

Abstract
Endotoxin-induced vascular hyporeactivity to phenylephrine (PE) is well described in rodent aorta, but has not been investigated in smaller vessels in vitro.
Segments of rat superior mesenteric artery were incubated in culture medium with or without foetal bovine serum (10%) for 6, 20 or 46 h in the presence or absence of bacterial lipopolysaccharide (LPS; 1 – 100 μg ml−1).
Contractions to PE were measured with or without nitric oxide synthase (NOS) inhibitors: L-NAME (300 μM), aminoguanidine (AMG; 400 μM) 1400W (10 μM) and GW273629 (10 μM); the guanylyl cyclase inhibitor, ODQ (3 μM); the COX-2 inhibitor, NS-398 (10 μM). Contractile responses to the thromboxane A2 mimetic, U46619 were also assessed.
In the presence of serum, LPS induced hyporeactivity at all time points. In its absence, hyporeactivity only occurred at 6 and 20 h.
L-NAME and AMG fully reversed hyporeactivity at 6 h, whereas they were only partially effective at 20 h and not at all at 46 h. In contrast partial reversal of peak contraction was observed with 1400W (62% at 46 h), GW273629 (57% at 46 h) and ODQ (75% at 46 h). COX-2 inhibition produced no reversal.
In contrast to PE, contractions to U46619 were substantially less affected by LPS.
We describe a well-characterized reproducible model of LPS-induced hyporeactivity, which is largely mediated by the NO-cyclic GMP-dependent pathway. Importantly, long-term (2-day) production of NO via iNOS is demonstrated. Moreover, conventional doses of L-NAME and AMG became increasingly ineffective over time. Thus, the choice of inhibitor merits careful consideration in long-term models.
Keywords: Vascular hyporeactivity, endotoxin, nitric oxide, phenylephrine, organ culture, NOS inhibitors, guanylyl cyclase inhibition, endothelium, thromboxane A2
Introduction
Sepsis is the leading cause of morbidity and mortality in critically ill patients with a mortality rate exceeding 50% (Deitch, 1998). Gram-negative bacterial infections are responsible for approximately 50% of cases of septic shock (Thiemermann, 1997). This condition is characterized by an exaggerated host systemic inflammatory response usually leading to a high cardiac output, low systemic vascular resistance circulation with a low blood pressure (Parillo, 1993). In such patients, therapy with an α1 adrenergic agonist is often required to reverse the hypotension. However, hypotension may be resistant to high doses of vasopressor agents, a state known as vascular hyporeactivity. Administration of endotoxin (lipopolysaccharide; LPS), a cell wall component ubiquitous to Gram-negative bacteria, to animals and human volunteers has been used extensively to mimic Gram-negative sepsis (Thiemermann, 1997; Deitch, 1998).
In many models, endotoxin-induced vascular hyporeactivity is associated with enhanced formation of nitric oxide (NO) within the blood vessel, involving activation of mainly inducible (iNOS) but also constitutive (eNOS) isoforms of NO synthase (NOS) (Julou-Schaeffer et al., 1990; Thiemermann, 1994). Once formed, NO can activate soluble guanylyl cyclase resulting in vascular smooth muscle relaxation through the formation of guanosine 3′-5′ cyclic-monophosphate (cyclic GMP). Consistent with this, inhibition of NOS or soluble guanylyl cyclase has been shown to reverse vascular hyporeactivity in vivo and in vitro, either partially (Yen et al., 1995; Mitolo-Chieppa et al., 1996; Wu et al., 1998) or completely (Julou-Schaeffer et al., 1990; Hall et al., 1996; Scott et al., 1996). Endotoxin is known to induce the expression of other enzymes, including cyclo-oxygenase (COX-2) (Bishop-Bailey et al., 1997) suggesting that additional mechanisms are likely to contribute to endotoxin-induced changes in vascular reactivity. For example, increased prostanoid production and enhanced potassium channel activity have been reported to contribute to the development of hypotension and vascular hyporeactivity (Wu et al., 1995b; Fatehi-Hassanabad et al., 1996; Clapp & Tinker, 1998).
The majority of in vitro investigations into mechanisms of vascular hyporeactivity have used aortic tissues from rodents (Julou-Schaeffer et al., 1990; Wu et al., 1995a; Scott et al., 1996, Hall et al., 1996). However, this is a large conduit vessel that only makes a small contribution to systemic vascular resistance. In contrast, inducing hyporeactivity in smaller rodent blood vessels has proved very difficult. Using vessels harvested from endotoxaemic animals (ex-vivo) or vessels superfused with endotoxin, Mitchell et al. (1993) and Glembot et al. (1995) were unable to demonstrate diminished responses to vasoconstrictor agents. However, hyporeactivity could be demonstrated in small mesenteric and femoral vessels ex vivo, but only if the bathing medium contained L-arginine, the precursor for NO synthesis. (Schneider et al., 1992; 1994; Mitolo-Chieppa et al., 1996).
The ex vivo and in vitro models described to date differ in the degree of hyporeactivity obtained and in the response to blockade of NO synthesis. These differences may relate to the species or origin of blood vessel, the endotoxin model under investigation, the dose and duration of incubation with LPS, or the type of NOS inhibitor used. We sought to develop a reproducible in vitro model of hyporeactivity in a smaller artery and chose to study a mesenteric vessel since the mesenteric circulation is an important contributor to vascular tone, receiving 30 – 40% of the total cardiac output. Using an organ culture method, we investigated the effect of LPS on vascular reactivity to the α1 agonist, phenylephrine (PE) and the thromboxane A2 mimetic, U46619 in rat superior mesenteric artery. We sought to characterize the response to PE in terms of the dose and duration of LPS incubation, the effect of serum and the role of the endothelium. The contribution of the NO pathway was also assessed using a variety of NOS inhibitors. Preliminary results have been presented in abstract form (O'Brien et al., 1999; 2000a, 2000b).
Methods
Model
Male Sprague-Dawley rats (280 – 300 g body weight) were killed via cervical dislocation. The superior mesenteric artery was dissected out and placed in sterile Hanks Balanced Salts Solution. The artery was cleaned of connective tissue and cut into four segments or eight rings. For fresh controls, rings were immediately mounted in the organ bath containing physiological saline solution (PSS) (the composition of which was in mM: NaCl 112, KCl 5, NaHCO3 25, MgCl2 1, KH2PO4 0.5, NaH2PO4 0.5 and phenol red 0.03) gassed with 95% O2/5% CO2 at 37°C. In a few further experiments vessels were incubated for 6 h in PSS containing 1 μg ml−1 LPS (Salmonella typhosa). Otherwise segments were incubated in sterile Dulbecco's Modified Eagle's Medium (DMEM) for 6, 20 or 46 h in an atmosphere of 95% air/5% CO2. In all experiments (except where stated) the culture medium was supplemented with foetal bovine serum (FBS, 10% v v−1). LPS was added to the appropriate segments at doses ranging from 1 – 100 μg ml−1. Following incubation in culture medium, the tissues were transferred to an organ bath chamber containing PSS. Rings of mesenteric artery were attached to an isometric force transducer coupled to a recorder. Tissues were subjected to a tension of 1.25 g and permitted to relax to a resting tension of 0.8 g, the optimum tension found in preliminary experiments. An equilibration period of 1 h was allowed during which time tissues were washed at 15 min intervals. Where appropriate, LPS was added to the organ baths for the duration of the experiment. Endothelial function was assessed by monitoring relaxation to 5 μM acetylcholine (Ach) in rings precontracted with either 1 μM phenylephrine (PE) or 0.1 μM U46619. Endothelium was considered present if there was greater than 50% relaxation to Ach, and these responses to Ach were maintained up to 46 h in culture medium. In some experiments the endothelium was removed by gently passing a small tungsten wire through the lumen of the ring prior to mounting it in the organ bath.
