

Abstract
The wild-type β2-adrenoceptor and a constitutively active mutant of this receptor were C-terminally tagged with luciferase from the sea pansy Renilla reniformis.
C-terminal addition of Renilla luciferase did not substantially alter the levels of expression of either form of the receptor, the elevated constitutive activity of the mutant β2-adrenoceptor nor the capacity of isoprenaline to elevate cyclic AMP levels in intact cells expressing these constructs.
Treatment of cells expressing constitutively active mutant β2-adrenoceptor-Renilla luciferase with antagonist/inverse agonist ligands resulted in upregulation of levels of this polypeptide which could be monitored by the elevated luciferase activity.
The pEC50 for ligand-induced luciferase upregulation and ligand affinity to bind the receptor were highly correlated.
Similar upregulation could be observed following sustained treatment with agonist ligands.
These effects were only observed at a constitutively active mutant of the β2-adrenoceptor. Co-expression of the wild-type β2-adrenoceptor C-terminally tagged with the luciferase from Photinus pyralis did not result in ligand-induced upregulation of the levels of activity of this luciferase.
Co-expression of the constitutively active mutant β2-adrenoceptor-Renilla luciferase and an equivalent mutant of the α1b-adrenoceptor C-terminally tagged with green fluorescent protein allowed pharmacological selectivity of adrenoceptor antagonists to be demonstrated.
This approach offers a sensitive and convenient means, which is amenable to high throughput analysis, to monitor ligand binding to a constitutively active mutant receptor.
As no prior knowledge of receptor ligands is required this approach may be suitable to identify ligands at orphan G protein-coupled receptors.
Keywords: Constitutive activity, luciferase, adrenoceptor, ligand screening, G protein-coupled receptor
Introduction
Mutations in G protein-coupled receptors (GPCRs) which impart higher levels of agonist-independent, constitutive regulation of second messenger levels than are produced by the wild-type receptors have been studied extensively in recent years (Lefkowitz et al., 1993; Scheer & Cotecchia, 1997; Leurs et al., 1998; Pauwels & Wurch, 1998). This has been, in part, to explore whether such mutants can provide insights into the conformational changes which must occur in a GPCR upon agonist binding to allow activation of G proteins. Many wild-type receptors also display agonist-independent function which can often be detected as increases in a functional endpoint measure in parallel with higher levels of expression of the receptor. Both of these approaches have been useful in demonstrations that many ligands, previously described as antagonists, have a capacity to dampen such constitutive activity (Lefkowitz et al., 1993; Milligan et al., 1995). As such ligands are thought to favour the presence of inactive or ground states of GPCRs they are generally now referred to as ‘inverse' agonists because they mirror the ability of agonist ligands to favour the enrichment of GPCRs in active conformational states (Milligan et al., 1995; Chen et al., 2000).
Recently, the constitutive activity of a GPCR has been suggested to provide a useful strategy in drug discovery which might be amenable for both GPCRs with known natural ligands and for so-called ‘orphan' GPCRs for which the natural ligands remain unidentified (Chen et al., 2000). One of the most studied constitutively active mutant (CAM) GPCRs is a form of the human β2-adrenoceptor in which a small segment of the distal region of the third intracellular loop was replaced by the equivalent sequence from the α1b-adrenoceptor (Samama et al., 1993; 1994; Pei et al., 1994; MacEwan & Milligan, 1996; Gether et al., 1997a; Javitch et al., 1997; McLean et al., 1999). As well as producing significantly greater levels of cyclic AMP in an agonist-independent manner than the wild-type β2-adrenoceptor, this mutant displays a substantially higher affinity to bind agonist but not antagonist/inverse agonist ligands. Furthermore, a Cys residue in transmembrane helix VI which can provide a monitor of agonist binding (Gether et al., 1997b) is closer to the ligand binding pocket in the CAM β2-adrenoceptor than in the wild-type receptor (Javitch et al., 1997). This CAM receptor has also been reported to be more structurally unstable and to denature more easily than the wild-type receptor (Gether et al., 1997a). These effects can be reversed by ligand binding to the CAM receptor (Gether et al., 1997a). Herein, we take advantage of this feature to demonstrate ligand-induced upregulation of a form of the CAM β2-adrenoceptor which has luciferase from the sea pansy Renilla reniformis linked in-frame to its C-terminal tail. Ligand-induced upregulation of the CAM receptor thus results in elevated cellular levels of luciferase activity which can be measured easily in formats appropriate to high throughput ligand screening programmes. The pEC50 for antagonist/inverse agonist-induced upregulation of the CAM β2-adrenoceptor-Renilla luciferase construct was well correlated with the pKi of the ligands, suggesting ligand occupancy of the GPCR binding site was the basis for the effect.
Methods
[3H]-dihydroalprenolol, ([3H]-DHA, 64 Ci mmol−1), [3H]-adenine and [3H]-cyclic AMP were purchased from Amersham Pharmacia Biotech. All reagents for cell culture were from Life Technologies (Paisley, Strathclyde, U.K.). Receptor ligands were from RBI (Gillingham, Dorset, U.K.). All other reagents were from Sigma or Fisons and were of the highest purity available.
Construction of wild-type and CAM β2-adrenoceptor/luciferase fusion proteins
Both human wild-type and CAM β2-adrenoceptor-Renilla luciferase fusion proteins were generated. A β2-adrenoceptor fragment was generated via PCR amplification of an existing β2-adrenoceptor DNA in pcDNA3. Generation of the CAM β2-adrenoceptor-fragment was also via the same PCR amplification of the CAM β2-adrenoceptor in pcDNA3. The mutations which comprise the CAM consist of four amino acid substitutions in intracellular loop 3 of the receptor. The primers used were as follows: 5′ forward primer 5′-AAA AAG CTT GCC ACC ATG GGG CAA CCC GGG AA-3′ which incorporates both a 5′ HindIII cloning site and Kozak sequence. The 3′ reverse primer had sequence 5′-CCT CTC GAG CAG TGA GTC ATT-3′ which incorporates an XhoI cloning site to allow linkage to the Renilla luciferase gene. Introducing the XhoI site results in an insertion of a glutamate residue between the β2-adrenoceptor DNA and the Renilla luciferase. It also resulted in alteration of the last nucleotide in the amino acid coding sequence of β2-adrenoceptor (G→C). This did not alter the amino acid sequence as the new codon CTC still encodes leucine. Renilla luciferase was similarly generated via PCR amplification of a Renilla luciferase DNA cloned into plasmid pRLCMV (Promega). The primers used for amplification were as follows: 5′ forward primer 5′-TCG CTC GAG ACT TCG AAA GTT TAT G-3′ which incorporates an XhoI site at the 5′ end of the gene to allow linkage to either the β2-adrenoceptor or the CAM β2-adrenoceptor fragment. The reverse primer had the sequence 5′-GCG TCT AGA TTA TTG TTC ATT TT-3′ which incorporates an XbaI site into the 3′ end of the gene immediately downstream of the stop codon.
