

Abstract
Serine proteinases elicit profound cellular effects in various tissues mediated by activation of proteinase-activated receptors (PAR). In the present study, we investigated the vascular effects of cathepsin G, a serine proteinase that is present in the azurophil granules of leukocytes and is known to activate several cells that express PARs.
In prostaglandin F2α (3 μM)-precontracted rings from porcine pulmonary arteries with intact endothelium, cathepsin G caused concentration-dependent relaxant responses (pEC50=9.64±0.12). The endothelium-dependent relaxant effect of cathepsin G could also be demonstrated in porcine coronary arteries (pEC50=9.23±0.07). In pulmonary arteries the cathepsin G-induced relaxation was inhibited after blockade of nitric oxide synthesis by L-NAME (200 μM) and was absent in endothelium-denuded vessels. Bradykinin- and cathepsin G-induced relaxant effects were associated with a 5.7 fold and 2.4 fold increase in the concentration of cyclic GMP, respectively.
Compared with thrombin and trypsin, which also produced an endothelium-dependent relaxation in pulmonary arteries, cathepsin G was 2.5 and four times more potent, respectively. Cathepsin G caused only small homologous desensitization. In cathepsin G-challenged vessels, thrombin was still able to elicit a relaxant effect.
The effects of cathepsin G were blocked by soybean trypsin inhibitor (IC50=0.043 μg ml−1), suggesting that proteolytic activity is essential for induction of relaxation. Recombinant acetyl-eglin C proved to be a potent inhibitor (IC50=0.14 μg ml−1) of the cathepsin G effect, whereas neither indomethacin (3 μM) nor the thrombin inhibitor hirudin (5 ATU ml−1) elicited any inhibitory activity. Due to their polyanionic structure defibrotide (IC50=0.11 μg ml−1), heparin (IC50=0.48 μg ml−1) and suramin (IC50=1.85 μg ml−1) diminished significantly the relaxation in response to the basic protein cathepsin G.
In conclusion, like thrombin and trypsin, cathepsin G is able to induce endothelium-dependent vascular relaxation. It can be released from activated leukocytes at sites of vascular injury and inflammation and, therefore, sufficiently high concentrations might be reached locally in the vascular space to induce vasodilatation.
Keywords: Porcine pulmonary artery, cathepsin G, thrombin, cyclic GMP, heparin, eglin C, endothelium-dependent relaxation
Introduction
Serine proteinases are involved in several physiological and pathophysiological processes, particularly in response to injury. They elicit profound cellular effects in a variety of tissues by activation of proteinase-activated receptors (PARs), which represent a new family of G-protein coupled receptors containing seven transmembrane domains (Dery et al., 1998). Activation of PARs is based on the cleavage of a peptide from the extracellular N-terminus. The newly generated N-terminus functions as a tethered ligand to activate the receptor. Cathepsin G is a serine proteinase that exists primarily in azurophilic granules of leukocytes but also as a proteolytically active, membrane-bound form. The amino acid sequence and the crystal structure of cathepsin G are known (Hof et al., 1996). Cathepsin G is released from activated granulocytes during inflammation, wound repair, immunologic processes, vascular injury, and thrombotic events. The enzyme mediates stimulant effects in several cells that express thrombin receptors such as platelets and endothelial cells. Cathepsin G causes aggregation, secretion, and cleavage of glycoprotein Ib in platelets (Selak et al., 1988; Evangelista et al., 1991; La Rosa et al., 1994; Cerletti et al., 1995; Si-Tahar et al., 1996; Rotondo et al., 1997; Kinlough-Rathbone et al., 1999). It activates factor V, inactivates factor VII, and enhances plasmin-induced fibrinolysis (Turkington, 1993; Anderssen et al., 1993; Allen & Tracy, 1995; Adams et al., 1995). Thrombin-induced PGI2 formation in endothelial cells may be inhibited by cathepsin G (Weksler et al., 1989). In cultured porcine endothelial cells, cathepsin G enhances the release of growth factors (Totani et al., 1994). Moreover, cathepsin G stimulates [3H]-Thymidine incorporation in human lymphocytes (Hase-Yamazaki & Aoki, 1995). In cathepsin G-deficient mice, there were no overt abnormalities in blood clotting and no defects in phagocytotic and chemotactic functions of neutrophils (MacIvor et al., 1999).