Cumulative concentration-response curves were constructed to PE (10−9 – 10−5 M) or U44619 (9,11-dideoxy-9α, 11α-methanoepoxy prostaglandin F2α) (10−9 – 10−6 M), with increasing doses added at 5 min intervals.
Experimental protocols
At least five superior mesenteric rings, taken from a minimum of three animals, were used for each experimental group. Except where indicated, all concentration-response curves were constructed to PE using endothelium-intact rings.
Dose of LPS
LPS, at doses of 1, 10 or 100 μg ml−1 were added to the culture medium and tissues incubated overnight for a total of 20 h.
Duration of incubation
Segments were incubated in the presence or absence of LPS (1 μg ml−1) for 6, 20 and 46 h prior to mounting in the organ bath.
Effect of foetal bovine serum
Rings were incubated with and without LPS (1 μg ml−1) for 6, 20 and 46 h in the presence and absence of 10% FBS in the culture medium.
Role of endothelium
Endothelium-intact or denuded rings were incubated with and without LPS (1 μg ml−1) for 20 h.
Nitric oxide synthase inhibition (a) Addition of Nω-nitro-L-arginine methyl ester (L-NAME)
Rings were incubated with and without LPS (1 μg ml−1) for 6, 20 and 46 h. The non-specific NOS inhibitor, L-NAME (300 μM) was added 25 min prior to addition of PE.
(b) Addition of aminoguanidine
Rings were incubated with and without LPS (1 μg ml−1) for 6, 20 and 46 h. The iNOS inhibitor, aminoguanidine (400 μM) was added 25 min prior to addition of PE.
(c) Addition of N-(3-(aminomethyl)benzyl)acetamidine (1400W) or GW273629
Rings were incubated with and without LPS (1 μg ml−1) for 20 and 46 h. The highly specific iNOS inhibitors, 1400 W (10 μM) and GW273629 (10 μM) were added 25 min prior to addition of PE.
Guanylyl cyclase inhibition
Rings were incubated with and without LPS (1 μg ml−1) for 20 and 46 h. The selective soluble guanylyl cyclase inhibitor, 1H-(1,2,4)oxadiazole(4,3-a)quinoxalin-1-one (ODQ; 3 μM) was added 25 min prior to addition of PE.
Cyclo-oxygenase-2 (COX-2) inhibition
Rings were incubated with and without LPS (1 μg ml−1) for 20 h. The selective cyclo-oxygenase-2 inhibitor, NS-398 N-(2-(cyclohexyloxy)-4-nitrophenyl)-methanesulphonamide (10 μM) was added 25 min prior to addition of PE.
Effect of LPS on responses to the thromboxane A2 mimetic
Rings, either endothelium-denuded or intact, were incubated for 20 h with and without LPS (1 μg ml−1) prior to constructing concentration-response curves to U46619 (10−9 – 10−6 M).
Concentration-response curves to the NO donor, SNAP
Concentration-response curves to S-nitroso-N-acetyl-D,L-penicillamine (SNAP) (10−10 – 10−5 M) were constructed following precontraction with either 1 μM PE or 0.1 μM U46619. Doses of SNAP were added in cumulative fashion once contraction to the agonist had stabilized.
Reagents and solutions
Hanks Balanced Salt Solution (Gibco, Paisley, U.K.) was supplemented with 5 mM HEPES, 1 mM NaHCO3, 50 units ml−1 penicillin and 50 μg ml−1 streptomycin. Dulbecco's Modified Eagle's Medium was supplemented with penicillin-streptomycin solution (100 units ml−1 and 100 μg ml−1, respectively) and 2 mM L-glutamine (Gibco). Physiological salt solution (PSS) used in tension experiments contained in mM: NaCl 112, KCl 5, NaHCO3 25, MgCl2 1, KH2PO4 0.5, NaH2PO4 0.5 and phenol red 0.03. L-NAME, aminoguanidine, LPS (Salmonella typhosa), acetylcholine and PE were all obtained from Sigma Chemical Company (Poole, Dorset, U.K.). Foetal bovine serum was obtained from Gibco, U46619 from Affiniti-Research (Exeter, U.K.) and 1400 W, ODQ, NS-398 and SNAP from Alexis Corporation (Nottingham, U.K.). GW 273629 was kindly donated by Glaxo Wellcome (Stevenage, U.K.).
Statistics
All data are represented as mean±standard error of the mean (s.e.mean) of n observations. Statistical analysis was performed using two way ANOVA with repeated measures and where appropriate, corrected for multiple comparisons against the control group (Bonferroni) or all groups (Student-Newman Keuls) (SigmaStat, Jandel corporation, Chicago, U.S.A.). The concentration of agonist causing a 50% contraction or relaxation of the maximal response is expressed as the log EC50 value and was calculated using the Origin 6.0 program (Microcal, Northampton, MA, U.S.A.). The log EC50 values were compared using a one way ANOVA (with Bonferroni or Student-Newman Keuls correction as appropriate). A P value <0.05 was considered statistically significant.
Results
Effect of endotoxin on the contractile responses to phenylephrine
A typical cumulative concentration-response curve to phenylephrine (PE) in rat superior mesenteric artery (RMA) incubated in the presence and absence of 1 μg ml−1 LPS for (a) 6 h and (b) 20 h is shown in Figure 1. LPS induced marked hyporeactivity to PE, causing a depression in the maximal response and a shift to the right of the concentration-response curve compared to control tissues. In contrast, we were unable to induce significant hyporeactivity to PE if tissues were incubated in PSS for 6 h in the organ bath in the presence of 1 μg ml−1 LPS (n=5; P>0.05, two-way ANOVA).
Figure 1.
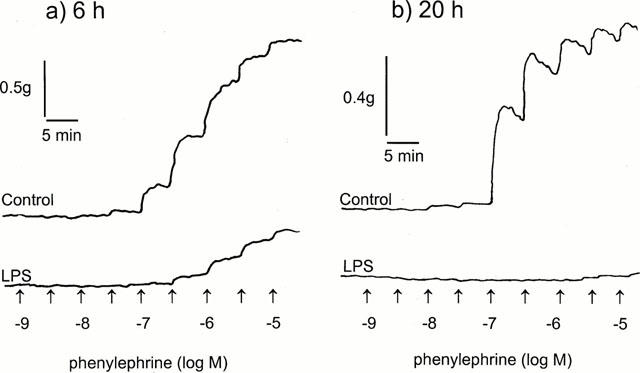
Representative traces of the contractile responses to phenylephrine in rat superior mesenteric artery rings. Tissues were incubated in the absence (control) or presence of 1 μg ml−1 LPS for either (a) 6 h or (b) 20 h.
The effect of varying the LPS concentration was investigated in RMA incubated for 20 h in culture medium supplemented with serum. LPS (1 – 100 μg ml−1) induced significant hyporeactivity to PE (P<0.001, two-wayANOVA; Figure 2) with the maximal response in the presence of 1 μg ml−1 LPS being suppressed from 1.66±0.27 – 0.7±0.14 g (n=5 – 7). In addition, LPS caused a rightward shift of the log EC50 from −6.54±0.09 (control) to −5.81±0.05, −5.64±0.07, −5.61±0.05, for 1, 10 and 100 μg ml−1, respectively (n=5 – 7; P<0.001, one-wayANOVA). However, the degree of hyporeactivity induced at the different concentrations of LPS was similar.
Figure 2.