Following PCR reactions the resultant fragments were digested with the appropriate enzymes and subsequently gel purified. pcDNA3 was digested with HindIII and XbaI to provide a recipient vector for the ligated fragments. Ligations were performed using Fast-link DNA ligation kit (CAMBIO) in which digested pcDNA3, β2-adrenoceptor (or CAM β2-adrenoceptor fragment) and Renilla luciferase fragments were mixed and ligated together.
Construction of a GFP-tagged form of the 3CAMα1b-adrenoceptor
Production and subcloning of the 3CAM hamster α1b-adrenoceptor-GFP fusion protein (Stevens et al., 2000) was performed in two separate stages. In the first step the coding sequence of a modified form of GFP (Zernicka-Goetz et al., 1997) was modified by polymerase chain reaction (PCR) amplification. Using the amino-terminal primer 5′-GGAAGGTACCAGTAAAGGAGAAGAACTT-3′ the initiating Met of GFP was removed and both a KpnI restriction site (underlined) and a 2-amino acid spacer (Gly-Asn) were introduced. Using the carboxy-terminal primer 5′-TGCTCTAGATTATTTGTATAGTTCATCCATGCATG-3′ an XbaI restriction site (underlined) was introduced downstream of the stop codon of GFP. The amplified fragment of GFP digested with KpnI and XbaI was subcloned into similarly digested pcDNA3 expression vector (Invitrogen). To obtain the 3CAM α1b-adrenoceptor-GFP fusion protein, the coding sequence of the 3CAM α1b-adrenoceptor was amplified by PCR. Using the amino-terminal primer 5′-GACGGTACCTCTAAAATGAATCCCGAT-3′, a KpnI restriction site (underlined) was introduced upstream of the initiator Met. Using the carboxy-terminal primer 5′-GTCCCTGGTACCAAAGTGCCCGGGTG-3′, a KpnI restriction site (underlined) was introduced immediately upstream of the stop codon. Finally, the GFP construct in pcDNA3 was digested with KpnI and ligated together with the PCR product of the α1b-adrenoceptor amplification also digested with KpnI.
Transient transfection of HEK293 cells
HEK293 cells were maintained in Minimum Essential Medium supplemented with 0.292 g L−1 L-glutamine, and 10% newborn calf serum at 37°C. Cells were grown to 60 – 80% confluency prior to transient transfection. Transfection was performed using LipofectAMINE reagent (Life Technology, Inc.) according to manufacturer's instructions. To generate cell lines stably expressing the various constructs, cells were seeded/diluted 2 days after transfection and maintained in DMEM supplemented with 1 mg ml−1 Geneticin sulphate (Life Technologies, Inc.). The medium was replaced every 3 days with DMEM containing 1 mg ml−1 Geneticin sulphate.
[3H]-ligand binding studies
Cells were grown in 6 cm dishes and treated with or without isoprenaline for various times. After treatment the cells were washed three times with ice-cold phosphate-buffered saline (PBS (mM): KCl 2.7, NaCl 137, KH2PO4 1.5, Na2HPO4 8, pH 7.4). Cells were then detached from plates with PBS/0.5 mM EDTA, pelleted and resuspended in ice cold TE buffer (10 mM Tris HCl, 0.1 mM EDTA pH 7.5) and lysed with 2×10 s bursts of a polytron. The homogenate was centrifuged at 500×g to remove unbroken cells and nuclei. The supernatant fraction was then centrifuged at 48,000×g for 30 min and the pellet resuspended in TE buffer and stored at −80°C until use.
[3H]-DHA binding assays were performed at 30°C for 45 min in TEM buffer (mM): Tris HCl 75, EDTA 1, MgCl2 12.5, pH 7.4. Parallel studies with 10 μM propranolol allowed assessment of non-specific binding. All experiments were terminated by rapid filtration through Whatman GF/C filters followed by three washes with ice-cold TE.
Intact cell adenylyl cyclase activity measurements
Were performed essentially as described in (Wong, 1994; Merkouris et al., 1997). Cells were split into wells of a 12-well plate and the cells were allowed to reattach. Cells were then incubated in medium containing [3H]-adenine (1.5 μCi well−1) for 16 – 24 h. The generation of [3H]-cyclic AMP in response to treatment of the cells with various ligands and other reagents was then assessed.
Luciferase activity assay
A stably transfected cell line of HEK293 cells expressing the CAM β2-adrenoceptor C-terminally tagged with Renilla luciferase was used to seed 96 well microtitre plates in 90 μl of medium. The 96 well plate was then incubated overnight and on the following day the cells were about 80% confluent. Drug dilutions were then prepared in phenol red-free medium and 10 μl from each of 10 concentration stock solutions were then added to wells of the 96 well plate. These plates were then incubated for 24 h and the medium pipetted off from each well.
Fifty μl of phenol red-free medium was then added to each well plus 50 μl of LucLite luciferase assay solution (Packard Biosciences). Fifty μl of 15 μM coelenterazine in phenol red-free medium was added (to produce a final concentration of 5 μM). The plates were then assayed immediately on a top count luminometer to determine the light intensity in relative light units.
Results
cDNAs encoding both the wild-type and a previously well characterized, constitutively active mutant (CAM), form of the human β2-adrenoceptor were modified to remove the stop codon and allow in-frame ligation of the luciferases from either the sea pansy Renilla reniformis or the firefly Photinus pyralis. As demonstrated previously (MacEwan & Milligan, 1996), transient expression of the unmodified, wild-type, β2-adrenoceptor resulted in higher levels of expression of this protein than was achieved for the C-terminally unmodified CAM β2-adrenoceptor when this was monitored by the specific binding of the β-adrenoceptor antagonist [3H]-dihydroalprenolol ([3H]-DHA) (Figure 1). The Renilla luciferase-tagged forms of both the wild-type and CAM β2-adrenoceptor were expressed at similar levels as the unmodified forms (Figure 1) but for both forms of the β2-adrenoceptor, C-terminal addition of Photinus luciferase significantly reduced levels of expression as monitored by specific [3H]-DHA binding (data not shown) and thus these constructs were only used in a limited series of further experiments (see later). Saturation [3H]-DHA binding studies demonstrated also that the Kd for this ligand (0.3±0.1 nM) was little affected by addition of Renilla luciferase to the C-terminal tail of the CAM β2-adrenoceptor (data not shown).