In previous studies, we have demonstrated that thrombin and trypsin cause an endothelium-dependent, receptor-mediated relaxation in porcine pulmonary arteries (Glusa & Paintz, 1994; Glusa et al., 1997). The aim of the present study was to investigate whether cathepsin G also elicits an endothelium-dependent relaxation of porcine pulmonary arteries. To exclude that the action of cathepsin G might be restricted to pulmonary arteries, we additionally studied its effects in porcine coronary arteries. Various compounds such eglin C (Evangelista et al., 1991), soybean trypsin inhibitor (SBTI) (Evangelista et al., 1991), heparin (Evangelista et al., 1992b; Ferrer-Lopez et al., 1992; Ermolieff et al., 1994), defibrotide (Evangelista et al., 1992a), and suramin (Cadene et al., 1997), which are known to suppress cathepsin G-evoked responses of platelets and other cells, were used to inhibit the action of the enzyme. In addition both the cyclo-oxygenase inhibitor indomethacin and the selective thrombin inhibitor hirudin were investigated. A preliminary report of some of these data has been published previously (Röbenack et al., 1998).
Methods
Experimental protocol
Pig lungs and hearts were obtained from the local slaughterhouse. Small branches of the pulmonary artery and the left circumflex coronary artery were dissected and carefully cleaned of parenchyma and connective tissue. Rings (2 – 3 mm in length, 1.5 – 2 mm diameter) were mounted on L-shaped platinum hooks and placed in 10-ml organ baths filled with modified Krebs – Henseleit solution of the following composition (mM): NaCl 118, KCl 4.7, CaCl2 2.5, MgSO4 1.2, NaHCO3 25, KH2PO4 1.2, and D-glucose 11. The solution was continuously gassed with a mixture of 95% O2 and 5% CO2 and warmed to a constant temperature of 37°C. ‘Calcium-free' medium was Krebs – Henseleit solution without calcium chloride. The rings were connected to an isometric force transducer (Hugo Sachs Elektronik, March, Germany) and changes in tension were recorded continuously. Resting tension was adjusted to 2 g at the beginning of each experiment. During an initial stabilization period of 60 min, the bathing medium was changed every 20 min and the tension repeatedly readjusted to 2 g. The tissues were stimulated at intervals of 45 min once with KCl (45 mM) and three times with prostaglandin F2α (PGF2α; 3 μM) until the contractile response was reproducible. The integrity of the endothelium was assessed functionally by measuring the extent of endothelium-dependent relaxation of PGF2α (3 μM)-precontracted rings following application of bradykinin (10 nM). Relaxation was absent after mechanical removal of the endothelium by gently rubbing the intimal surface with a roughened plastic stick. The relaxant responses to the enzymes and other agonists were studied after the third PGF2α (3 μM)-induced contraction had stabilized. Concentration-response curves to cathepsin G were constructed from mean relaxant responses to single concentrations as described previously for thrombin and trypsin (Glusa & Paintz, 1994; Glusa et al., 1997). Relaxant effects were expressed as a percentage of PGF2α-induced contraction. L-NAME and indomethacin were added to the organ bath 15 min prior to the administration of PGF2α. The inhibitors were added to the bathing medium when the PGF2α-induced contraction had nearly reached its maximum, and cathepsin G was administered to the organ bath 10 min later.
