-42 induced 31% decrease in acetylcholine level in the cortex, which was tended to be ameliorated by ferulic acid.
In addition, Aβ1-42 increased immunoreactivities of the astrocyte marker glial fibrillary acidic protein (GFAP) and interleukin-1β (IL-1β) in the hippocampus, effects also suppressed by pretreatment with ferulic acid.
Administration of ferulic acid per se unexpectedly induced a transient and slight increase in GFAP and IL-1β immunoreactivity in the hippocampus on day 14, which returned to basal levels on day 28. A slight (8%) decrease in alternation behaviour was observed on day 14.
These results demonstrate that long-term administration of ferulic acid induces resistance to Aβ1-42 toxicity in the brain, and suggest that ferulic acid may be a useful chemopreventive agent against Alzheimer's disease.
Keywords: β-amyloid peptide (Aβ), in vivo toxicity, ferulic acid, chemoprevention, Alzheimer's disease
Introduction
A pathologic hallmark of Alzheimer's disease is the formation of senile plaques (Glenner & Wong, 1984). β-Amyloid peptide (Aβ), a 39 – 43 amino acid peptide, is a major component of these plaques. Aβ was shown to have the potential to induce oxidative stress and inflammation in the brain, which are postulated to play important roles in the pathogenesis of Alzheimer's disease (Behl, 1999; McGeer & McGeer, 1999). For example, Aβ induces the production of hydrogen peroxide and lipid peroxide in neurons (Behl et al., 1994). In addition, Aβ has been reported to induce superoxide (Mcdonald et al., 1997) and proinflammatory cytokines (Araujo & Cotman, 1992; Gitter et al., 1995) in astrocytes as well as in microglial cells. Antioxidants such as α-tocopherol protect against cytotoxicity in vitro (Behl et al., 1992) as well as learning and memory deficits induced by Aβ (Yamada et al., 1999). Furthermore, α-tocopherol and anti-inflammatory agents such as indomethacin reportedly slow the progression of Alzheimer's disease (Sano et al., 1997; Rogers et al., 1993).
Ferulic acid (4-hydroxy-3-methoxycinnamic acid), a phenolic compound present in a variety of plants, has potent antioxidant (Graf, 1992; Scott et al., 1993) and anti-inflammatory activities (Fernandez et al., 1998; Ozaki, 1992). Therefore, we hypothesized that ferulic acid may have beneficial effects in Alzheimer's patients. To address this hypothesis, we investigated the potential preventive effect of long-term oral administration of ferulic acid against Aβ1-42-induced toxicity in vivo.
Methods
Materials
Male ICR mice weighing 18 – 26 g at the beginning of experiments (Myung-Jin, Inc., Seoul, Korea) were used in all the experiments. The mice were housed five per cage in a room maintained at 22±1°C with an alternating 12 h light-dark cycle. Food was available ad libitum. Aβ1-42 (American Peptide Company, U.S.A.) and Aβ42-1 (Bachem, Switzerland) were prepared as stock solutions at a concentration of 37 μg/μl in sterile 0.1 M phosphate-buffered saline (pH 7.4) and aliquots were stored at −20°C until use. Ferulic acid (Sigma) was dissolved each day in tap water at concentrations of 0.002%, 0.004%, and 0.006% (w v−1). Assay with high performance liquid chromatography with electrochemical detection showed 11% loss of ferulic acid (0.006% w v−1) in tap water in 24 h at room temperature.
Experimental design
Mice were allowed free access to normal drinking water or ferulic acid solution for 1, 2, 3 or 4 weeks. Subsequently, Aβ1-42 was administered via intracerebroventricular injection and the behavioural tests were started 1 day after injection (see Figure 2). Control animals were injected with Aβ42-1. Each group consisted of 10 mice. Learning and memory capacity was assessed using three separate tests: passive avoidance, Y-maze and Morris water maze tests.
Figure 2.
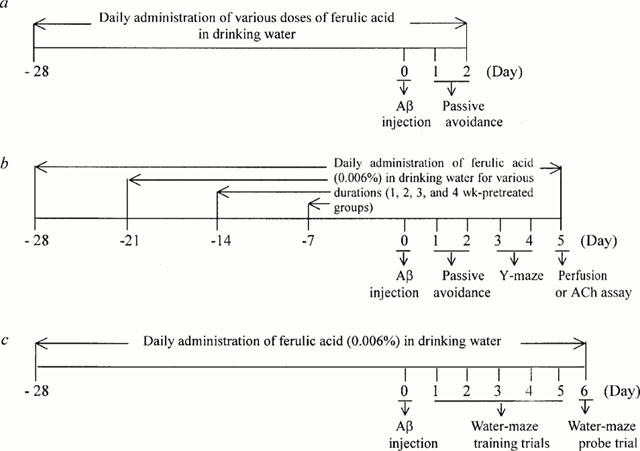
Experimental schedules.
Intracerebroventricular injection of β-amyloid peptide
The administration of Aβ1-42 was performed according to the procedure established by Laursen & Belknap (1986). Briefly, each mouse was injected at bregma with a 50 μl Hamilton microsyringe fitted with a 26-gauge needle that was inserted to a depth of 2.4 mm. The injection volume was 5 μl.
Passive avoidance performance
One or 7 days after Aβ1-42 injection, mice were trained on a one-trial step-through passive avoidance task. The passive avoidance box was divided into two compartments, one illuminated and one dark, equipped with a grid floor. During the training trial, each mouse was placed in the lighted compartment; as soon as it entered the dark compartment, the door was closed and the mouse received an inescapable shock (0.25 mA, 1 s). In the testing trial, given 1 day after the training trial, the mouse was again placed in the lighted compartment and the time until it re-entered the dark compartment was measured (the step-through latency maximum testing limit was 300 s).