Figure 1.
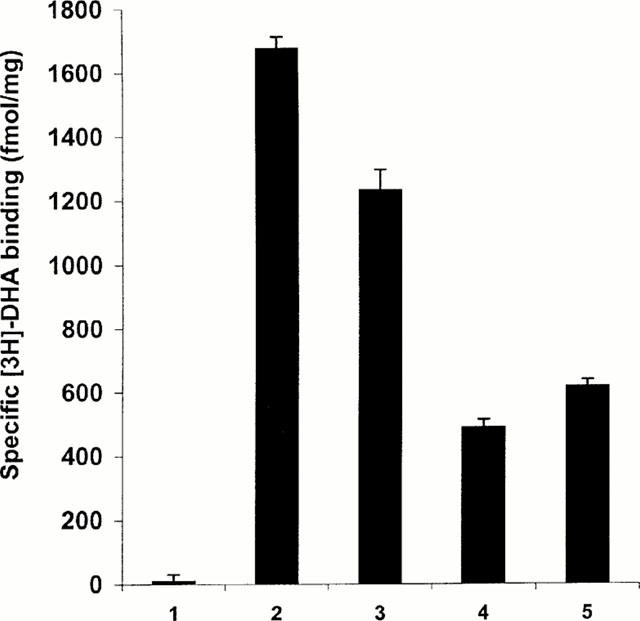
Expression levels of forms of the CAM β2-adrenoceptor are lower than equivalent forms of the wild-type β2-adrenoceptor. HEK293 cells were mock transfected (1) or transfected transiently with cDNAs encoding wild-type β2-adrenoceptor (2), β2-adrenoceptor-Renilla luciferase (3), CAM β2-adrenoceptor (4) or CAM β2-adrenoceptor-Renilla luciferase (5). Forty-eight hours later membranes were prepared and the specific binding of [3H]-DHA (2 nM) measured. Data are means±s.e.mean values of triplicate assays from a representative transfection. Two further experiments produced similar results.
Although levels of expression are low (Figure 1), HEK293 cells express the β2-adrenoceptor endogenously. Thus, in intact cell adenylyl cyclase assays, addition of the β-adrenoceptor agonist isoprenaline (10 μM) to mock transfected cells resulted in a significant stimulation of cyclic AMP generation (Figure 2). However, in cells transfected with either the unmodified wild-type β2-adrenoceptor or the Renilla luciferase-tagged form of this receptor both basal cyclic AMP levels and the stimulation produced by isoprenaline was substantially greater (Figure 2). Following expression of either the C-terminally unmodified CAM β2-adrenoceptor or the equivalent Renilla luciferase-tagged form, basal cyclic AMP levels were greater than observed with expression of the equivalent wild-type receptor forms. This is the basic characteristic of CAM forms of GPCRs and confirmed that C-terminal addition of Renilla luciferase did not suppress the CAM nature of this construct nor indeed the spontaneous, agonist-independent, signalling capacity of the wild-type β2-adrenoceptor. Addition of isoprenaline now further increased cyclic AMP generation to levels similar to those obtained for the wild-type receptors (Figure 2).
Figure 2.
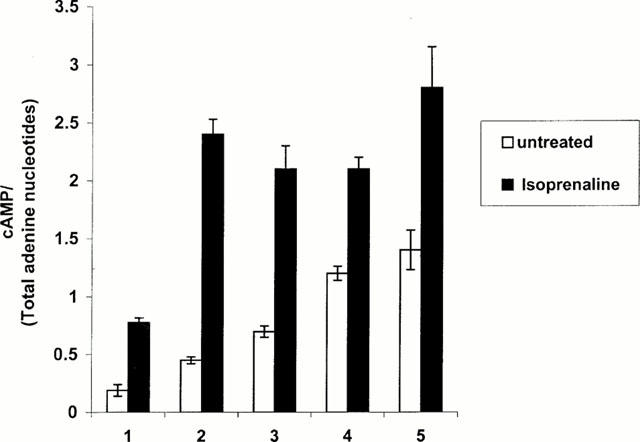
C-terminal addition of Renilla luciferase neither inhibits constitutive activity of the CAM β2-adrenoceptor nor the capacity of agonist to stimulate cyclic AMP production. HEK293 cells were mock transfected (1) or transfected transiently with cDNAs encoding wild-type β2-adrenoceptor (2), β2-adrenoceptor-Renilla luciferase (3), CAM β2-adrenoceptor (4) or CAM β2-adrenoceptor-Renilla luciferase (5). Twenty-four hours later cells were labelled with 3H-adenine and after a further 24 h cyclic AMP generation measured as in Methods in the absence or presence of the β-adrenoceptor agonist isoprenaline (10 μM). Results are taken from a representative experiment of three performed.