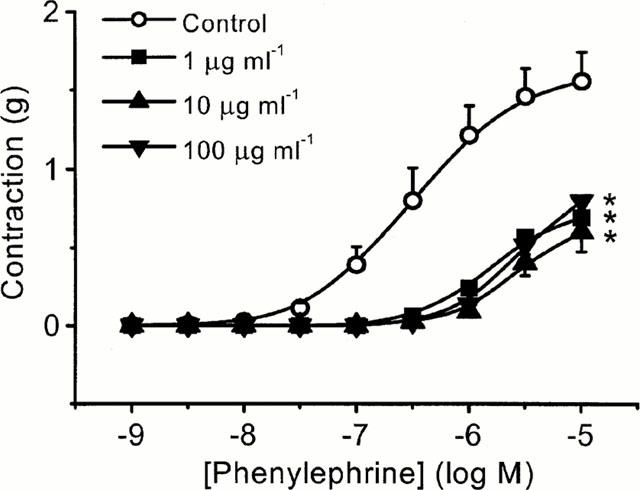
Lipopolysaccharide (LPS) induces vascular hyporeactivity in rat mesenteric artery. Tissues were incubated at 37°C in culture medium (DMEM) containing 10% serum for 20 h in the absence (control) and presence of either 1, 10 or 100 μg ml−1 LPS. Concentration-response curves to phenylephrine were obtained in endothelium intact arterial rings and comparisons made in tissues taken from the same animal. Data are expressed as the mean±s.e.mean of 5–6 observations from six animals. *=P<0.001 (ANOVA with Bonferroni correction) when compared to controls.
Effect of the duration of incubation and the presence of serum
Control tissues were incubated in the presence and absence of FBS (10% v v−1) for 6, 20 or 46 h. Over this time course, peak responses to PE were comparable over the whole concentration range to those observed in fresh controls (P>0.05, two-way ANOVA; Figure 3a,c). However, contractions elicited from tissues incubated for 46 h in culture medium with or without serum were much less sustained than was observed at either 6 or 20 h. For example, at 1 μM PE, peak contractions over a 5 min period declined by 2% at 6 h, 9% at 20 h and 42% at 46 h. Moreover, when tissues were incubated in the presence of LPS, there appeared to be a differential effect on the contractile responses to PE. In serum-free conditions, substantial hyporeactivity to LPS (1 μg ml−1) was evident at 6 and 20 h time points (maximal response 0.47±0.13 g, and 0.75±0.09 g, respectively compared to 1.56±0.16 g in controls; n=5 – 6, P<0.001, two-way ANOVA), but not at 46 h (maximal response 1.55±0.17 g) (Figure 3d). However, in tissues incubated in serum, LPS induced significant hyporeactivity at all time points including 46 h (maximal response 0.21±0.05 g compared to 1.56±0.14 g in controls; n=7, P<0.001, two-way ANOVA) (Figure 3b).
Figure 3.
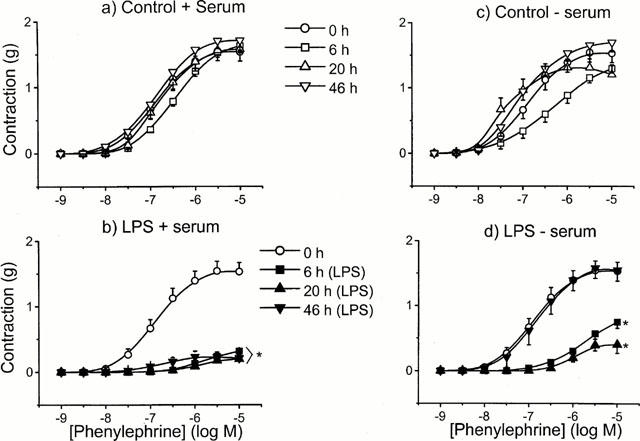
The effect of incubation time and serum on the hyporeactivity induced by LPS. Tissues were incubated in the absence (controls, open symbols) or presence (closed symbols) of 1 μg ml−1 LPS for 0–46 h, with (a,b) or without (c,d) serum in endothelium intact rings. Data are expressed as mean±s.e.mean of 6–10 observations from 8–12 animals. *=P<0.001 (ANOVA with Bonferroni correction) when compared to control.
Effect of endothelium on contractile responses to phenylephrine
In the absence of LPS, the presence of endothelium produced a rightward shift in the concentration-response to PE although the maximum contraction observed was similar in both cases (Figure 4). The log EC50 was shifted from −6.35±0.29 in endothelium-intact rings to −7.73±0.27 in endothelium-denuded rings (n=6; P<0.01, two-way ANOVA). However, following 20 h incubation in LPS (1 μg ml−1), the same level of hyporeactivity was observed regardless of whether the endothelium was present or not (Figure 4).
Figure 4.
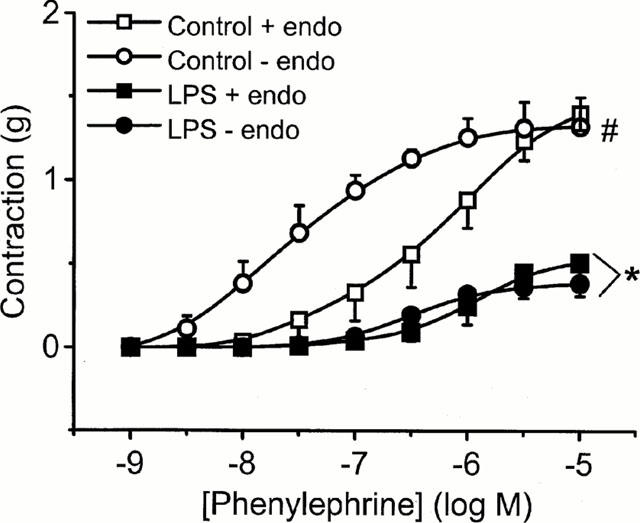
The effect of endothelium (endo) on contractile responses to phenylephrine in rat mesenteric artery. Tissues were incubated in culture medium with serum for 20 h with or without 1 μg ml−1 LPS. Data are expressed as mean±s.e.mean of 7–9 observations from 10–12 animals. *=P<0.001 when compared to their respective controls and #=P<0.05 when phenylephrine control responses are compared (ANOVA with Student-Newman Keuls correction for multiple comparisons).
Effect of nitric oxide synthase inhibitors on contractile responses to PE in LPS treated tissue
To investigate the role of NOS enzymes, we used L-NAME (a non-selective NOS inhibitor), aminoguanidine (a more selective iNOS inhibitor) and the highly selective and potent iNOS inhibitors 1400 W (Garvey et al., 1997) and GW273629 (Dr Richard Knowles, Glaxo Wellcome, personal communication). At 6 h, the addition of either aminoguanidine (400 μM) or L-NAME (300 μM) fully reversed LPS-induced hyporeactivity (n=7 – 9) (Figure 5a). However, at 20 h no reversal was seen with L-NAME. By contrast, partial reversal was achieved by aminoguanidine and 1400W (10 μM), the latter being more effective above 0.3 μM PE (P<0.01, two-way ANOVA) (Figure 5b). At 46 h partial reversal of hyporeactivity was only seen with 1400W, whereas aminoguanidine and L-NAME had no effect (Figure 5c). In control tissues, small but significant (P<0.05, two-way ANOVA; n=5 – 7) increases in the contractile responses to PE were seen in the presence of aminoguanidine (400 μM) at all time points, increasing the maximal response by 14, 26 and 19% to 1.75±0.16 g, 1.75±0.22 g and 2.06±0.12 g at 6, 20 and 46 h, respectively. Aminoguanidine had no effect on PE contractions in fresh control tissues (data not shown). The differential effects of these NOS inhibitors prompted us to conduct a series of further investigations using GW273629, another specific iNOS inhibitor, and ODQ, the highly selective guanylyl cyclase inhibitor. Similar effects to 1400W were seen at 20 and 46 h with both compounds (Figure 6a,b).