Y-maze task
Immediate working memory performance was assessed by recording spontaneous alternation behaviour in a Y-maze (Sarter et al., 1988). The Y-maze task was carried out on days 3 and 4 after Aβ1-42 administration. The maze was made of black-painted wood and each arm was 25 cm long, 14 cm high, 5 cm wide and positioned at equal angles. Mice were placed at the end of one arm and allowed to move freely through the maze during an 8-min session. The series of arm entries was recorded visually and arm entry was considered to be completed when the hind paws of the mouse were completely placed in the arm. Alternation was defined as successive entries into the three arms on overlapping triplet sets. The percentage alternation was calculated as the ratio of actual to possible alternations (defined as the total number of arm entries minus two), multiplied by 100.
Water maze task
The Morris water maze was performed as described previously (Morris, 1984). The experimental apparatus consisted of a circular water tank (diameter=100 cm; height=35 cm), containing water at 23°C to a depth of 15 cm and rendered opaque by adding powdered milk. A platform (diameter=4.5 cm; height=14.5 cm) was submerged 0.5 cm below the water surface and placed at the midpoint of one quadrant. The pool was located in a test room containing various prominent visual cues. Three training trials per day were conducted for 5 consecutive days post Aβ1-42 injection, with an inter-trial interval of 5 min. Mice were placed in the pool at one of four starting positions. In each training trial, the time required to escape onto the hidden platform was recorded. Mice that found the platform were allowed to remain on the platform for 10 s and were then returned to the home cage during the inter-trial interval. Mice that did not find the platform within 120 s were placed on the platform for 10 s at the end of trial. The probe test was carried out on day 6 post-injection. For this test, the platform was removed from the pool and the trial was performed with the cutoff time of 90 s. The time spent in the quadrant that previously contained the platform was recorded as a percentage of the total time in the pool.
Measurement of acetylcholine levels
Mice were sacrificed 5 days after Aβ1-42 injection. Sections of cortex were collected, rapidly frozen and stored at −80°C. Acetylcholine levels were determined as described by Israel & Lesbats (1982). Briefly, brain tissue was extracted with 5% trichloroacetic acid, washed in ether to denature proteins and centrifuged. After allowing the residual ether to evaporate, the protein pellets were dissolved in 1 M NaOH and the solution neutralized and buffered. Fifty μl of sample and reaction buffer (100 μl of 250 units ml−1 choline oxidase, 50 μl of 2 mg ml−1 peroxidase and 100 μl of 1 mM luminol) were mixed together. Subsequently, 5 μl acetylcholinesterase was added to trigger the hydrolysis of acetylcholine and light emission was measured with a luminometer (Lumat LB 9507).
Immunocytochemistry
Five days after injection of Aβ, all mice were transcardially perfused and post-fixed for 3 h in 4% paraformaldehyde. Brains were cryoprotected in 30% sucrose, sectioned coronally (45 μm) on a freezing microtome and collected in cryoprotectant for storage at −20°C. Floating sections of brains were processed as described previously (Baker & Farbman, 1993). After five 10-min rinses in PBS, sections were placed in cryoprotectant, pre-incubated for 30 min in 0.1 M PBS with 1% bovine serum albumin and 0.2% Triton X-100 and incubated overnight with the following primary antisera: rabbit anti-GFAP (1 : 2000; Chemicon) and rabbit anti-IL-1β (1 : 250; R&D). On the following day, sections were incubated for 1 h in biotinylated rabbit secondary antibody obtained from Vector laboratories. After incubation with the Vector Elite ABC kit, antigens were detected with 3,3-diaminobenzidine tetrahydrochloride (DAB) as the chromogen.
Statistical analysis
Statistical analysis was carried out by two-way analysis of variance (Figures 1 and 3e) or one-way analysis of variance (Figures 3a – d,f, and 4); the Newman-Keuls test was used for post hoc comparisons with P<0.05 considered to indicate statistical significance.
Figure 1.
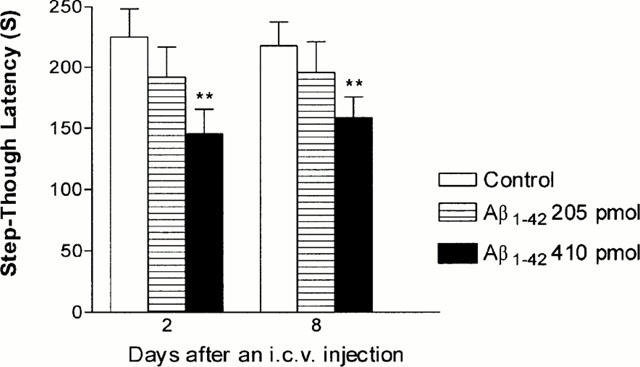
Effects of Aβ1-42 given by intracerebroventricular injection on passive avoidance performance in mice. Various doses of Aβ1-42 were injected and on day 1 post-injection, mice were subjected to the training trial; testing trials were conducted on day 2 and day 8 post-injection. Control animals were injected with Aβ42-1 (410 pmol per mouse). The data are expressed as mean±s.e.mean, with n=10 – 20 mice per group. ** P<0.01 compared to Aβ42-1-treated control.
Figure 3.