Sustained (24 h) treatment of HEK293 cells transiently expressing the untagged or Renilla luciferase-tagged CAM β2-adrenoceptor with the β-adrenoceptor antagonist/inverse agonist betaxolol (10 μM) resulted in the presence of substantially increased levels of [3H]-DHA binding sites in membranes prepared from these cells (Figure 3A). Saturation [3H]-DHA binding studies confirmed this to represent a true upregulation of the Renilla luciferase-tagged CAM β2-adrenoceptor (Figure 3B). Parallel measures of the upregulation of [3H]-DHA binding sites and Renilla luciferase luminescence produced by treatment with varying concentrations of betaxolol showed these parameters to overlap (Figure 3C). This suggested a novel strategy to identify antagonist/inverse agonist ligands at this receptor which would potentially be amenable to high throughput analysis. HEK293 clones stably expressing the Renilla luciferase-tagged CAM β2-adrenoceptor were thus isolated and characterized for expression of the construct based on a combination of [3H]-DHA binding and the presence of Renilla luciferase activity. A single clone was selected for detailed study. Following plating on wells of a 96 well microtitre plate, the cells were incubated with a range of concentrations of betaxolol at 37°C for 24 h. The luciferase activity of plate wells was subsequently monitored using 5 μM colenterazine. Betaxolol produced a concentration-dependent upregulation of Renilla luciferase activity with EC50=1.2±0.26×10−7 M (Figure 4A). The maximal effect of betaxolol was between 2 – 3 fold in individual experiments. Equivalent experiments with propranolol produced equivalent results except with EC50=3.9±0.2×10−9 M (Figure 4A). Parallel binding experiments confirmed upregulation of [3H]-DHA binding sites (data not shown) which would be expected to be in a 1 : 1 ratio with the increase in Renilla luciferase activity based on the 1 : 1 stoichiometry of the receptor and luciferase in the fusion construct. Betaxolol was able to compete with [3H]-DHA for binding to the CAM β2-adrenoceptor-Renilla luciferase with a Ki=6.9×10−8 M (Figure 5b). Equivalent experiments with propranolol (Figure 4b) resulted in a Ki=6.8×10−10 M. A range of other β-adrenoceptor antagonists/inverse agonists, including ICI118551, CGP12177A and sotolol produced similar concentration-dependent upregulation of Renilla luciferase activity. These compounds had varying affinity to compete with [3H]-DHA in binding experiments. However, a strong correlation was observed between the pKi values of these ligands to bind to the Renilla luciferase-tagged CAM β2-adrenoceptor and their pEC50 values for upregulation of this construct (Figure 5). Interestingly, equivalent long-term treatment with the agonists isoprenaline, salbutamol and salmeterol also produced significant, concentration-dependent, upregulation of the Renilla luciferase activity of cells expressing the tagged CAM β2-adrenoceptor (Figure 6).
Figure 3.
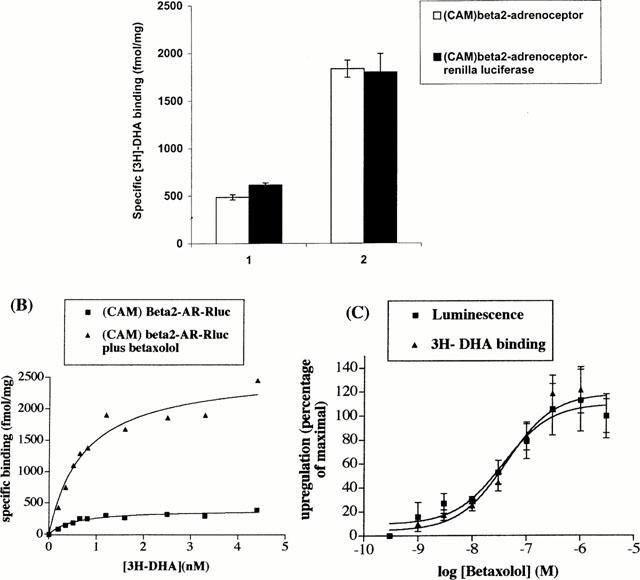
Sustained treatment with betaxolol upregulates levels of the untagged and Renilla luciferase-tagged forms of the CAM β2-adrenoceptor. (A) HEK293 cells were transfected transiently with cDNAs encoding CAM β2-adrenoceptor or CAM β2-adrenoceptor-Renilla luciferase. Twenty-four hours later the cells were exposed to betaxolol (10 μM) (2) or vehicle (1). Twenty-four hours later membranes were prepared and the specific binding of [3H]-DHA (2 nM), measured. (B) HEK293 cells were transfected transiently to express the CAM β2-adrenoceptor-Renilla luciferase construct. Twenty-four hours later the cells were exposed to betaxolol (10 μM) or vehicle. Twenty-four hours later membranes were prepared and the specific binding of a wide range of [3H]-DHA concentrations measured. Results are representative of three independent experiments. (C) HEK293 cells were transfected transiently to express the CAM β2-adrenoceptor-Renilla luciferase construct. Twenty-four hours later the cells were exposed to varying concentrations of betaxolol. The specific binding of [3H]-DHA (2 nM) and the luminescence from Renilla luciferase were measured in parallel 24 h later. Data are presented as per cent of maximal effect with results derived from three independent experiments.
Figure 4.
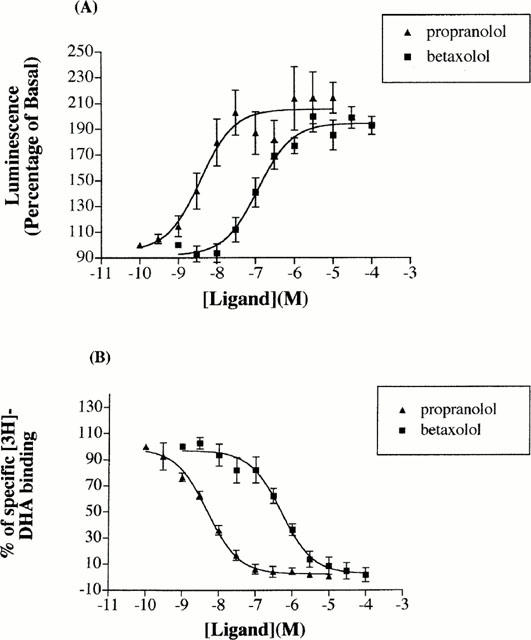
EC50 for betaxolol-induced upregulation of CAM β2-adrenoceptor-Renilla luciferase is similar to ligand Ki. (A) Cells of a clone of HEK293 cells stably expressing CAM β2-adrenoceptor-Renilla luciferase were grown in a 96 well microtitre plate and exposed to various concentrations of betaxolol or propranolol for 24 h. Renilla luciferase activity was then monitored as described in Methods. (B) The capacity of various concentrations of betaxolol or propranolol to compete with [3H]-DHA (2 nM) to bind to CAM β2-adrenoceptor-Renilla luciferase in membranes of the clone used in (A) was assessed. Data represent means±s.e.mean from three experiments.
Figure 5.