Determination of cyclic GMP
When the relaxant effect in response to cathepsin G or bradykinin in PGF2α-precontracted pulmonary rings had reached the near-maximum level (usually after 90 s), the vessels were rapidly removed from the organ bath and frozen in liquid nitrogen. In those cases where the relaxation was suppressed by inhibitors, the vessels were frozen after 90 s. The frozen samples were powdered by means of a dismembrator and then treated with 0.5 ml distilled water and 0.5 ml of 10% HClO4 at 4°C for 60 min. The homogenates were centrifuged at 10,000×g at 4°C for 5 min. The precipitated proteins were dissolved in NaOH (1 M) for protein determination using bovine serum albumin as a standard. To 400 μl supernatant, 100 μl EDTA (10 mM, pH 7.5) and 450 μl of a mixture of freon/trioctylamine (1 : 1) were added. After centrifugation at 350×g at 4°C for 2 min 400 μl of the aqueous upper phase were lyophilized. The samples were then dissolved in 0.1 ml of a buffer (pH 6.3) used for radioimmunoassay ([125I]-RIA-Kit, DuPont NEN®, Life Sciences, Brussels, Belgium). The results are expressed as pmol cyclic GMP formed per mg protein.
Data analysis
Data are presented as mean±s.e.mean for n separate experiments using vessels from different animals. Concentration-effect curves were fitted using the computer program Origin (Microcel Software, Inc., Northampton, MA, U.S.A.). Agonist potencies were expressed as pEC50 values (negative logarithm of the molar concentration of agonist producing 50% of the maximum response). The IC50 (μg ml−1) values represent the concentration of the inhibitors that reduces the relaxant response to cathepsin G (0.55 nM) by 50%. Comparison of means was made using Student's t-test modified according to the Bonferroni method. Differences were considered statistically significant at P<0.05.
Drugs
The following substances were used: cathepsin G (from human neutrophils, 2 u mg−1 protein, Calbiochem, Bad Soden, Germany), thrombin (bovine thrombin, specific activity 2023 u mg−1, was prepared and purified according to Walsmann (1968)), trypsin (bovine pancreatic trypsin, 42 u mg−1, Serva Heidelberg, Germany), recombinant hirudin (HbW 13300 ATU mg−1, Hoechst, Frankfurt, Germany), indomethacin, prostaglandin F2α (PGF2α) and bradykinin triacetate (Serva, Heidelberg, Germany), NG-nitro-L-arginine methyl ester (L-NAME, Sigma, Deisenhofen, Germany), soybean trypsin inhibitor (SBTI, 7000 BAEE inhibitor u mg−1, Biomol, Hamburg, Germany), eglin C (recombinant acetyl eglin C, Bachem, Heidelberg, Germany), suramin (Merck, Darmstadt, Germany), defibrotide (Crinos, Como, Italy), heparin (Calciparin™ 25.000 u ml−1, Sanofi, Munich, Germany). The purified bovine thrombin was kindly provided by Dr J. Stürzebecher (Center for Vascular Biology and Medicine, Erfurt).
Results
Relaxant response to cathepsin G
In porcine pulmonary arteries with intact endothelium, the mean contractile response to PGF2α (3 μM) was 2.14±0.09 g (n=46). Bradykinin (10 nM) relaxed the precontracted vessels by 92.4±1.6% (n=40) (Figure 1). The addition of cathepsin G to PGF2α-precontracted vascular rings resulted in concentration-dependent transient relaxation, which is shown in Figure 1. The pEC50 value for cathepsin G was 9.64±0.12 (n=8). In preliminary experiments an endothelium-dependent relaxant response to cathepsin G was also observed in porcine coronary arteries (pEC50 9.23±0.07; Figure 2). In the coronary arteries PGF2α (3 μM) induced a precontraction of 2.18±0.26 g (n=4). Previous studies have shown that in PGF2α-stimulated porcine pulmonary arteries with intact endothelium, thrombin and trypsin caused reversible, concentration-dependent relaxation (Glusa & Paintz, 1994; Glusa et al., 1997). Concentration-response relationships for the relaxant action of cathepsin G in comparison to those for thrombin and trypsin are shown in Figure 3.
Figure 1.