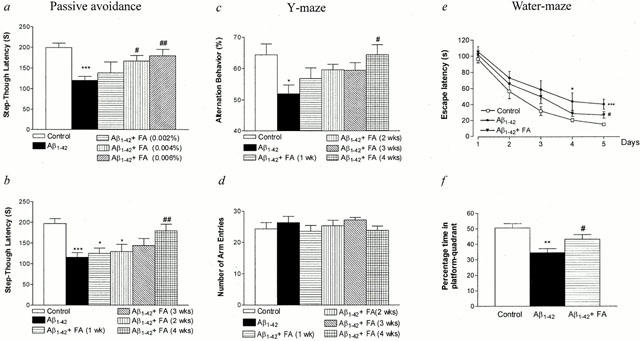
Protective effect of ferulic acid on the Aβ1-42-induced impairment in learning and memory in mice. After injection of Aβ (410 pmol per mouse), each behavioural test was performed as shown in Figure 2. Passive avoidance task (a,b): On day 1 post-injection, mice were trained on a one-trial step-through passive avoidance task. The testing trial was given 1 day after the training trial. (a) Dose-dependent effect of ferulic acid. (b) Time-dependent effect of ferulic acid. Y-maze task: Spontaneous alternation behaviour (c) and the number of arm entries (d) were measured during an 8-min session. Water-maze task: The training trials (e) and probe trial (f) were carried out on days 1 – 5 and on day 6 after Aβ injection, respectively. The latency showed the mean of a block of three trials per day (e). The data are presented as means±s.e.mean (n=10 – 20). Control mice were injected with Aβ42-1 (410 pmol per mouse). * P<0.05, ** P<0.01, *** P<0.001 vs Aβ42-1-treated control, # P<0.05, ## P<0.01 vs Aβ1-42 alone.
Figure 4.
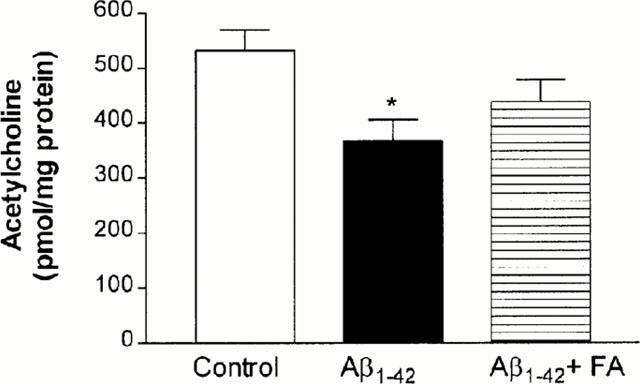
Effect of ferulic acid on the Aβ1-42-induced decrease in acetylcholine levels. Mice were allowed free access to the drinking water containing ferulic acid (0.006%) for up to 4 weeks prior to 410 pmol Aβ1-42 or Aβ42-1 injection. Acetylcholine level in the cortex was measured on day 5 after Aβ injection. Control animals were injected with 410 pmol Aβ42-1. The data are means±s.e.mean (n=10). * P<0.05 vs Aβ42-1-treated control.
Results
Effect of intracerebroventricular Aβ1-42 on passive avoidance performance
Intracerebroventricular administration of Aβ causes memory deficits (Flood et al., 1991; Maurice et al., 1996; Nabeshima & Nitta, 1994; Nitta et al., 1994) and decreases choline acetyltransferase activity (Nitta et al., 1994; 1997); therefore, this method of Aβ exposure is a useful in vivo model for Aβ toxicity. To determine the dose of Aβ1-42 sufficient to cause memory impairment in mice, two different doses (205 and 410 pmol per mouse) of Aβ1-42 were injected intracerebroventricularly and mice were subjected to a passive avoidance task on days 2 and 8 post-injection. Control animals were injected with non-toxic reverse fragment Aβ42-1. As shown in Figure 1, mice injected with 410 pmol Aβ1-42 exhibited a significant reduction (35% decrease; P<0.01) in the step-through escape latency on day 2 post-injection compared to Aβ42-1 treated controls. This effect persisted for up to 8 days post-injection. Thus, the passive avoidance task was performed on day 2 post-injection of 410 pmol Aβ1-42 in subsequent experiments.
Effect of ferulic acid on Aβ1-42-induced memory impairment
In a separate experimental series, mice were treated with ferulic acid in the drinking water at various concentrations for up to 4 weeks prior to Aβ1-42 administration. This treatment protocol is summarized in Figure 2. Because the average water intake per mouse per day was approximately 6 to 8 ml, the amount of ferulic acid consumed by mice receiving 0.006% ferulic acid in the drinking water ranged from 360 to 480 μg mouse−1 day−1, which is approximately 14 to 19 mg kg−1 day−1. All mice treated with ferulic acid gained body weight normally (32.2±0.9 g for control mice versus 31.8±1.3 g for mice treated with ferulic acid for 4 weeks) and did not show any signs of toxicity during the experiment (data not shown).
As shown in Figure 3a, treatment of mice with ferulic acid for 4 weeks (Figure 2a) attenuated the Aβ1-42-induced impairment of passive avoidance performance in a dose-dependent manner with maximal effects observed at a concentration of 0.006%. Thus, this concentration was used in the subsequent experiments. To determine the length of ferulic acid pretreatment necessary to yield optimal protection, a time-course study was performed (Figure 2b). The results indicated that 4 weeks of pretreatment effectively prevented the Aβ1-42-induced decrements in the passive performance test (Figure 3b).
Spontaneous alternation behaviour, which is regarded as a measure of spatial memory (Sarter et al., 1988), was investigated using the Y-maze test. Mice injected with Aβ1-42 displayed significantly impaired spatial working memory (19% decrease in alternation behaviour), when measured on day 4 post-injection (Figure 3c); pretreatment with ferulic acid blunted the Aβ1-42-induced decrease in alternation behaviour with the greatest protection observed in mice pretreated for 4 weeks (Figure 3c). In contrast, the number of arm entries did not change among all the experimental groups, demonstrating that general locomotor activity was not affected by Aβ1-42 (Figure 3d).