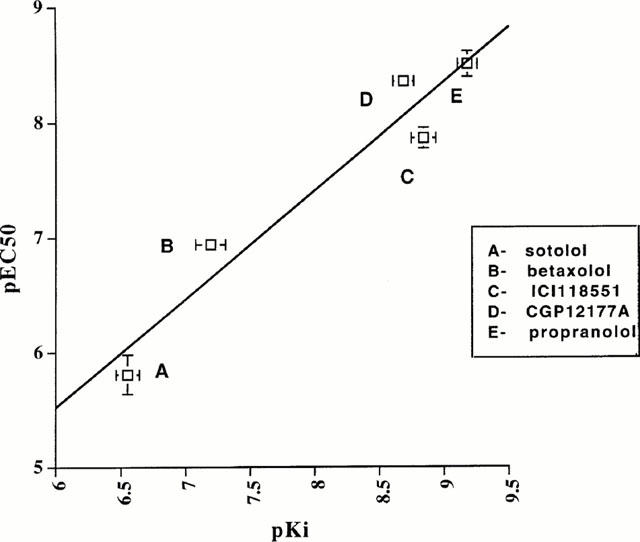
Correlation of pEC50 for ligand-induced upregulation and pKi for CAM β2-adrenoceptor-Renilla luciferase activity for a range of β2-adrenoceptor antagonists/inverse agonists. Experiments similar to those described in Figure 4 were conducted using varying concentrations of sotolol (A), betaxolol (B), ICI118551 (C), CGP12177A (D) and propranolol (E). Measured EC50 values for upregulation and estimated Ki values from binding studies were then compared. Data represent means±s.e.mean from three experiments.
Figure 6.
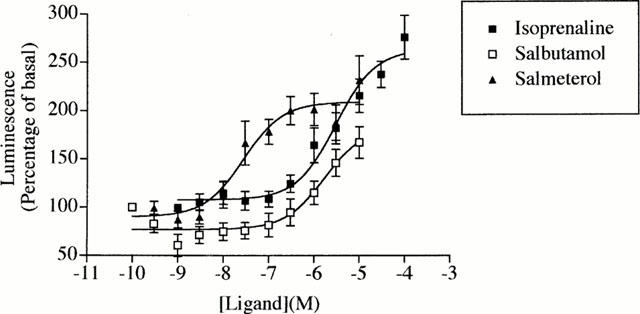
Sustained treatment with agonists produces concentration-dependent increases in CAM β2-adrenoceptor-Renilla luciferase activity. Equivalent experiments to those of Figure 4A were performed using varying concentrations of isoprenaline, salbutamol and salmeterol. Data represent means±s.e.mean from three experiments.
To ascertain the importance of the CAM nature of the β2-adrenoceptor to observe ligand-mediated upregulation of luciferase activity we transiently co-expressed CAM β2-adrenoceptor-Renilla luciferase and wild-type β2-adrenoceptor-Photinus luciferase in HEK293 cells. Following sustained treatment of the cells with or without betaxolol we sequentially monitored Photinus luciferase and Renilla luciferase activity using a dual luciferase assay system. Photinus luciferase activity, as a monitor of levels of the wild-type β2-adrenoceptor, was barely increased by betaxolol treatment whereas Renilla luciferase activity was increased substantially (Figure 7). Betaxolol substantially increased the Renilla/Photinus activity ratio and therefore the ratio of expression of CAM to wild-type β2-adrenoceptors.
Figure 7.
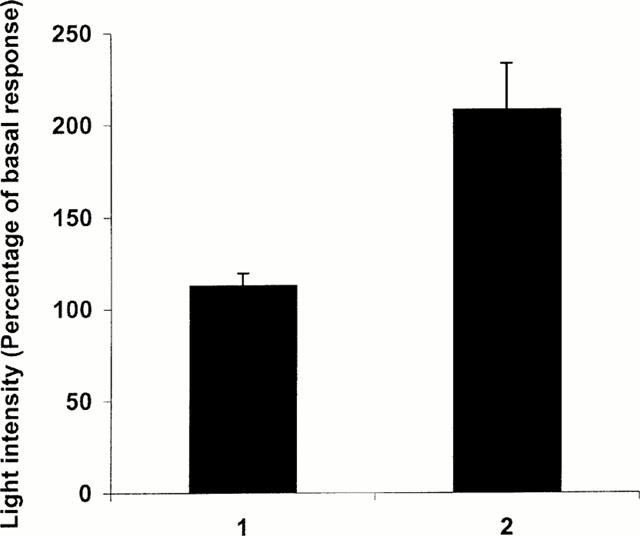
Ligand-induced upregulation of β2-adrenoceptor-luciferase activity requires the CAM form of the receptor. HEK293 cells were transfected with a combination of CAM β2-adrenoceptor-Renilla luciferase and wild-type β2-adrenoceptor-Photinus luciferase constructs. Cells were then treated 24 h later without or with betaxolol (10 μM) for a further 24 h. Photinus (1) and Renilla (2) luciferase activities were then measured in parallel. Data represent means±s.e.mean from three experiments.
To demonstrate pharmacological specificity of ligand-induced upregulation we established a HEK293 clone stably expressing a C-terminally GFP-tagged form of a previously characterized CAM form of the α1b-adrenoceptor which we designate 3CAM (Stevens et al., 2000). These cells were then transiently transfected with CAM β2-adrenoceptor-Renilla luciferase and the cells subsequently treated with either betaxolol or the α1-adrenoceptor antagonist/inverse agonist phentolamine (each at 10 μM, 24 h). These cells were then monitored for either Renilla luciferase activity or GFP fluorescence. Betaxolol increased levels of luciferase activity without a significant effect on levels of GFP whereas phentolamine had the opposite effect, increasing GFP fluorescence but not luciferase activity (Figure 8).
Figure 8.
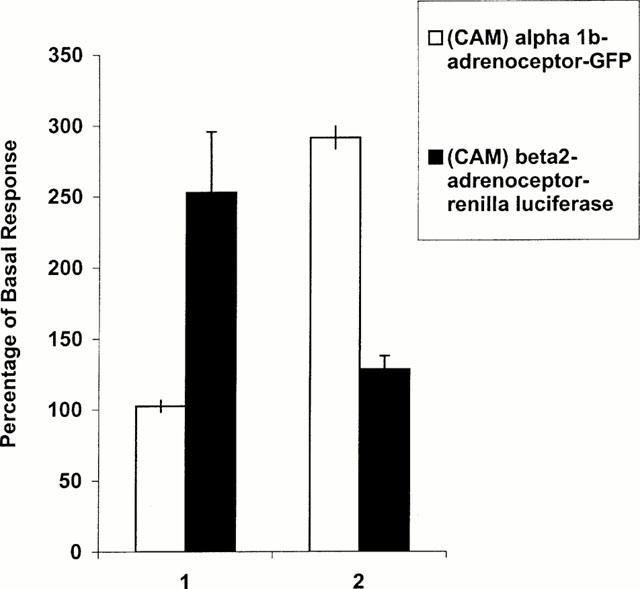
Pharmacological specificity of ligand-induced upregulation of CAM adrenoceptors. A clone of HEK293 cells stably expressing a CAM α1b-adrenoceptor-GFP construct (Stevens et al., 2000) was transiently transfected for 24 h to express CAM β2-adrenoceptor-Renilla luciferase. The cells were then exposed to either betaxolol (10 μM) (1) or to the α1-adrenoceptor antagonist/inverse agonist phentolamine (10 μM) (2) for 24 h. Renilla luciferase activity (as bioluminescence) and GFP fluorescence were then monitored in parallel. Results are presented as per cent of the signals obtained by treatment of the cells with vehicle and represent means±s.e.mean from three independent experiments.