Figure 5.
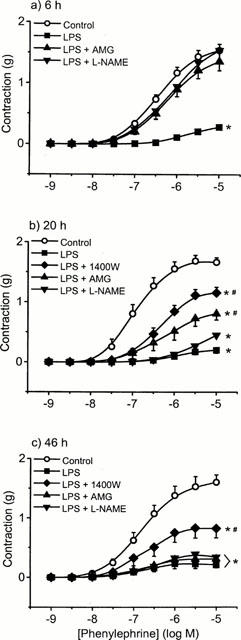
The effect of the NOS inhibitors, aminoguanidine (AMG), Nω-nitro-L-arginine methyl ester (L-NAME), and N-(3-(aminomethyl)benzyl)acetamidine (1400W) on LPS-induced hyporeactivity in endothelium-intact tissue. Rings were incubated in culture medium containing serum for (a) 6 h (b) 20 h and (c) 46 h in the absence (open symbols) or presence of 1 μg ml−1 LPS (closed symbols)±NOS inhibitor. Data are expressed as mean±s.e.mean of 8–10 observations from 14–16 animals. *P<0.001 when compared to control and #=P<0.05 when compared to LPS alone (ANOVA with Student-Newman Keuls correction for multiple comparisons).
Figure 6.
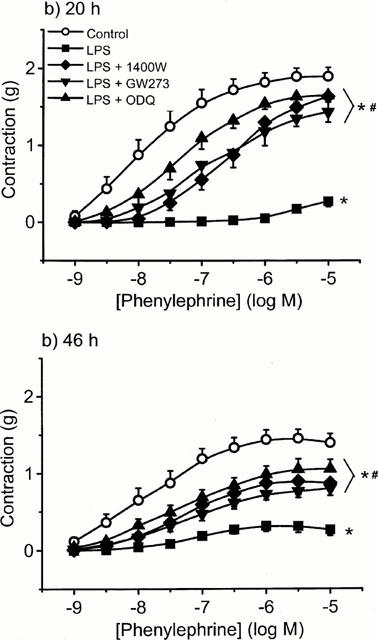
The effect of the specific iNOS inhibitors, 1400 W and GW273629 and the selective soluble guanylyl cyclase inhibitor, ODQ on LPS-induced hyporeactivity in endothelium-intact tissue. Rings were incubated in culture medium containing serum for (a) 20 h and (b) 46 h in the absence or presence of 1 μg ml−1 LPS±iNOS/ guanylyl cyclase inhibitor. Data are expressed as mean±s.e.mean of 8–10 observations from 14–16 animals. *=P<0.001 when compared to control and #=P<0.001 when compared to LPS alone (ANOVA with Student-Newman Keuls correction for multiple comparison).
Comparison between the dose-response curves to U46619 and phenylephrine
There was no significant hyporeactivity to U46619 following 20 h incubation with LPS in endothelium-intact tissue, although some hyporeactivity was induced by LPS in endothelium-denuded tissue (Figure 7). When compared to PE, the maximum response obtained to U46619 in LPS-treated tissues was significantly greater in either the presence (1.31±0.13 vs 0.51±0.11 g; Figure 7a) or absence (1.01±0.07 g vs 0.38±0.08 g; Figure 7b) of endothelium (n=6 – 10; P<0.001, two-way ANOVA). In order to examine whether the differential effect of LPS on these two contractile agonists might relate to differences in the ability of NO to inhibit responses, we examined the effect of the NO donor, SNAP. Endothelium-intact tissues were precontracted with either 1 μM PE (mean contraction 1.09±0.05 g; n=6) or 0.1 μM U46619 (mean contraction 1.33±0.08 g; n=8). Figure 8 shows that SNAP relaxed PE and U46619 induced contractions with a similar potency, the log EC50 values being −6.41±0.13 and −6.32±0.19 for U46619 and PE, respectively (n=6).
Figure 7.
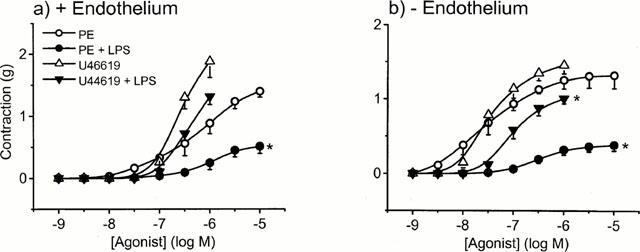
The effect of LPS on phenylephrine or U46619 (thromboxane A2 agonist) induced contractions in the presence (a) and absence (b) of endothelium. Tissues were incubated in culture medium containing serum for 20 h in the absence (open symbols) or presence of 1 μg ml−1 LPS (closed symbols). Data are expressed as the mean±s.e.mean of 8–10 observations from 12–14 animals. *=P<0.001 (ANOVA with Student-Newman Keuls test for multiple comparison) when compared to their respective controls.
Figure 8.
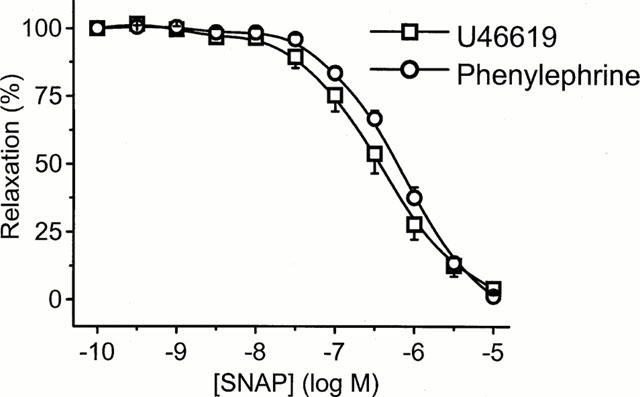
Mean concentration-response curves for S-nitroso-N- acetyl-D,L-penicillamine (SNAP) evoked relaxation of phenylephrine (1 μM) and U-46619 (0.1 μM) contractions in endothelium-intact rings. Data are expressed as the mean±s.e.mean of eight observations from six animals.
Effect of COX-2 inhibition on endotoxin-induced hyporeactivity
Since we did not achieve full reversal of endotoxin-induced hyporeactivity using NOS inhibitors, we investigated the effect of the COX-2 inhibitor NS-398. As shown in Figure 9, NS-398 had no effect on the degree of vascular hyporeactivity to PE following incubation with LPS for 20 h.
Figure 9.
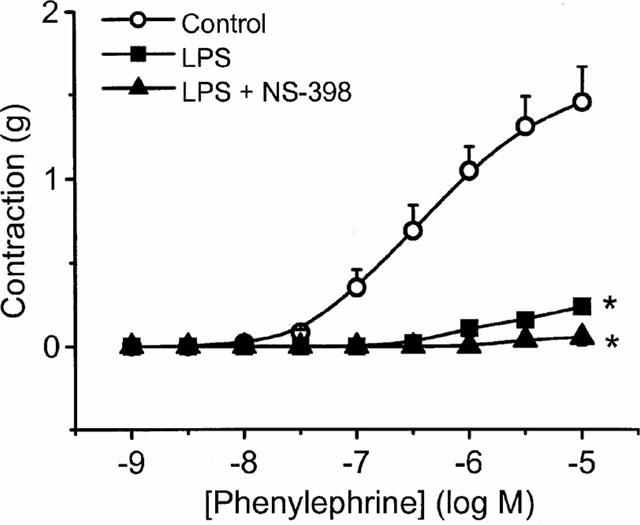
The effect of the specific COX-2 inhibitor, NS-398 on LPS-induced hyporeactivity in endothelium-intact tissue. Rings were incubated in culture medium containing serum for 20 h in the absence or presence of 1 μg ml−1 LPS±NS-398 (10 μM). Data are expressed as mean±s.e.mean of six observations from seven animals. *=P<0.001 when compared to control (ANOVA with Student-Newman Keuls test for multiple comparisons).