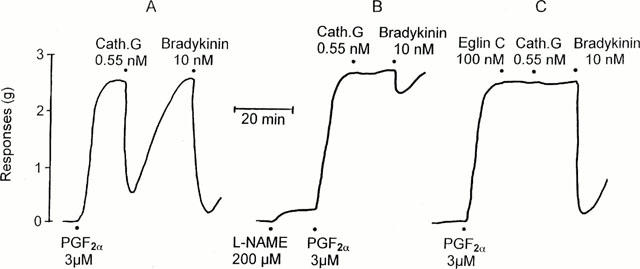
Representative experiments (ring A, B, C) for the cathepsin G (0.55 nM)-induced endothelium-dependent relaxation of PGF2α (3 μM)-precontracted rings from porcine pulmonary arteries and the inhibitory effect of L-NAME and eglin C. To assess the integrity of the endothelium, the response to bradykinin was compared.
Figure 2.
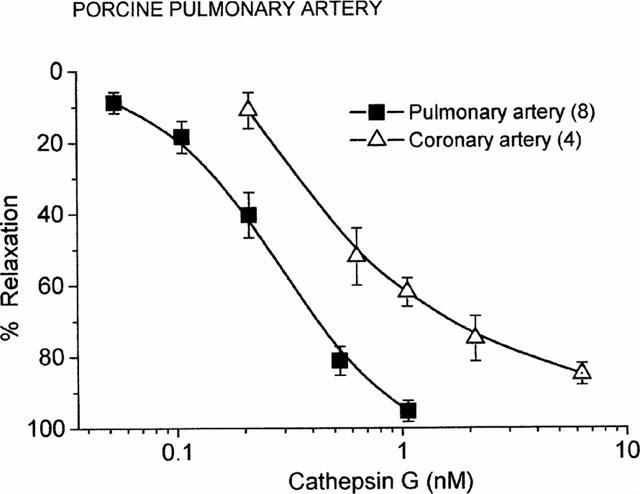
Concentration-response curves for the endothelium-dependent relaxation of PGF2α (3 μM)-precontracted porcine pulmonary artery and coronary artery induced by cathepsin G. Points are mean values and vertical bars represent the standard error of the mean for the number of tissues indicated in parentheses.
Figure 3.
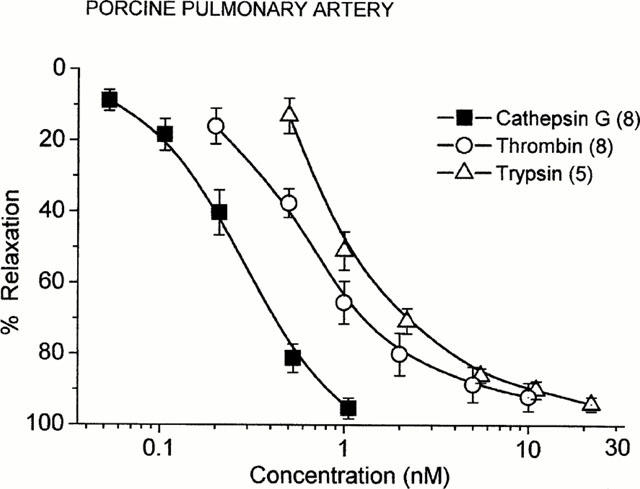
Concentration-response relationships for the endothelium-dependent relaxation of PGF2α (3 μM)-precontracted porcine pulmonary arteries induced by cathepsin G, thrombin, and trypsin. Points are mean values and vertical bars represent the standard error of the mean for the number of tissues indicated in parentheses. Data for thrombin and trypsin are from previous studies (Glusa & Paintz, 1994; Glusa et al., 1997).
The cathepsin G-induced relaxation was dependent on extracellular Ca2+. After incubation of the rings in Ca2+-free Krebs – Henseleit solution for a period of 15 min, the maximum contractile response induced by PGF2α was reduced by 10 – 15% (not significant), but the cathepsin G (0.55 nM)-induced relaxation was very small or completely absent. It should be emphasized that the bradykinin-induced relaxation was reduced by more than 75%. When the bath medium was replaced by Ca2+-containing Krebs – Henseleit, however, the relaxant response to both cathepsin G and bradykinin were completely restored within 45 min (data not shown).