Discussion
A number of CAM forms of GPCRs appear to be physically destabilized compared to the wild-type receptors (MacEwan & Milligan, 1996; Lee et al., 1997; Gether et al., 1997b). Following either transient or stable transfection antagonist/inverse agonist binding to such CAM GPCRs can result in their upregulation without alteration in mRNA levels (MacEwan & Milligan, 1996; Lee et al., 1997; McLean et al., 1999; see Milligan & Bond, 1997 for review). Such results have added to the concept that such CAM GPCRs may provide a useful model of conformationally active, R*, states of the GPCR with antagonist/inverse agonist binding resulting in a relaxation of the CAM receptor to ground state, inactive, R, forms. Such effects of antagonist/inverse agonist ligands on CAM GPCRs have been examined using approaches as diverse as alterations in fluorescence of a conformation-sensitive reporter in the purified receptor (Gether et al., 1997b) and analysis of increased levels of receptor binding sites present in cells following treatment with appropriate ligands (MacEwan & Milligan, 1996; Lee et al., 1997).
In recent times, C-terminal addition of the 27 kDa green fluorescent protein to a wide range of GPCRs has been employed to monitor their cellular expression, distribution and trafficking in response to stimulation (see Milligan, 1999; Kallal & Benovic, 2000 for reviews). In general, such modification does not appear to prevent interaction of the GPCR with G proteins nor to substantially alter the ligand binding properties of the GPCR. Treatment of a cell line stably expressing a CAM β2-adrenoceptor-GFP construct with a range of ligands with β2-adrenoceptor antagonist/inverse agonist pharmacology resulted in upregulation of the fusion polypeptide and the cells becoming markedly more fluorescent over time (McLean et al., 1999). In essence such an approach should be more amenable to quantitative analysis if an easy to assay enzyme was linked in frame to the C-terminus of the GPCR providing that the modified GPCR functioned essentially as the unmodified. Herein, we have used forms of luciferase derived from either the firefly Photinus pyralis or the sea pansy Renilla reniformis and linked either of these to both the wild-type and a CAM β2-adrenoceptor. Assays of Photinus luciferase activity are generally more widely employed than Renilla (Rees et al., 1999). However, presumably simply because Photinus luciferase is a relatively large (61 kDa) polypeptide, transient expression levels of the GPCRs tagged with this luciferase were relatively low. This restricted the use we made of these constructs. By contrast, C-terminal tagging with the 31 kDa Renilla luciferase had little effect on levels of transient expression of either the wild-type or CAM β2-adrenoceptor. Both the wild-type β2-adrenoceptor-Renilla luciferase and the CAM β2-adrenoceptor version of this construct were able to stimulate cyclic AMP levels upon addition of the agonist isoprenaline. As such, addition of the luciferase to the C-terminal tail of these GPCR forms does not prevent coupling of the receptors to GS and thence to adenylyl cyclase. Equally, agonist stimulation of adenylyl cyclase activity by the β2-adrenoceptor is not prevented by addition of GFP to the C-terminal tail (Barak et al., 1997; Kallal et al., 1998; McLean et al., 1999). Interestingly, both the wild-type β2-adrenoceptor-Renilla luciferase and the CAM β2-adrenoceptor version of this construct possessed the capacity to elevate cellular cyclic AMP levels in an agonist-independent manner (Figure 2). However, as expected from the untagged version of these GPCRs, the agonist-independent effect of the CAM β2-adrenoceptor-Renilla luciferase was substantially more pronounced when expression levels were taken into consideration.
Upregulation of the CAM β2-adrenoceptor-Renilla luciferase construct stably expressed in HEK293 cells was produced in a concentration-dependent manner by all the antagonist/inverse agonist ligands we tested. This was also the case in our previous studies using the CAM β2-adrenoceptor-Renilla luciferase construct (McLean et al., 1999). However, as luciferase activity is both technically easy to measure and provides a robust and reproducible marker this system was ideally suited to the studies being performed in 96 well microtitre plates. The pEC50 for ligand upregulation of the CAM β2-adrenoceptor-Renilla luciferase construct was well correlated with their pKi to bind to the construct in intact cell binding assays (Figure 5). This suggests strongly that ligand occupancy is the key determinant which produces the upregulation. It has been shown previously that inverse agonist-induced upregulation of an unmodified CAM β2-adrenoceptor occurs without alteration in levels of mRNA encoding the protein (MacEwan & Milligan, 1996). As such, although the activity of luciferases is most usually employed in transcriptionally-induced ‘reporter gene' assays (Rees et al., 1999), the current studies do not require transcriptional modulation. Rather, they are based on ligand-induced protein stabilization of the construct. Coupled with unchanged rates of protein synthesis this results in time-dependent upregulation of these constructs.