Discussion
We have demonstrated that in vitro incubation of rat superior mesenteric artery in culture medium with LPS reproducibly induces substantial vascular hyporeactivity to the α1-agonist, phenylephrine (PE). In contrast, contractions to the thromboxane mimetic, U46619 were only weakly affected by LPS under similar experimental conditions. Depression of the contractile response to PE was unaffected by the concentration of LPS used (1 – 100 μg ml−1), although responses were dependent upon incubation time. We noted that serum prolonged the effects of LPS, causing marked depression of the contractile response even at 46 h whereas, without serum, responses had fully recovered by this time. Moreover, hyporeactivity did not depend upon the presence of an intact endothelial layer. We investigated the contribution of the NOS pathway using inhibitors with varying selectivity towards the different NOS isoforms. While these inhibitors fully reversed the LPS-induced hyporeactivity at 6 h, the effects of these agents varied thereafter. Inhibition of soluble guanylyl cyclase substantially reversed (75 – 85%) the hyporeactivity at 20 and 46 h, while inhibition of the COX-2 pathway had no effect.
A variety of models have been developed to study vascular hyporeactivity in sepsis utilizing LPS, Gram-negative, Gram-positive bacteria, or pro-inflammatory cytokines (Fink & Heard, 1990; Deitch, 1998). Examples include in vivo models (e.g. Julou-Schaeffer et al., 1990; Rees et al., 1998) and organ bath models using vessels either incubated in LPS (Mckenna, 1990; Hall et al., 1996), or harvested from endotoxaemic animals (Schneider et al., 1992; Yen et al., 1995; Wu et al., 1995a) and septic patients (Tsuneyoshi et al., 1996). Although vascular hyporeactivity is a consistent feature, highly variable responses are seen in the level of hyporeactivity achieved and its reversal by inhibitors targeted against either NO, prostanoids or K+ channels. The mechanisms underlying this highly complex condition are yet to be fully clarified. A reproducible laboratory model of hyporeactivity would assist investigation; we thus sought to develop such a model and to characterize the response to incubation with LPS.
Substantial evidence suggests that NO is an important mediator of vascular hyporeactivity in sepsis (Thiemermann, 1994; 1997). In septic patients, elevated levels of nitrate/nitrite (breakdown products of NO) have been measured (Ochoa et al., 1991), and administration of the non-selective NOS inhibitor, L-NMMA produced significant elevations in blood pressure with a concomitant 60 – 80% reduction in cathecolamine requirements (Grover et al., 1999). Similar findings have also been reported in rodent models (Thiemermann, 1994; 1997; Rees et al., 1998). Enhanced NO production may result from activation of eNOS in the early stages of the septic insult (<1 h) followed by expression of iNOS commencing after 2 – 3 h (Thiemermann, 1994; 1997). Consistent with a major role for iNOS, vascular hyporeactivity in arteries removed from animals treated with LPS for 12 h was only observed in wild-type but not iNOS-deficient mice (Gunnett et al., 1998). Moreover, long-term exposure to LPS results in changes in iNOS, but not eNOS expression (Bishop-Bailey et al., 1997).
An important new finding in our in vitro model was the variable response over time to NOS inhibitors. Complete reversal was achieved with NOS inhibitors at 6 h, although sensitivity to these agents decreased over time. L-NAME completely lost its effect at 20 h, though 1400W, GW273629 and aminoguanidine remained effective. At 46 h, only1400W and GW273629 produced a partial reversal of LPS-induced hyporeactivity. In addition, the guanylyl cyclase inhibitor, ODQ reversed the hyporeactivity at the two later timepoints to a similar extent. Partial reversal implies that either incomplete NO blockade is occurring or that NO-independent mechanisms produce hyporeactivity at 20 and 46 h. The former may be due to either inadequate NOS inhibition, NOS-independent generation of NO, or release of NO from nitrosylated thiols (e.g. albumin). An inadequate dose of AMG or L-NAME may explain the lack of effect at the later timepoints. However, we did observe full reversal with both these agents at 6 h using the same doses. These are well above the reported IC50 values for inhibition of NOS, being 30 μM and 11 μM, respectively for aminoguanidine and L-NAME against iNOS and 0.6 μM for L-NAME against eNOS (Wu et al., 1995a). We recently found that a combination of NOS inhibitor and NO scavenger was required to inhibit NO production in rat mesenteric and hepatic arteries (Chauhan et al., 2000). Furthermore, NO release induced by LPS in rat aortic segments after 48 h in culture medium was found to be only partially inhibited by 1 mM L-NAME, though fully by a protein synthesis inhibitor (Bishop-Bailey et al., 1997). While this latter observation suggests that NO production was probably occurring through iNOS, continued production of NO and/or its metabolites may lead to the formation of NO stores within the blood vessel (Muller et al., 1998). The generation of nitrated/nitrosylated compounds by reaction of NO or its metabolites with either components of the tissue bathing medium or complexes in blood vessels is well recognized (Dowell & Martin, 1998; Muller et al., 1998; Stubauer et al., 1999; Amirmansour et al., 1999). This may also be responsible for the greater hyporeactivity seen when serum was added to the incubation medium. Such a mechanism was recently proposed to explain L-NAME-resistant vascular hyporeactivity (Muller et al., 1998).
We were surprised to find a continuing effect, albeit partial, of 1400W and GW273629 at 46 h, whereas no reversal was observed with either AMG or L-NAME. This could simply reflect the greater potency of these agents, the IC50 for GW273629 being 2 μM (R Knowles, personal communication) and the apparent inhibitory constant for 1400W being 2 nM (Garvey et al., 1997). Although we cannot exclude other actions of these agents. However, the similar effect of ODQ, which at 3 μM is considered to be a specific inhibitor of soluble guanylyl cyclase, suggests that the continued hyporeactivity is largely mediated by NO activating guanylyl cyclase and generating cyclic GMP. A further factor may be that 1400W is an irreversible inhibitor of iNOS (Garvey et al., 1997).
As we were unable to achieve full reversal with NO pathway inhibitors at the later timepoints, we investigated the effect of NS-398 a selective COX-2 inhibitor (Futaki et al., 1994). We failed to show any reversal of hyporeactivity using this agent in tissues incubated with LPS for 20 h. The production of prostaglandins E2 and F2 has been demonstrated in vitro in LPS-treated rat aortic tissues, and this was successfully blocked by the same dose of NS-398 (Bishop-Bailey et al., 1997). Although blockade of the inducible and/or constitutive cyclo-oxygenase pathways rarely reverses hypotension (Bernard et al., 1997; Leach et al., 1998), it does partially reverse LPS-induced hyporeactivity (Mckenna, 1990; Gunnett et al., 1998). In terms of other proposed mechanisms, K+ channel inhibitors can reverse some LPS effects, both in vitro (Hall et al., 1996; Muller et al., 1998) and in vivo (Wu et al., 1995b), though the type of K+ channel involved depends on the model (Clapp & Tinker, 1998).