In the water maze test (Figure 2c), Aβ1-42 impaired place learning and this change was significantly different from Aβ42-1 treated controls after 5 days of training (Figure 3e); this effect was also ameliorated by ferulic acid. Similarly, Aβ1-42 adversely affected performance in the probe test (32% decrease in percentage time in platform-quadrant) (Figure 3f); ferulic acid effectively inhibited reductions (14% decrease) in this parameter.
Aβ1-42 decreased acetylcholine levels by 31% (P<0.05) in the cortex (Figure 4). Pretreatment with ferulic acid tended to inhibit the Aβ1-42-induced decrease in acetylcholine levels, but the inhibition did not reach statistical significance.
Effect of ferulic acid on Aβ1-42-induced increase in GFAP and IL-1β immunoreactivity
Glial fibrillary acidic protein (GFAP) and interleukin-1β (IL-1β) immunoreactivities in the hippocampus were markedly enhanced on day 5 post-injection of Aβ1-42 (Figure 5b,e). The IL-1β-immunoreactive cells appeared to be astrocytes. In animals pretreated with ferulic acid for 4 weeks, the Aβ1-42-induced increases in hippocampal GFAP and IL-1β immunoreactivities were effectively blocked (Figure 5c,f). To determine if Aβ1-42 induced apoptotic neuronal death, the TdT-mediated dUTP nick-end labelling (TUNEL) procedure was performed in the hippocampus. Apoptosis was not induced by Aβ1-42 (data not shown). Furthermore, there was no acid fuchsin-stained degenerating neuronal cells in mice injected with Aβ1-42 (data not shown).
Figure 5.
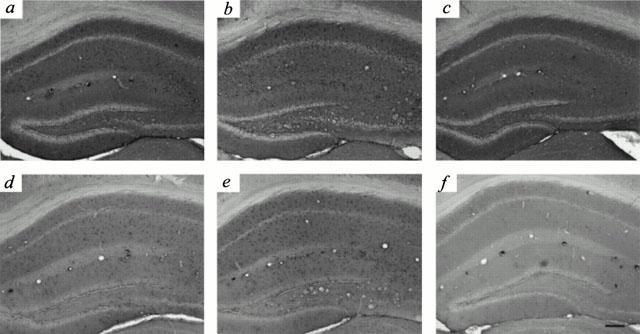
Blockade of the Aβ1-42-induced increase in GFAP and IL-1β immunoreactivity by ferulic acid. Mice were allowed free access to normal drinking water (a,b,d,e), or water containing ferulic acid (0.006%) (c,f) for up to 4 weeks prior to Aβ injection. GFAP (a – c) and IL-1β (d – f) immunoreactivities in the hippocampus were examined on day 5 after injection of 410 pmol Aβ42-1 (a,d) or Aβ1-42 (b,c,e,f). Mice injected with 410 pmol Aβ42-1 (a,d) served as controls. Scale bar, 100 μm.
Effect of ferulic acid per se on the immunoreactivity of various neurotrophic factors, GFAP, and IL-1β
To study the possible mechanisms of the protective effect of ferulic acid against Aβ1-42 toxicity, expressions of various neurotrophic factors in the hippocampus were examined at various time points (1, 5, 14, and 28 days) after administration of ferulic acid per se. Administration of ferulic acid did not affect the immunoreactivity of nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), and basic fibroblast growth factor (bFGF) (data not shown).
Unexpectedly, a transient and slight increase in GFAP immunoreactivity in the hippocampus was observed in the ferulic acid-treated animals. The ferulic acid-induced increase in GFAP immunoreactivity, which was much less intense than that induced by Aβ1-42 injection, was apparent on day 5, peaked on day 14, and returned to basal levels on day 28 (Figure 6a – e). Similarly, hippocampal IL-1β immunoreactivity was transiently and slightly increased in the ferulic acid-treated animals on day 14, which returned to basal levels on day 28 (Figure 6f – j). The IL-1β-immunoreactive cells appeared to be astrocytes. A slight but not significant decrease in avoidance performance was noted on day 14 (200±16 s for control mice versus 171±17 s for mice treated with ferulic acid for 14 days, P=0.19, n=20), which had recovered to control levels on day 28 (211±18 s for control mice versus 203±23 s for mice treated with ferulic acid for 28 days). A slight decrease (8%; P<0.05) in alternation behaviour in the Y-maze test was noted on day 14 (72±2% for control mice versus 66±2% for mice treated with ferulic acid for 14 days).
Figure 6.
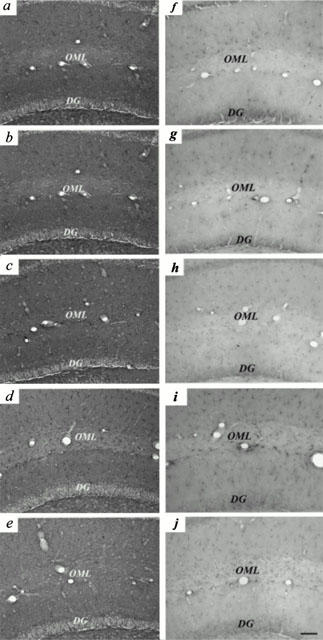
Ferulic acid induces transient increase in GFAP and IL-1β immunoreactivities in hippocampus. Mice were allowed free access to water containing ferulic acid (0.006%) for 1 (b,g), 5 (c,h), 14 (d,i) and 28 (e,j) days before being examined for GFAP (a – e) and IL-1β (f – j) immunoreactivities in hippocampus. Mice not exposed to ferulic acid (a,f) served as controls. OML, outer molecular layer, DG, dentate gyrus. Scale bar, 50 μm.