To demonstrate that the ligand effect required the structural variation present in the CAM receptor, we performed experiments in which the CAM β2-adrenoceptor-Renilla luciferase was co-expressed with the Photinus luciferase tagged wild-type β2-adrenoceptor. Initial transfection ratios were arranged such that dual luciferase assay produced similar levels of light output from the two forms of the enzyme. Now following sustained treatment with betaxolol the activity of Photinus luciferase was essentially unchanged although that of Renilla luciferase was substantially increased. We have recently demonstrated that a similar antagonist/inverse agonist-induced upregulation can be observed using a C-terminally GFP-tagged form of an equivalent CAM mutant of the α1b-adrenoceptor (Stevens et al., 2000). We thus reasoned that a parallel monitor of ligand regulation of GFP fluorescence and Renilla luciferase luminescence could provide monitors of pharmacological selectivity of ligands following co-expression of the CAM α1b-adrenoceptor-GFP and the CAM β2-adrenoceptor-Renilla luciferase. As anticipated, the β-blocker betaxolol upregulated luciferase activity but not the GFP signal whereas the α1-adrenoceptor antagonist/inverse agonist phentolamine produced the opposite profile (Figure 8). We were somewhat surprised to observe that a series of β-agonists were also able to cause upregulation of the CAM β2-adrenoceptor-Renilla luciferase construct. When these experiments were performed, our only real expectation of agonists producing the upregulation we have observed was based on the results of Gether et al. (1997a). They have argued convincingly that the occupancy of the CAM β2-adrenoceptor will produce protein stabilization, no matter the efficacy of the ligand. However, their experiments were performed with the purified CAM β2-adrenoceptor. Thus, our initial hypothesis was that agonists would cause internalization of the CAM β2-adrenoceptor-Renilla luciferase as we have previously noted for a GFP-tagged form of this receptor (McLean et al., 1999) and that with long-term agonist treatment downregulation of the construct would occur, as is often observed for the wild-type β2-adrenoceptor (McLean & Milligan, 2000). This would be expected to result in lower levels of the constitutively active mutant β2-adrenoceptor-Renilla luciferase protein and that consequent lower levels of luciferase activity would provide an assay for agonist. However, this is not what was observed. HEK293 cells find it significantly more difficult to destroy such tagged forms of receptors than they do the unmodified forms (McLean & Milligan, 2000). It is not important to the basis of the luciferase assay where the protein is located at the end of the treatment because addition of the luciferase assay reagents disrupts cell integrity. We thus envisage that the agonist-induced upregulation represents a balance between ligand-induced stabilization which favours upregulation of the construct and the poor capacity of the cells to destroy the tagged receptor. This, however, must remain speculation at this point. Recent studies on two mutated, constitutively active, forms of the rat histamine H2 receptor have, however, also demonstrated substantial upregulation of these to be produced by sustained treatment with both an agonist and an inverse agonist ligand at the receptor (Alewijnse et al., 2000). By contrast, levels of wild-type rat histamine H2 receptor were reduced by sustained exposure to the agonist and the upregulatory effect of the inverse agonist, although statistically significant, was much less pronounced than for the CAM forms (Alewijnse et al., 2000). Interestingly, Zhu et al. (2000) have recently observed that sustained treatment with a variety of ligands which function as inverse agonist at a CAM form of the human α1a-adrenoceptor produces substantial upregulation of the mutant but not the wild-type receptor. Moreover, they reported a lack of upregulation of this CAM form when using KMD-3213, which they indicated to be a neutral antagonist (Zhu et al., 2000), suggesting a possible means to discriminate between neutral and inverse agonist ligands. Although intriguing, the basis for these differences were not established and the efficacy of KMD-3213 was very similar to that of BMY7378, which did produce strong upregulation (Zhu et al., 2000).
It also appears that all CAM mutants of the same receptor are not equivalent. Stevens et al. (2000) demonstrated that only certain CAM forms of the GFP-tagged hamster α1b-adrenoceptor are strongly upregulated by sustained exposure to antagonist/inverse agonist ligands and that this capacity was correlated with the level of constitutive activity imbued by distinct mutations.
The constitutive activity of GPCRs has been suggested to provide means of developing and enhancing drug discovery programmes (Chen et al., 2000). It has also been noted that mutations at different regions of a GPCR which result in constitutive activity can produce synergistic effects when combined (Hwa et al., 1997).
These studies provide a potential strategy for the identification of ligands which interact with a wide range of GPCRs, including those for which the natural ligands are currently unknown, if ligand-regulated protein stabilization either segregates with receptor constitutive activity or can be induced by limited mutation. These two issues will form the basis for future work, as will the concept that different cell types with varying overall rates of receptor internalization and recycling may be identified to optimise ligand regulation of the steady state levels of such CAM receptors.
Acknowledgments
D. Ramsay thanks the Biotechnology and Biosciences Research Council for a studentship. Financial support for this work was provided by the Medical Research Council, the Biotechnology and Biosciences Research Council and the European Union Biomed II programme ‘Inverse agonism: Implications for drug design'.
Abbreviations
- CAM
constitutively active mutant
- DHA
dihydroalprenolol
- DMEM
Dulbecco's Minimum Essential Medium
- GFP
green fluorescent protein
- GPCR
G protein-coupled receptor
- PBS
phosphate buffered saline
References
- ALEWIJNSE A.E., TIMMERMAN H., JACOBS E.H., SMIT M.J., ROOVERS E., COTECCHIA S., LEURS R. The effect of mutations in the DRY motif on the constitutive activity and structural instability of the histamine H2 receptor. Mol. Pharmacol. 2000;57:890–898. [PubMed] [Google Scholar]
- BARAK L.S., FERGUSON S.S., ZHANG J., MARTENSON C., MEYER T., CARON M.G. Internal trafficking and surface mobility of a functionally intact beta2-adrenergic receptor-green fluorescent protein conjugate. Mol. Pharmacol. 1997;51:177–184. doi: 10.1124/mol.51.2.177. [DOI] [PubMed] [Google Scholar]