The continued presence of LPS within the incubation medium prolonged the duration of vascular hyporeactivity. Animal models given a bolus of LPS have demonstrated shorter-lived effects. In mice given i.v. LPS, maximum haemodynamic effects were seen at 12 h, which corresponded with peak iNOS activity (Rees et al., 1998). Surviving animals showed some recovery by 18 h, which corresponded with a fall in iNOS activity. In rats receiving intraperitoneal LPS, levels of mRNA for iNOS peaked at 4 – 8 h, decreasing markedly thereafter (Lui et al., 1997). In similar models, maximum induction of iNOS occurred at 6 h returning almost to control levels by 24 h (Mitchell et al., 1993; Fricker et al., 1997). However, arterial and venous mesenteric vascular beds removed from these animals at 6 h were not hyporeactive to PE, U44619, endothelin-1 or 5-HT. In contrast, using an in vitro organ culture model in rat aorta, the expression of iNOS and COX-2 remained elevated over the entire 10-day duration in the presence of LPS (Bishop-Bailey et al., 1997). Although vascular hyporeactivity was not assessed, nitrate/nitrite levels peaked on day 2 and 9. There was hyporeactivity at 46 h in our model when LPS was added to culture medium containing serum. Thus we speculate that continued presence of LPS in the incubation media prolongs the period of iNOS induction in blood vessels compared to a bolus dose administered to rodents. We have been unable to find an organ bath experiment utilizing vessels incubated in LPS or harvested from endotoxaemic rats that have extended beyond 24 h exposure.
We and others have previously demonstrated that 6 h incubation of aortic rings with LPS in the organ bath is sufficient to produce substantial hyporeactivity to PE (Hall et al., 1996). We were unable to reproduce this effect in mesenteric rings unless we incubated tissue in culture media. This implies that the medium contains a factor(s) which promotes/enhances LPS activation of cells within the blood vessel. To date, no in vitro model of hyporeactivity has been described using mesenteric vessels. Studies have demonstrated hyporeactivity ex vivo, albeit inconsistently, where the mesenteric artery is harvested from rats 4 – 6 h after treatment with LPS (Mitchell et al., 1993; Schneider et al., 1992). We acknowledge that this artery is not a resistance vessel however, using our model, we have successfully demonstrated vascular hyporeactivity in third order human mesenteric arteries (unpublished observations).
The addition of serum to the incubation medium enhanced and prolonged the effects of LPS. Serum contains LPS binding protein and soluble CD14 which have key roles in LPS-induced cellular activation and production of pro-inflammatory cytokines (Schletter et al., 1995). LPS binding protein transfers LPS to soluble CD14 and the newly formed LPS-CD14 complex can, in turn, activate endothelial and vascular smooth muscle cells to release transcription factors responsible for the induction of various proteins (Arditi et al., 1993; Loppnow et al., 1995). The situation is somewhat different in macrophages and monocytes which already contain membrane bound CD14 receptors that can be activated directly by LPS (Schletter et al., 1995). However, CD14-independent mechanisms also contribute to LPS induction of iNOS expression and NO production in macrophages (Matsuno et al., 1998). Such a mechanism may well account for the profound hyporeactivity we observed with LPS in the absence of serum.
We found no dose-dependent effect of LPS in our model, implying complete activation of mechanisms inducing hyporeactivity at the lowest dose used (1 μg ml−1). While the LPS concentrations we used are similar to most other in vitro studies (Glembot et al., 1995; Scott et al., 1996; Muller et al., 1998), they still exceed those found in the plasma (mean peak 0.5 ng ml−1) of septic humans (Danner et al., 1991). Levels can rise up to 10 ng ml−1 in septic patients with meningitis (Brandtzaeg et al., 1992). There has been one study of concentration-dependent depression of vascular contractility at much lower concentrations of LPS (1 – 100 ng ml−1) in aortic rings incubated for 16 h (Mckenna, 1990).
Denuding vessels of endothelium did not affect the level of vascular hyporeactivity we observed with LPS, suggesting that the medial and/or adventitial layers are principally responsible for continued hyporeactivity once the vessel has been transferred to the organ bath. This is consistent with previous studies showing that an intact endothelium is not necessary for endotoxin-mediated vascular suppression (McKenna 1990; Julou-Schaeffer et al., 1990; Hall et al., 1996). Interestingly, recent data in rat aorta suggest that the adventitia is responsible for the majority of iNOS expression, NO production and medial hyporeactivity following exposure to LPS (Kleschyov et al., 1998; Zhang et al., 1999).
As observed previously in pig mesenteric and pulmonary artery (Perez-Vizcaino et al., 1996), we also found that the thromboxane A2 mimetic, U46619 was substantially less affected by LPS compared to PE. In an attempt to explain these differences, we postulated that contractions to U46619 would be less sensitive to the relaxing actions of NO. However, this was not the case as the NO donor, SNAP was equipotent against PE and U46619. Alternatively, enhanced sensitivity of LPS-treated tissues to thromboxane A2 may counter-balance the over-production of NO or other mediators. Consistent with this notion, increased sensitivity to U46619 was observed in the presence of L-NAME in the perfused mesenteric bed taken from rats treated with LPS (Mitchell et al., 1993). The relevance of these findings is unclear, but increased production and/or sensitivity of thromboxane A2 may account for the vasoconstriction seen in some vascular beds following endotoxin treatment, and for the development of pulmonary hypertension in early septic shock (Hales et al., 1981).
In summary, we have developed a reproducible model using rat superior mesenteric artery that demonstrates in vitro vascular hyporeactivity to LPS. This is largely mediated by the NO-cyclic GMP pathway. The continued responsiveness to iNOS inhibitors suggests expression of iNOS for at least 2 days. Our results highlight the importance of both incubation time and serum on the degree of hyporeactivity. Furthermore, the differential effect of the various NOS inhibitors tested emphasises the importance of selecting the appropriate agent. Immunohistochemical staining can be used to localise temporal and anatomical expression of the different NOS isoforms following LPS exposure. We conclude that organ culture may be a useful way to study the mechanisms of vascular hyporeactivity in smaller vessels over a wide time range.
Acknowledgments
This work was supported by the Medical Research Council, U.K. Dr O'Brien is an MRC Clinical Training Fellow and Dr Clapp is an MRC Senior Fellow in Basic Science. GW273629 was kindly donated by Dr Richard Knowles, Glaxo Wellcome, Stevenage, U.K.