Effect of post-treatment with ferulic acid on Aβ1-42-induced impairment of passive avoidance performance
To examine the therapeutic effect of ferulic acid on the Aβ1-42-induced impairment of passive avoidance performance, ferulic acid began to be administered to mice either immediately, 2 days, or 8 days after an intracerebroventricular injection of Aβ1-42 (410 pmol per mouse). As shown in Figure 7, post-treatment of ferulic acid tended to alleviate the Aβ1-42-induced impairment of passive avoidance performance, especially for the group treated with ferulic acid immediately after Aβ1-42 injection, but the inhibition did not reach statistical significance.
Figure 7.
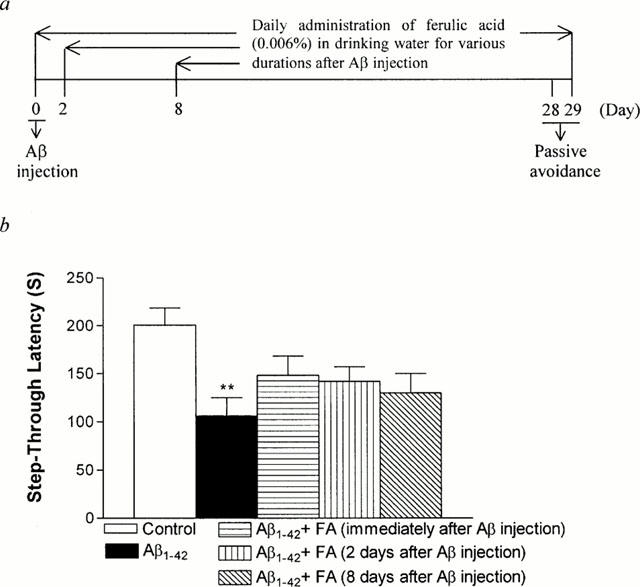
Therapeutic effect of ferulic acid on the Aβ1-42-induced impairment in passive avoidance performance in mice. After injection of Aβ (410 pmol per mouse), ferulic acid was administered to mice after either immediately, 2 days, or 8 days after an i.c.v. injection of Aβ1-42. On day 28 post-injection, mice were trained on a one-trial step-through passive avoidance task. The testing trial was given 1 day after the training trial (a, Experimental schedule). The data are presented as means±s.e.mean (n=10 – 11). Control mice were injected with Aβ42-1 (410 pmol per mouse).
Discussion
An intracerebroventricular injection of low nanomolar doses of Aβ25-35 (Maurice et al., 1996) or Aβ1-28 (Flood et al., 1991; Maurice et al., 1996) impairs avoidance behaviour and Y-maze alternation behaviour in mice. Similarly, continuous intracerebroventricular infusion of Aβ1-40 (Nitta et al., 1994) or Aβ1-42 (Yamada et al., 1999) induces learning and memory impairment in rats. In this study, a single intracerebroventricular injection of a picomolar dose of Aβ1-42 effectively impaired learning and memory behaviour in mice. Furthermore, this behavioural abnormality was accompanied by decrease in cortical acetylcholine levels and increase in hippocampal GFAP immunoreactivity, supporting previous reports in rats that continuous infusion of Aβ1-40 blunts choline acetyltransferase activity and increases GFAP immunoreactivity (Nitta et al., 1994; 1997). Further, this study showed that Aβ1-42 injection induced a marked increase in IL-1β immunoreactivity in the hippocampus. Aβ has been reported to induce IL-1 release from astrocytes in culture (Araujo & Cotman, 1992). IL-1β inhibits long-term potentiation in the hippocampus (Katsuki et al., 1990; Bellinger et al., 1993; Cunningham et al., 1996; Murray & Lynch, 1998). Thus, in addition to the decreased acetylcholine levels, Aβ1-42-induced increase in IL-1β immunoreactivity may have contributed to learning and memory deficit observed in this study. Taken together, results from the present study illustrate the validity of this animal model for at least some aspects of Alzheimer's disease.
The results presented here demonstrated that long-term administration of ferulic acid effectively protected against Aβ1-42 toxicity in vivo. Because ferulic acid has both antioxidant (Graf, 1992; Scott et al., 1993) and anti-inflammatory activities (Chawla et al., 1987; Fernandez et al., 1998; Hirabayashi et al., 1995; Ozaki, 1992), it should be ideal for protection against the oxidative stress and inflammation induced by Aβ1-42. Interestingly, long-term administration of ferulic acid per se induced transient activation of astrocytes in hippocampus on day 14, as shown by GFAP immunostaining. Transient and slight increase in IL-1β immunoreactivity in hippocampus was also noted on day 14, when animals displayed a slight decrease in Y-maze performance. Correlation between the number of astrocytes immunoreactive to GFAP and the impairment of spatial memory has been reported (Onozuka et al., 2000). Because IL-1β inhibits long-term potentiation in the hippocampus (Katsuki et al., 1990; Bellinger et al., 1993; Cunningham et al., 1996; Murray & Lynch, 1998), the slight increase in IL-1β may be related to the slight impairment in Y-maze performance.
The time required for hippocampal astrocytes to be stimulated and then to return to the basal state took 4 weeks, which was required for full protection against in vivo Aβ1-42 toxicity by ferulic acid. Thus, the protective effect of ferulic acid against in vivo Aβ1-42 toxicity may be related, at least in part, to the ferulic acid-induced activation and subsequent recovery of hippocampal astrocytes. Although the hippocampal astrocytes returned to the apparently basal state in 4 weeks, these cells may have been altered to show suppressed response to Aβ1-42 injection. The ferulic acid-induced biochemical changes which are responsible for the resistance to Aβ1-42 toxicity remain to be identified.