- CHEN G., WAY J., ARMOUR S., WATSON C., QUEEN K., JAYAWICKREME C.K., CHEN W.J., KENAKIN T. Use of constitutive G protein-coupled receptor activity for drug discovery. Mol. Pharmacol. 2000;57:125–134. [PubMed] [Google Scholar]
- GETHER U., BALLESTEROS J.A., SEIFERT R., SANDERS-BUSH E., WEINSTEIN H., KOBILKA B.K. Structural instability of a constitutively active G protein-coupled receptor. Agonist-independent activation due to conformational flexibility. J. Biol. Chem. 1997a;272:2587–2590. doi: 10.1074/jbc.272.5.2587. [DOI] [PubMed] [Google Scholar]
- GETHER U., LIN S., GHANOUNI P., BALLESTEROS J.A., WEINSTEIN H., KOBILKA B.K. Agonists induce conformational changes in transmembrane domains III and VI of the beta2 adrenoceptor. EMBO J. 1997b;16:6737–6747. doi: 10.1093/emboj/16.22.6737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HWA J., GAIVIN R., PORTER J.E., PEREZ D.M. Synergism of constitutive activity in alpha 1-adrenergic receptor activation. Biochemistry. 1997;36:633–639. doi: 10.1021/bi962141c. [DOI] [PubMed] [Google Scholar]
- JAVITCH J.A., FU D., LIAPAKIS G., CHEN J. Constitutive activation of the beta2 adrenergic receptor alters the orientation of its sixth membrane-spanning segment. J. Biol. Chem. 1997;272:18546–18549. doi: 10.1074/jbc.272.30.18546. [DOI] [PubMed] [Google Scholar]
- KALLAL L., BENOVIC J.L. Using green fluorescent proteins to study G-protein-coupled receptor localization and trafficking. Trends Pharmacol. Sci. 2000;21:175–180. doi: 10.1016/s0165-6147(00)01477-2. [DOI] [PubMed] [Google Scholar]
- KALLAL L., GAGNON A.W., PENN R.B., BENOVIC J.L. Visualization of agonist-induced sequestration and down-regulation of a green fluorescent protein-tagged β2-adrenergic receptor. J. Biol. Chem. 1998;273:322–328. doi: 10.1074/jbc.273.1.322. [DOI] [PubMed] [Google Scholar]
- LEE T.W., COTECCHIA S., MILLIGAN G. Up-regulation of the levels of expression and function of a constitutively active mutant of the hamster alpha1B-adrenoceptor by ligands that act as inverse agonists. Biochem. J. 1997;325:733–739. doi: 10.1042/bj3250733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEFKOWITZ R.J., COTECCHIA S., SAMAMA P., COSTA T. Constitutive activity of receptors coupled to guanine-nucleotide regulatory proteins. Trends Pharmacol. Sci. 1993;14:303–307. doi: 10.1016/0165-6147(93)90048-O. [DOI] [PubMed] [Google Scholar]
- LEURS R., SMIT M.J., ALEWIJNSE A.E., TIMMERMAN H. Agonist-independent regulation of constitutively active G-protein-coupled receptors. Trends Biochem. Sci. 1998;23:418–422. doi: 10.1016/s0968-0004(98)01287-0. [DOI] [PubMed] [Google Scholar]
- MACEWAN D.J., MILLIGAN G. Inverse agonist-induced upregulation of the human β2-adrenoceptor in transfected neuroblastoma×glioma hybrid cells. Mol. Pharmacol. 1996;50:1479–1486. [PubMed] [Google Scholar]
- MCLEAN A.J., BEVAN N., REES S., MILLIGAN G. Visualizing differences in ligand regulation of wild type and constitutively active mutant β2-adrenoceptor-green fluorescent protein fusion proteins. Mol. Pharmacol. 1999;56:1182–1191. doi: 10.1124/mol.56.6.1182. [DOI] [PubMed] [Google Scholar]
- MCLEAN A.J., MILLIGAN G. Ligand regulation of green fluorescent protein-tagged forms of the human β1- and β2-adrenoceptors; comparisons with the unmodified receptors. Br. J. Pharmacol. 2000;130:1825–1832. doi: 10.1038/sj.bjp.0703506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MERKOURIS M., MULLANEY I., GEORGOUSSI Z., MILLIGAN G. Regulation of spontaneous activity of the δ-opioid receptor: Studies of inverse agonism in intact cells. J. Neurochem. 1997;69:2115–2122. doi: 10.1046/j.1471-4159.1997.69052115.x. [DOI] [PubMed] [Google Scholar]
- MILLIGAN G. Exploring the dynamics of regulation of G protein-coupled receptors using green fluorescent protein. Br. J. Pharmacol. 1999;128:501–510. doi: 10.1038/sj.bjp.0702824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MILLIGAN G., BOND R.A. Inverse agonism and the regulation of receptor number. Trends Pharmacol. Sci. 1997;18:468–474. doi: 10.1016/s0165-6147(97)01139-5. [DOI] [PubMed] [Google Scholar]
- MILLIGAN G., BOND R.A., LEE M. Inverse agonism: pharmacological curiosity or potential therapeutic strategy. Trends Pharmacol. Sci. 1995;16:10–13. doi: 10.1016/s0165-6147(00)88963-4. [DOI] [PubMed] [Google Scholar]
- PAUWELS P.J., WURCH T. Review: amino acid domains involved in constitutive activation of G-protein-coupled receptors. Mol. Neurobiol. 1998;17:109–135. doi: 10.1007/BF02802027. [DOI] [PubMed] [Google Scholar]
- PEI G., SAMAMA P., LOHSE M., WANG M., CODINA J., LEFKOWITZ R.J. A constitutively active mutant beta 2-adrenergic receptor is constitutively desensitized and phosphorylated. Proc. Natl. Acad. Sci. U.S.A. 1994;91:2699–2702. doi: 10.1073/pnas.91.7.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REES S., BROWN S., STABLES J.Reporter gene systems for the study of G protein-coupled receptor signal transduction in mammalian cells Signal Transduction: A Practical Approach 2nd Edition 1999Oxford: Oxford University Press; 171–221.ed. Milligan, G. pp [Google Scholar]
- SAMAMA P., COTECHHIA S., COSTA T., LEFKOWITZ R.J. A mutation-induced activated state of the β2-adrenergic receptor–extending the ternary complex model. J. Biol. Chem. 1993;268:4625–4636. [PubMed] [Google Scholar]
- SAMAMA P., PEI G., COSTA T., COTECCHIA S., LEFKOWITZ R.J. Negative antagonists promote an inactive conformation of the beta 2-adrenergic receptor. Mol. Pharmacol. 1994;45:390–394. [PubMed] [Google Scholar]
- SCHEER A., COTECCHIA S. Constitutively active G protein-coupled receptors: potential mechanisms of receptor activation. J. Recept. Signal Transduct. Res. 1997;17:57–73. doi: 10.3109/10799899709036594. [DOI] [PubMed] [Google Scholar]
- STEVENS P.A., BEVAN N., REES S., MILLIGAN G. Resolution of inverse agonist-induced upregulation from constitutive activity of mutants of the α1b-adrenoceptor. Mol. Pharmacol. 2000;58:438–448. doi: 10.1124/mol.58.2.438. [DOI] [PubMed] [Google Scholar]
- WONG Y.G. Gi assays in transfected cells. Methods Enzymol. 1994;238:81–94. doi: 10.1016/0076-6879(94)38008-2. [DOI] [PubMed] [Google Scholar]
- ZERNICKA-GOETZ M., PINES J., MCLEAN HUNTER S., DIXON J.P.C., SIEMERING K., HASELOFF J., EVANS M.J. Following cell fate in the living mouse embryo. Development. 1997;124:1133–1137. doi: 10.1242/dev.124.6.1133. [DOI] [PubMed] [Google Scholar]
- ZHU J., TANIGUCHI T., TAKAUJI R., SUZUKI F., TANAKA T., MURAMATSU I. Inverse agonism and neutral antagonism at a constitutively active alpha-1a adrenoceptor. Br. J. Pharmacol. 2000;131:546–552. doi: 10.1038/sj.bjp.0703584. [DOI] [PMC free article] [PubMed] [Google Scholar]