Abbreviations
- Ach
acetylcholine
- AMG
aminoguanidine
- cyclic GMP
guanosine 3′-5′ cyclic-monophosphate
- DMEM
Dulbecco's modified Eagle's medium
- ENDO
endothelium
- FBS
foetal bovine serum
- 1400W
N-(3-(aminomethyl)benzyl)acetamidine
- GW
- L-NAME
Nω-nitro-L-arginine methyl ester
- LPS
lipopolysaccharide
- NO
nitric oxide
- NOS
nitric oxide synthase
- NS-398
N-(2-(cyclohexyloxy)-4-nitrophenyl)-methanesulphonamide
- ODQ
1H-(1,2,4)oxadiazole(4,3-a)quinoxalin-1-one
- PE
phenylephrine
- RMA
rat mesenteric artery
- SNAP
S-nitroso-N-acetyl-D,L-penicillamine
- U46619
9,11-dideoxy-9α, 11α-methanoepoxy prostaglandin F2α
References
- AMIRMANSOUR C., VALLANCE P., BOGLE R.G. Tyrosine nitration in blood vessels occurs with increasing nitric oxide concentration. Br. J. Pharmacol. 1999;127:788–794. doi: 10.1038/sj.bjp.0702590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARDITI M., ZHOU J., DORIO R., RONG G.W., GOYERT S.M., KIM K.S. Endotoxin-mediated endothelial cell injury and activation: role of soluble CD14. Infect. Immun. 1993;60:3049–3156. doi: 10.1128/iai.61.8.3149-3156.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERNARD G.R., WHEELER A.P., RUSSEL J.A., SCHEIN R., SUMMER W.R., STEINBERG K.P., FULKERSON W.J., WRIGHT P.E., CHRISTMAN B.W., DUPONT W.D., HIGGINS S.B., SWINDELL B.B. The effects of ibuprofen on the physiology and survival of patients with sepsis. The Ibuprofen in Sepsis Study Group. N. Engl. J. Med. 1997;336:912–918. doi: 10.1056/NEJM199703273361303. [DOI] [PubMed] [Google Scholar]
- BISHOP-BAILEY D., LARKIN S.W., WARNER T.D., CHEN G., MITCHELL J.A. Characterisation of the induction of nitric oxide synthase and cyclo-oxygenase in rat aorta in organ culture. Br. J. Pharmacol. 1997;121:125–133. doi: 10.1038/sj.bjp.0701100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRANDTZAEG P., OVSTEBOO R., KIERULF P. Compartmentalization of lipopolysaccharide production correlates with clinical presentation in meningococcal disease. J. Infect. Dis. 1992;66:650–652. doi: 10.1093/infdis/166.3.650. [DOI] [PubMed] [Google Scholar]
- CHAUHAN S.D., MACALLISTER R.J., CLAPP L.H., AHLUWALIA A.Evidence that NO may contribute to “EDHF-like” responses in rat mesenteric and hepatic small arteries Br. J. Pharmacol. 2000129IP (abstract) [Google Scholar]
- CLAPP L.H., TINKER A. Potassium channels in the vasculature. Curr. Opin. Nephrol. Hypertens. 1998;7:91–98. doi: 10.1097/00041552-199801000-00015. [DOI] [PubMed] [Google Scholar]
- DANNER R.L., ELIN R.J., HOSSEINI J.M., WELSEY R.A., REILLY J.M., PARILLO J.E. Endotoxaemia in human septic shock. Chest. 1991;99:169–175. doi: 10.1378/chest.99.1.169. [DOI] [PubMed] [Google Scholar]
- DEITCH E.A. Animal models of sepsis and shock: a review and lessons learned. Shock. 1998;9:1–11. doi: 10.1097/00024382-199801000-00001. [DOI] [PubMed] [Google Scholar]
- DOWELL J.F., MARTIN W. Interaction between peroxynitrite and L-cysteine: effects on rat aorta. Eur. J. Pharmacol. 1998;344:183–190. doi: 10.1016/s0014-2999(97)01598-7. [DOI] [PubMed] [Google Scholar]
- FATEHI-HASSANABAD Z., MULLER B., ANDRIANSITOHAINA R., FURMAN R., PARRAT J.R., STOCLET J.C. Influence of indomethacin on the hemodynamic effects of lipopolysaccharide in rats. Fundam. Clin. Pharmacol. 1996;10:258–263. doi: 10.1111/j.1472-8206.1996.tb00304.x. [DOI] [PubMed] [Google Scholar]
- FINK M.P., HEARD S.O. Laboratory models of sepsis and septic shock. J. Surg. Res. 1990;49:186–196. doi: 10.1016/0022-4804(90)90260-9. [DOI] [PubMed] [Google Scholar]
- FRICKER S.P., SLADE E., POWELL N.A., VAUGHAN O.J., HENDERSON G.R., MURRER B.A., MEGSON I.L., BISLAND S.K., FLITNEY F.W. Ruthenium complexes as nitric oxide scavengers: a potential therapeutic approach to nitric-oxide mediated diseases. Br. J. Pharmacol. 1997;122:1441–1449. doi: 10.1038/sj.bjp.0701504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FUTAKI N., TAKAHASHI S., YOKOYAMA M., ARAI I., HIGUCHI S., OTOMO S. NS-398 a new anti-inflammatory agent, selectively inhibits prostaglandin snthase/cyclo-oxygenase (COX-2) activity in vitro. Prostaglandins. 1994;47:55–59. doi: 10.1016/0090-6980(94)90074-4. [DOI] [PubMed] [Google Scholar]
- GARVEY E.P., OPLINGER J.A., FURFINE E.S., KIFF R.J., LASZLO F., WHITTLE B.J.R., KOWLES R.G. 1400 W is a slow, tight binding, and highly selective inhibitor of inducible nitric oxide synthase in vitro and in vivo. J. Biol. Chem. 1997;272:4959–4963. doi: 10.1074/jbc.272.8.4959. [DOI] [PubMed] [Google Scholar]
- GLEMBOT T., BRITT L.D., HILL M.A. Lack of direct endotoxin-induced vasoactive effects on isolated skeletal muscle arterioles. Shock. 1995;3:216–223. doi: 10.1097/00024382-199503000-00010. [DOI] [PubMed] [Google Scholar]
- GROVER R., ZACCARDELLI D., COLICE G., GUNTUPALLI K., WATSON D., VINCENT J.L. An open-label dose escalation study of the nitric oxide synthase inhibitor, NG-methyl-L-arginine hydrochloride (546C88), in patients with septic shock. Glaxo Wellcome International Septic Shock Study Group. Crit. Care Med. 1999;27:913–922. doi: 10.1097/00003246-199905000-00025. [DOI] [PubMed] [Google Scholar]
- GUNNETT C.A., CHU Y., HEISTAD D.D., LOIHL A., FARACI F.M. Vascular effects of LPS in mice deficient in expression of the gene for inducible nitric oxide synthase. Am. J. Physiol. 1998;275:H416–H421. doi: 10.1152/ajpheart.1998.275.2.H416. [DOI] [PubMed] [Google Scholar]
- HALES C.A., SONNE L., PETERSON M., MILLER M., WATKINS W.D. Role of thromboxane and prostacyclin in pulmonary vasomotor changes after endotoxin in dogs. J. Clin. Invest. 1981;68:497–506. doi: 10.1172/JCI110281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HALL S., TURCATO S., CLAPP L. Abnormal activation of K+ channels underlies relaxation to bacterial lipopolysaccharide in rat aorta. Biochem. Biophys. Res. Comm. 1996;224:184–190. doi: 10.1006/bbrc.1996.1005. [DOI] [PubMed] [Google Scholar]
- JULOU-SCHAEFFER G., GRAY G.A., FLEMING I., SCHOTT C., PARRATT J.R., STOCLET J. Loss of vascular responsiveness induced by endotoxin involves the L-arginine pathway. Am. J. Physiol. 1990;259:H1038–H1043. doi: 10.1152/ajpheart.1990.259.4.H1038. [DOI] [PubMed] [Google Scholar]