It is well established that preconditioning of the brain with mild stresses, including sublethal ischaemia (Kitagawa et al., 1990), hyperthermia (Chopp et al., 1989; Kitagawa et al., 1991), systemic administration of lipopolysaccharide (Tasaki et al., 1997), and cortical spreading depression (Kawahara et al., 1995; Kobayashi et al., 1995) induces tolerance to the subsequent ischaemic insult. Activation of astrocytes by preconditioning stimulus was postulated to be responsible for the induction of ischaemic tolerance (Kato et al., 1994; Matsushima et al., 1998). This phenomenon is apparently similar to the results of the present study. Thus it is suggested that activation and subsequent recovery of hippocampal astrocytes by ferulic acid somehow induces resistance to the subsequent Aβ1-42 insult to the brain. Because astrocytes are source of various neurotrophic factors, activation of astrocytes with accompanying expression of neurotrophic factors was postulated to be responsible for the induction of ischaemic tolerance (Kato et al., 1994; Kawahara et al., 1999). However, when we examined the immunostaining of NGF, BDNF, and bFGF, ferulic acid did not induce significant changes in any of these neurotrophic factors. Thus, it is suggested that other neurotrophic factors or other mechanisms may be involved in the ferulic acid-induced protection against Aβ1-42-induced toxicity in vivo. Further study is needed for the molecular mechanisms of the ferulic acid-induced activation and recovery of astrocytes and subsequent neuroprotection to Aβ1-42.
Although less effective than pretreatment, post-treatment with ferulic acid tended to ameliorate the Aβ1-42-induced toxicity in vivo. Because there is a progressive accumulation of Aβ in brains of Alzheimer's disease, long-term administration of ferulic acid may effectively inhibit toxic effects of newly accumulated Aβ and consequently blunt the slope of deterioration of dementia.
In conclusion, we propose a novel prophylactic strategy against in vivo Aβ1-42 toxicity. Long-term administration of ferulic acid induces a transient and slight increase in GFAP and IL-1β immunoreactivity in the hippocampus with slight impairment in memory behaviour, which returned to control levels in 28 days, despite continued administration. This transient activation of astrocytes may be related to the subsequent resistance to Aβ1-42 toxicity. How long does the resistance persist after discontinuation of ferulic acid remains to be determined. In fact, ferulic acid has been reported to have various other beneficial effects, such as chemopreventive effects on carcinogenesis (Huang et al., 1988), hepatoprotective effects (Wang & Peng, 1994), antithrombotic effects (Gao & Chen, 1988), and beneficial effects on human sperm motility and viability (Zheng & Zhang, 1997). Further, the finding that long-term administration of ferulic acid protected mice against Aβ1-42-induced toxicity in vivo suggests that ferulic acid may be a potential chemopreventive agent against Alzheimer's disease, extending the list of beneficial effects of this agent.
Acknowledgments
This study was supported in part by Ministry of Health and Welfare (HMP-99-O-11-0008-C), Brain Korea 21, and KOSEF through the Brain Disease Research Center at Ajou University. We thank Drs Moo-Ho Won and Jae-Chul Lee for acid fuchsin staining and Dr Choon-Gon Jang for statistical analysis.
Abbreviations
- Aβ
β-amyloid peptide
- GFAP
glial fibrillary acidic protein
- IL-1β
interleukin-1β
References
- ARAUJO D.M., COTMAN C.W. Beta-amyloid stimulates glial cells in vitro to produce growth factors that accumulate in senile plaques in Alzheimer's disease. Brain Res. 1992;569:141–145. doi: 10.1016/0006-8993(92)90380-r. [DOI] [PubMed] [Google Scholar]
- BAKER H., FARBMAN A.I. Olfactory afferent regulation of the dopamine phenotype in the fetal rat olfactory system. Neuroscience. 1993;52:115–134. doi: 10.1016/0306-4522(93)90187-k. [DOI] [PubMed] [Google Scholar]
- BEHL C. Alzheimer's disease and oxidative stress: implications for novel therapeutic approaches. Prog. Neurobiol. 1999;57:301–323. doi: 10.1016/s0301-0082(98)00055-0. [DOI] [PubMed] [Google Scholar]
- BEHL C., DAVIS J., COLE G.M., SCHUBERT D. Vitamin E protects nerve cells from amyloid beta protein toxicity. Biochem. Biophys. Res. Commun. 1992;186:944–950. doi: 10.1016/0006-291x(92)90837-b. [DOI] [PubMed] [Google Scholar]
- BEHL C., DAVIS J.B., LESLEY R., SCHUBERT D. Hydrogen peroxide mediates amyloid beta protein toxicity. Cell. 1994;77:817–827. doi: 10.1016/0092-8674(94)90131-7. [DOI] [PubMed] [Google Scholar]
- BELLINGER F.P., MADAMBA S., SIGGINS G.R. Interleukin 1β inhibits synaptic strength and long-term potentiation in the rat CA1 hippocampus. Brain Res. 1993;628:227–234. doi: 10.1016/0006-8993(93)90959-q. [DOI] [PubMed] [Google Scholar]