Blockade of prostaglandin synthesis by indomethacin (3 μM) did not change cathepsin G-induced relaxation. Mechanical removal of the endothelium, however, led to a complete loss of cathepsin G-induced relaxation. It should be mentioned that in unstimulated, endothelium-denuded vessels, cathepsin G did not elicit any contractile response at concentrations up to 25 nM.
Desensitization of relaxation
When vessels were exposed to a second challenge with cathepsin G of the same concentration (without washouts), the second relaxant response was not significantly reduced (Figure 4). On the other hand, the cathepsin G-exposed vessels were fully responsive to thrombin (0.2 u ml−1). In contrast, in thrombin-challenged vessels, which were refractory to the second addition of thrombin, the relaxant response to cathepsin G was not significantly reduced (Figure 4). When exposure of PGF2α-precontracted vascular rings to cathepsin G was repeated after washouts at an interval of 45 min, cathepsin G exhibited the same relaxant effect as observed after the first exposure (data not shown).
Figure 4.
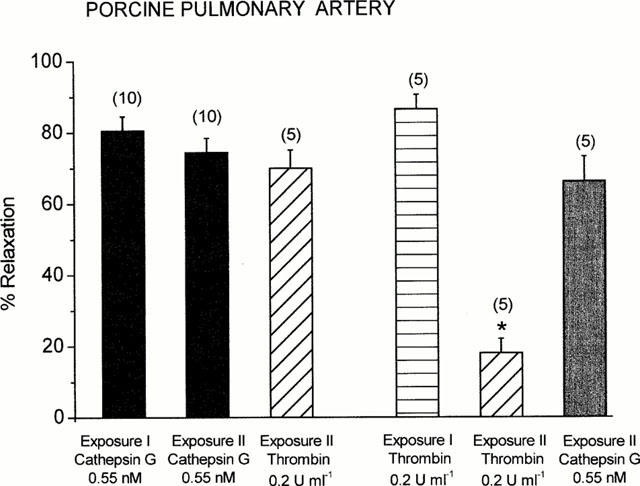
Studies on desensitization of the relaxant responses of PGF2α (3 μM)-precontracted porcine pulmonary arteries to cathepsin G and thrombin. After the first relaxant response to one enzyme (Exposure I), the porcine pulmonary vessels were exposed to a second challenge (Exposure II) by the same or another enzyme (about 25–35 min later). Columns are mean values and vertical bars represent the standard error of the mean for the number of tissues indicated in parentheses. *P<0.05 compared with thrombin (Exposure I).
Influence on cyclic GMP level
Pretreatment of the vessels with the NO synthase inhibitor L-NAME (200 μM) caused a small increase in basic tone (0.19±0.04 g, n=6). Under these conditions the combined action of L-NAME and 3 μM PGF2α resulted in a contractile effect of 2.59±0.26 g (n=6). In control rings from the same arteries the contractile response amounted to 2.07±0.17 g (n=6, not significant). L-NAME (200 μM) strongly attenuated the relaxant effect of cathepsin G, as shown in the representative tracings of Figure 1. To investigate whether cathepsin G-induced relaxation was mediated through the release of NO, cyclic GMP concentrations in rings from pulmonary arteries were determined. In unstimulated rings the basal cyclic GMP concentrations amounted to 0.52±0.04 pmol mg−1 protein (n=6) which was significantly (P<0.05) diminished to 0.12±0.03 pmol mg−1 protein (n=4) when the tissues were preincubated with L-NAME (200 μM). Compared to PGF2α-stimulated control rings, both bradykinin and cathepsin G caused a 5.7 fold and 2.4 fold increase in the vascular cyclic GMP concentration, respectively (Figure 5). The effect of cathepsin G was significantly reversed by both eglin C (0.1 μM) and L-NAME (200 μM; Figure 5).
Figure 5.