- KLESCHYOV A.L., MULLER B., SCHOTT C., STOCLET J.C. Role of adventitial nitric oxide in vascular hyporeactivity induced by lipopolysaccharide in rat aorta. Br. J. Pharmacol. 1998;124:623–626. doi: 10.1038/sj.bjp.0701916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEACH M., HAMILTON L.C., OLBRICH A., WRAY G.M., THIEMERMANN C. Effects of inhibitors of the activity of cyclo-oxygenase-2 on the hypotension and multiple organ dysfunction caused by endotoxin: A comparison with dexamethasone. Br. J. Pharmacol. 1998;124:586–592. doi: 10.1038/sj.bjp.0701869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOPPNOW H., STELTER F., SCHONBECK U., ERNST M., SCHUTT C., FLADD H.D. Endotoxin activates human vascular smooth muscle cells despite lack of expression of CD14 mRNA or endogenous membrane CD14. Infect. Immun. 1995;63:1020–1026. doi: 10.1128/iai.63.3.1020-1026.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUI S.F., BANES P.J., EVANS T.W. Time course and cellular localisation of lipopolysaccharide-induced inducible nitric oxide synthase messenger RNA expression in the rat in vivo. Crit. Care Med. 1997;25:512–518. doi: 10.1097/00003246-199703000-00022. [DOI] [PubMed] [Google Scholar]
- MATSUNO R., ARMAKI Y., ARIMA H., ADACHI Y., OHNO N., YADOMAE T., TSUCHIYA T. Contribution of CR3 to nitric oxide production from macrophages stimulated with high-dose of LPS. Biochem. Biophys. Res. Comm. 1998;244:115–119. doi: 10.1006/bbrc.1998.8231. [DOI] [PubMed] [Google Scholar]
- MCKENNA T.M. Prolonged exposure of rat aorta to low levels of endotoxin in vitro results in impaired contractility. J. Clin. Invest. 1990;86:160–168. doi: 10.1172/JCI114679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MITCHELL J.A., KOHLHAAS K.L., SORRENTINO R., WARNER T.D., MURAD F., VANE J.R. Induction by endotoxin of nitric oxide synthase in the rat mesentery: lack of effect of action of vasoconstrictors. Br J. Pharmacol. 1993;109:265–270. doi: 10.1111/j.1476-5381.1993.tb13563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MITOLO-CHIEPPA D., SERIO M., POTENZA M.A., MONTAGNANI M., MANSI G., PECE S., JIRILLO E., STOCLET J.C. Hyporeactivity of mesenteric vascular bed in endotoxin-treated rats. Eur. J. Pharmacol. 1996;309:175–182. doi: 10.1016/0014-2999(96)00347-0. [DOI] [PubMed] [Google Scholar]
- MULLER B., KLESCCHYOV A.L., MALBLANC S., STOCLET J.C. Nitric oxide-related cyclic GMP-independent relaxing effect of N-acetylcysteine in lipopolysaccharide-treated rat aorta. Brit. J. Pharmacol. 1998;123:1221–1229. doi: 10.1038/sj.bjp.0701737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'BRIEN A.J., SINGER M., CLAPP L.H. Effect of endotoxin on vasoconstrictor responses in rat mesenteric artery: role of inducible nitric oxide synthase. Intensive Care Med. 1999;25:S149. [Google Scholar]
- O'BRIEN A.J., SINGER M., CLAPP L.H. Comparison of nitric oxide synthase inhibitors in reversing endotoxin-induced vascular hyporeactivity. Intensive Care Med. 2000a;26:S173. [Google Scholar]
- O'BRIEN A.J., SINGER M., CLAPP L.H. Endotoxin-induced vascular hyporeactivity: does duration of incubation or addition of serum matter. Intensive Care Med. 2000b;26:S169. [Google Scholar]
- OCHOA J.B., UDEKWU A.O., BILLIAR T.R., CURRAN R.D., CERRA F.B., SIMMONS R.L., PEITZMAN A.B. Nitrogen oxide levels in patients after trauma and during sepsis. Ann. Surg. 1991;214:621–626. doi: 10.1097/00000658-199111000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PARILLO J.E. Pathogenetic mechanisms of septic shock. N. Engl. J. Med. 1993;328:1471–1477. doi: 10.1056/NEJM199305203282008. [DOI] [PubMed] [Google Scholar]
- PEREZ-VIZCAINO F., VILLAMOR E., FERNANDEZ DEL POZO B., MORO M., TAMARGO J. Lack of endotoxin-induced hyporesponsiveness to U46619 in isolated neonatal porcine pulmonary but not mesenteric arteries. J. Vasc. Res. 1996;33:249–257. doi: 10.1159/000159152. [DOI] [PubMed] [Google Scholar]
- REES D.D., MONKHOUSE J.E., CAMBRIDGE D., MONCADA S. Nitric oxide and the haemodynamic profile of endotoxin shock in the conscious mouse. Br. J. Pharmacol. 1998;124:540–546. doi: 10.1038/sj.bjp.0701815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHLETTER J., HEINE H., ULMER A.J., RIETSCHEL E.T. Molecular mechanisms of endotoxin activity. Arch. Microbiol. 1995;164:383–389. doi: 10.1007/BF02529735. [DOI] [PubMed] [Google Scholar]
- SCHNEIDER F., BUCHER B., SCHOTT C., ANDRE A., JULOU-SCHAEFFER G., STOCLET J.C. Effect of bacterial lipopolysaccharide on function of rat small femoral arteries. Am. J. Physiol. 1994;266:H191–H198. doi: 10.1152/ajpheart.1994.266.1.H191. [DOI] [PubMed] [Google Scholar]
- SCHNEIDER F., SCHOTT J.C., STOCLET J.C., JULOU-SCHAEFFER G. L-Arginine induces relaxation of small mesenteric arteries from endotoxin-treated rats. Eur. J. Pharmacol. 1992;211:269–272. doi: 10.1016/0014-2999(92)90539-g. [DOI] [PubMed] [Google Scholar]
- SCOTT J.A., MACHOUN M., MCCORMACK D.G. Inducible nitric oxide synthase and vascular reactivity in rat thoracic aorta: effect of aminoguanidine. J. Appl. Physiol. 1996;80:271–277. doi: 10.1152/jappl.1996.80.1.271. [DOI] [PubMed] [Google Scholar]
- STUBAUER G., GIUFFRE A., SARTI P. Mechanism of S-nitrosothiol formation and degradation mediated by copper ions. J. Biol. Chem. 1999;274:28128–28133. doi: 10.1074/jbc.274.40.28128. [DOI] [PubMed] [Google Scholar]
- THIEMERMANN C. The role of the L-arginine: Nitric oxide pathway. Adv. Pharmacol. 1994;28:45–79. doi: 10.1016/s1054-3589(08)60493-7. [DOI] [PubMed] [Google Scholar]
- THIEMERMANN C. Nitric oxide and septic shock. Gen. Phamacol. 1997;29:159–166. doi: 10.1016/s0306-3623(96)00410-7. [DOI] [PubMed] [Google Scholar]
- TSUNEYOSHI I., KANUMARA Y., YOSHIMURA N. Nitric oxide as a mediator of reduced arterial responsiveness in septic patients. Crit. Care Med. 1996;24:1083–1085. doi: 10.1097/00003246-199606000-00033. [DOI] [PubMed] [Google Scholar]
- WU C.C., CHEN S.J., SZABO C., THIEMERMANN C., VANE J.R. Aminoguanidine attenuates the delayed circulatory failure and improves survival in rodent models of endotoxic shock. Br. J. Pharmacol. 1995a;114:1666–1672. doi: 10.1111/j.1476-5381.1995.tb14955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WU C.C., CHEN S.J., YEN M.H. Nitric oxide-independent activation of soluble guanylyl cyclase contributes to endotoxin shock in rats. Am. J. Physiol. 1998;275:H1148–H1157. doi: 10.1152/ajpheart.1998.275.4.H1148. [DOI] [PubMed] [Google Scholar]
- WU C.C., THIEMERMANN C., VANE J.R. Glibenclamide-induced inhibition of the expression inducible nitric oxide synthase in cultured macrophages and in the anaesthetized rat. Br. J. Pharmacol. 1995b;114:1273–1281. doi: 10.1111/j.1476-5381.1995.tb13343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YEN M.H., CHEN S.J., WU C.C. Comparison of responses to aminoguanidine and Nomega-nitro-L-arginine methyl ester in the rat aorta. Clin. Exp. Pharmacol. Physiol. 1995;22:641–645. doi: 10.1111/j.1440-1681.1995.tb02081.x. [DOI] [PubMed] [Google Scholar]
- ZHANG H., COHEN R.A., CHOBANIAN A.V., BRECHER P. Adventitia as a source of inducible nitric oxide synthase in rat aorta. Am. J. Hypertens. 1999;12:467–475. doi: 10.1016/s0895-7061(98)00271-4. [DOI] [PubMed] [Google Scholar]