- CHAWLA A.S., SINGH M., MURTHY M.S., GUPTA M., SINGH H. Anti-inflammatory action of ferulic acid and its esters in carrageenan induced rat paw oedema model. Indian J. Exp. Biol. 1987;25:187–189. [PubMed] [Google Scholar]
- CHOPP M., CHEN H., HO K.L., DERESKI M.O., BROWN E., HETZEL F.W., WELCH K.M. Transient hyperthermia protects against subsequent forebrain ischemic cell damage in the rat. Neurology. 1989;39:1396–1398. doi: 10.1212/wnl.39.10.1396. [DOI] [PubMed] [Google Scholar]
- CUNNINGHAM A.J., MURRAY C.A., O'NEILL L.A., LYNCH M.A., O'CONNOR J.J. Interleukin-1β (IL-1β) and tumour necrosis factor (TNF) inhibit long-term potentiation in the rat dentate gyrus in vitro. Neurosci. Lett. 1996;203:1–4. doi: 10.1016/0304-3940(95)12252-4. [DOI] [PubMed] [Google Scholar]
- FERNANDEZ M.A., SAENZ M.T., GARCIA M.D. Anti-inflammatory activity in rats and mice of phenolic acids isolated from Scrophularia frutescens. J. Pharm. Pharmacol. 1998;50:1183–1186. doi: 10.1111/j.2042-7158.1998.tb03332.x. [DOI] [PubMed] [Google Scholar]
- FLOOD J.F., MORLEY J.E., ROBERTS E. Amnestic effects in mice of four synthetic peptides homologous to amyloid beta protein from patients with Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 1991;88:3363–3366. doi: 10.1073/pnas.88.8.3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAO S.W., CHEN Z.J. Effects of sodium ferulate on platelet aggregation and platelet thromboxane A2 in patients with coronary heart disease. Chung Hsi I Chieh Ho Tsa Chih. 1988;8:263–265. [PubMed] [Google Scholar]
- GITTER B.D., COX L.M., RYDEL R.E., MAY P.C. Amyloid beta peptide potentiates cytokine secretion by interleukin-1 beta-activated human astrocytoma cells. Proc. Natl. Acad. Sci. U.S.A. 1995;92:10738–10741. doi: 10.1073/pnas.92.23.10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GLENNER G.G., WONG C.W. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- GRAF E. Antioxidant potential of ferulic acid. Free Radic. Biol. Med. 1992;13:435–448. doi: 10.1016/0891-5849(92)90184-i. [DOI] [PubMed] [Google Scholar]
- HIRABAYASHI T., OCHIAI H., SAKAI S., NAKAJIMA K., TERASAWA K. Inhibitory effect of ferulic acid and isoferulic acid on murine interleukin-8 production in response to influenza virus infections in vitro and in vivo. Planta Med. 1995;61:221–226. doi: 10.1055/s-2006-958060. [DOI] [PubMed] [Google Scholar]
- HUANG M.T., SMART R.C., WONG C.Q., CONNEY A.H. Inhibitory effect of curcumin, chlorogenic acid, caffeic acid, and ferulic acid on tumor promotion in mouse skin by 12-O- tetradecanoylphorbol-13-acetate. Cancer Res. 1988;48:5941–5946. [PubMed] [Google Scholar]
- ISRAEL M., LESBATS B. Application to mammalian tissues of the chemiluminescent method for detecting acetylcholine. J. Neurochem. 1982;39:248–250. doi: 10.1111/j.1471-4159.1982.tb04727.x. [DOI] [PubMed] [Google Scholar]
- KATO H., KOGURE K., ARAKI T., ITOYAMA Y. Astroglial and microglial reactions in the gerbil hippocampus with induced ischemic tolerance. Brain Res. 1994;664:69–76. doi: 10.1016/0006-8993(94)91955-0. [DOI] [PubMed] [Google Scholar]
- KATSUKI H., NAKAI S., HIRAI Y., AKAJI K., KISO Y., SATOH M. Interleukin-1 beta inhibits long-term potentiation in the CA3 region of mouse hippocampal slices. Eur. J. Pharmacol. 1990;181:323–326. doi: 10.1016/0014-2999(90)90099-r. [DOI] [PubMed] [Google Scholar]
- KAWAHARA N., RUETZLER C.A., KLATZO I. Protective effect of spreading depression against neuronal damage following cardiac arrest cerebral ischaemia. Neurol Res. 1995;17:9–16. doi: 10.1080/01616412.1995.11740281. [DOI] [PubMed] [Google Scholar]
- KAWAHARA N., RUETZLER C.A., MIES G., KLATZO I. Cortical spreading depression increases protein synthesis and upregulates basic fibroblast growth factor. Exp. Neurol. 1999;158:27–36. doi: 10.1006/exnr.1999.7091. [DOI] [PubMed] [Google Scholar]
- KITAGAWA K., MATSUMOTO M., TAGAYA M., HATA R., UEDA H., NIINOBE M., HANDA N., FUKUNAGA R., KIMURA K., MIKOSHIBA K., KAMATA T. ‘Ischemic tolerance' phenomenon found in the brain. Brain Res. 1990;528:21–24. doi: 10.1016/0006-8993(90)90189-i. [DOI] [PubMed] [Google Scholar]
- KITAGAWA K., MATSUMOTO M., TAGAYA M., KUWABARA K., HATA R., HANDA N., FUKUNAGA R., KIMURA K., KAMADA T. Hyperthermia-induced neuronal protection against ischemic injury in gerbils. J. Cereb. Blood Flow Metab. 1991;11:449–452. doi: 10.1038/jcbfm.1991.86. [DOI] [PubMed] [Google Scholar]
- KOBAYASHI S., HARRIS V.A., WELSH F.A. Spreading depression induces tolerance of cortical neurons to ischemia in rat brain. J. Cereb. Blood Flow Metab. 1995;15:721–727. doi: 10.1038/jcbfm.1995.92. [DOI] [PubMed] [Google Scholar]
- LAURSEN S.E., BELKNAP J.K. Intracerebroventricular injections in mice. Some methodological refinements. J. Pharmacol. Methods. 1986;16:355–357. doi: 10.1016/0160-5402(86)90038-0. [DOI] [PubMed] [Google Scholar]