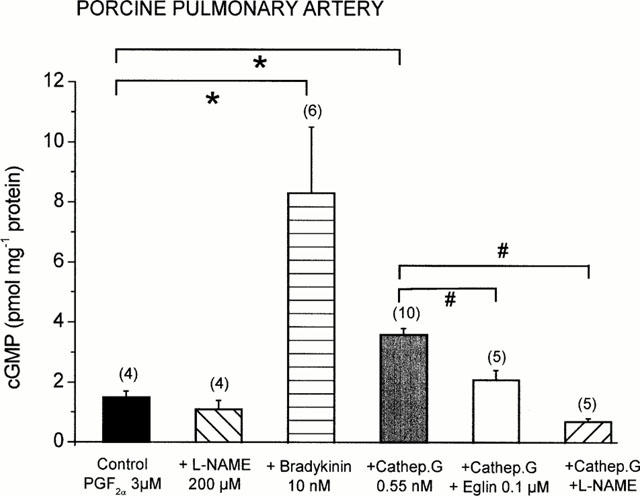
Changes in cyclic GMP concentrations in PGF2α (3 μM)-precontracted porcine pulmonary arteries after treatment with L-NAME, bradykinin, cathepsin G, and cathepsin G in the presence of L-NAME or eglin C. Columns are mean values and vertical bars represent the standard error of the mean for the number of tissues indicated in parentheses. *P<0.05 compared with the control; #P<0.05 compared with the cathepsin G values.
Inhibition of relaxation
The relaxation induced by cathepsin G (0.55 nM) was not influenced by the thrombin inhibitor hirudin (5 ATU ml−1) but was susceptible to SBTI, which reduced the relaxant effect of cathepsin G by 93.5±3.5% at 3 μg ml−1 (Figure 6). For SBTI an IC50 value of 0.043±0.005 μg ml−1 (n=5) was determined. In the presence of SBTI, when the cathepsin G-induced relaxation was nearly completely inhibited, the subsequent addition of thrombin (0.2 u ml−1) induced a relaxation that was not significantly different from that in control vessels. Under the same conditions the trypsin (0.1 μg ml−1)-induced relaxation was completely suppressed by SBTI.
Figure 6.
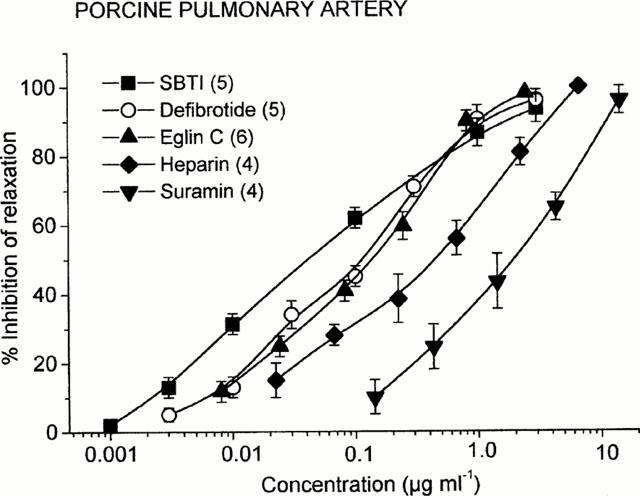
Inhibition of cathepsin G (0.55 nM)-induced relaxation of PGF2α (3 μM)-precontracted porcine pulmonary arteries by soybean trypsin inhibitor (SBTI), eglin C, heparin (Calciparin™), defibrotide, and suramin in porcine pulmonary arteries. Points are mean values and vertical bars represent the standard error of the mean for the number of tissues indicated in parentheses.