- MCDONALD D.R., BRUNDEN K.R., LANDRETH G.E. Amyloid fibrils activate tyrosine kinase-dependent signaling and superoxide production in microglia. J. Neurosci. 1997;17:2284–2294. doi: 10.1523/JNEUROSCI.17-07-02284.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCGEER P.L., MCGEER E.G. Inflammation of the brain in Alzheimer's disease: implications for therapy. J. Leukoc. Biol. 1999;65:409–415. doi: 10.1002/jlb.65.4.409. [DOI] [PubMed] [Google Scholar]
- MATSUSHIMA K., SCHMIDT-KASTNER R., HOGAN M.J., HAKIM A.M. Cortical spreading depression activates trophic factor expression in neurons and astrocytes and protects against subsequent focal brain ischemia. Brain Res. 1998;807:47–60. doi: 10.1016/s0006-8993(98)00716-1. [DOI] [PubMed] [Google Scholar]
- MAURICE T., LOCKHART B.P., PRIVAT A. Amnesia induced in mice by centrally administered beta-amyloid peptides involves cholinergic dysfunction. Brain Res. 1996;706:181–193. doi: 10.1016/0006-8993(95)01032-7. [DOI] [PubMed] [Google Scholar]
- MORRIS R. Developments of a water-maze procedure for studying spatial learning in the rat. J. Neurosci. Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- MURRAY C.A., LYNCH M.A. Evidence that increased hippocampal expression of the cytokine interleukin-1 beta is a common trigger for age- and stress-induced impairments in long-term potentiation. J. Neurosci. 1998;18:2974–2981. doi: 10.1523/JNEUROSCI.18-08-02974.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NABESHIMA T., NITTA A. Memory impairment and neuronal dysfunction induced by beta-amyloid protein in rats. Tohoku J. Exp. Med. 1994;174:241–249. doi: 10.1620/tjem.174.241. [DOI] [PubMed] [Google Scholar]
- NITTA A., FUKUTA T., HASEGAWA T., NABESHIMA T. Continuous infusion of beta-amyloid protein into the rat cerebral ventricle induces learning impairment and neuronal and morphological degeneration. Jpn. J. Pharmacol. 1997;73:51–57. doi: 10.1254/jjp.73.51. [DOI] [PubMed] [Google Scholar]
- NITTA A., ITOH A., HASEGAWA T., NABESHIMA T. beta-Amyloid protein-induced Alzheimer's disease animal model. Neurosci. Lett. 1994;170:63–66. doi: 10.1016/0304-3940(94)90239-9. [DOI] [PubMed] [Google Scholar]
- ONOZUKA M., WATANABE K., NAGASAKI S., JIANG Y., OZONO S., NISHIYAMA K., KAWASE T., KARASAWA N., NAGATSU I. Impairment of spatial memory and changes in astroglial responsiveness following loss of molar teeth in aged SAMP8 mice. Behav. Brain Res. 2000;108:145–155. doi: 10.1016/s0166-4328(99)00145-x. [DOI] [PubMed] [Google Scholar]
- OZAKI Y. Antiinflammatory effect of tetramethylpyrazine and ferulic acid. Chem. Pharm. Bull. (Tokyo) 1992;40:954–956. doi: 10.1248/cpb.40.954. [DOI] [PubMed] [Google Scholar]
- ROGERS J., KIRBY L.C., HEMPELMAN S.R., BERRY D.L., MCGEER P.L., KASZNIAK A.W., ZALINSKI J., COFIELD M., MANSUKHANI L., WILLSON P. Clinical trial of indomethacin in Alzheimer's disease. Neurology. 1993;43:1609–1611. doi: 10.1212/wnl.43.8.1609. [DOI] [PubMed] [Google Scholar]
- SANO M., ERNESTO C., THOMAS R.G., KLAUBER M.R., SCHAFER K., GRUNDMAN M., WOODBURY P., GROWDON J., COTMAN C.W., PFEIFFER E., SCHNEIDER L.S., THAL L.J. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer's disease. The Alzheimer's Disease Cooperative Study. N. Engl. J. Med. 1997;336:1216–1222. doi: 10.1056/NEJM199704243361704. [DOI] [PubMed] [Google Scholar]
- SARTER M., BODEWITZ G., STEPHENS D.N. Attenuation of scopolamine-induced impairment of spontaneous alteration behaviour by antagonist but not inverse agonist and agonist beta- carbolines. Psychopharmacology. 1988;94:491–495. doi: 10.1007/BF00212843. [DOI] [PubMed] [Google Scholar]
- SCOTT B.C., BUTLER J., HALLIWELL B., ARUOMA O.I. Evaluation of the antioxidant actions of ferulic acid and catechins. Free Radic. Res. Commun. 1993;19:241–253. doi: 10.3109/10715769309056512. [DOI] [PubMed] [Google Scholar]
- TASAKI K., RUETZLER C.A., OHTSUKI T., MARTIN D., NAWASHIRO H., HALLENBECK J.M. Lipopolysaccharide pre-treatment induces resistance against subsequent focal cerebral ischemic damage in spontaneously hypertensive rats. Brain Res. 1997;748:267–270. doi: 10.1016/s0006-8993(96)01383-2. [DOI] [PubMed] [Google Scholar]
- WANG H., PENG R.X. Sodium ferulate alleviated paracetamol-induced liver toxicity in mice. Chung Kuo Yao Li Hsueh Pao. 1994;15:81–83. [PubMed] [Google Scholar]
- YAMADA K., TANAKA T., HAN D., SENZAKI K., KAMEYAMA T., NABESHIMA T. Protective effects of idebenone and alpha-tocopherol on beta-amyloid-(1-42)-induced learning and memory deficits in rats: implication of oxidative stress in beta-amyloid-induced neurotoxicity in vivo. Eur. J. Neurosci. 1999;11:83–90. doi: 10.1046/j.1460-9568.1999.00408.x. [DOI] [PubMed] [Google Scholar]
- ZHENG R.L., ZHANG H. Effects of ferulic acid on fertile and asthenozoospermic infertile human sperm motility, viability, lipid peroxidation, and cyclic nucleotides. Free Radic. Biol. Med. 1997;22:581–586. doi: 10.1016/s0891-5849(96)00272-9. [DOI] [PubMed] [Google Scholar]