Recombinant eglin C, which has been shown to be a potent inhibitor of the catalytic activity of cathepsin G (Seemüller et al., 1981), inhibited significantly the cathepsin G-induced relaxation producing an IC50 value of 0.14±0.05 μg ml−1 (n=6, Figure 6). Neither the thrombin- nor the trypsin-induced relaxation was inhibited by eglin C. Cathepsin G is a basic protein whose activity can be inhibited by polyanionic glucosaminoglycans such as heparin (Evangelista et al., 1992a). In porcine pulmonary arteries, heparin caused a concentration-dependent inhibition of cathepsin G-induced relaxation (Figure 6). The IC50 value for heparin was 0.48±0.11 μg ml−1 (corresponding to IC50 of 0.07 u ml−1). At the concentrations used, heparin did not interfere with thrombin- and trypsin-mediated relaxant effects. Suramin, another polyanionic compound that inhibits the neutrophil proteinases elastase and cathepsin G (Cadene et al., 1997), significantly suppressed cathepsin G-induced vascular relaxation with an IC50 value of 1.85±0.45 μg ml−1 (Figure 6). In addition, defibrotide, which is known to inhibit platelet activation by cathepsin G (Evangelista et al., 1992b), blocked the cathepsin G-mediated relaxation of pulmonary arteries in a concentration-dependent manner (IC50=0.11±0.03 μg ml−1; Figure 6).
Discussion
The present study demonstrates for the first time that cathepsin G at nanomolar concentrations induces endothelium-dependent relaxation of precontracted porcine pulmonary arteries. The cathepsin-induced relaxant response could also be demonstrated in porcine coronary arteries. The nearly equipotent relaxant activity of cathepsin G in both types of arteries supported the suggestion that this activity was not restricted to the pulmonary vasculature but might also contribute to the circulation in other vascular beds.
In pulmonary arteries cathepsin G was 2.5 and four times more potent at causing relaxation than thrombin and trypsin, respectively. The relaxant effect was probably due to the release of endothelial NO because L-NAME blocked the effect and no relaxation was found in endothelium-denuded rings. In addition, the relaxant effect was accompanied by a 2.4 fold increase in cyclic GMP, consistent with the activation of guanylyl cyclase. On the other hand, indomethacin did not modify the cathepsin G-induced relaxation, indicating that prostaglandins were not involved. Cathepsin G did not directly affect smooth muscle cells, as it did not contract endothelium-denuded rings even at concentrations much higher than those sufficient to induce relaxation. This is in contrast to the action of thrombin and trypsin, both of which cause contractile effects in endothelium-denuded pulmonary arteries (Glusa et al., 1994; 1997).
The proteolytic activity of cathepsin G is necessary for its relaxant effects, as is the case for thrombin and trypsin. Inhibitors that block cathepsin G activity attenuated also the relaxant response. Besides soybean trypsin inhibitor, recombinant eglin C is a very potent selective inhibitor of the proteolytic activity of cathepsin G. Eglin C, a protein from the leech Hirudo medicinalis, forms a 1 : 1 molar inactive complex with cathepsin G with Ki values in the range of 0.01 – 0.1 nM (Seemüller et al., 1981). In the present study, it was also a potent inhibitor of the vascular effects of cathepsin G but failed to inhibit the thrombin- or trypsin-induced relaxation. When the catalytic activity of cathepsin G was measured with the specific chromogenic substrate N-succinyl-Ala-Ala-Pro-Phe-p-nitroanilide, recombinant eglin C inhibited the enzymatic activity in a concentration-dependent manner. The range of the effective concentrations was comparable to that observed for inhibition of vascular relaxation (unpublished data).
Cathepsin G is a very basic protein due to the positively charged arginine residues (Hof et al., 1996). Therefore, polyanionic substances such as heparin, defibrotide, or suramin may reduce the catalytic activity of cathepsin G. The inhibitory potency of heparin is not related to its anticoagulant properties (Evangelista et al., 1992b). It has been shown that cathepsin G and a low molecular weight heparin form a tightly bound 1 : 1 complex that is inhibited only extremely slowly by α1-antichymotrypsin and α1-protease inhibitor and is resistant to inhibition by eglin C (Ermolieff et al., 1994). Heparin inhibits cathepsin G-induced platelet aggregation and secretion in a concentration-dependent manner between 0.01 and 0.07 u ml−1 (Ferrer-Lopez et al., 1992). Inhibition of cathepsin G-induced vascular relaxation occurred at heparin concentrations that were in same range as used in studies with platelets (Ferrer-Lopez et al., 1992; Evangelista et al., 1992b). Similarly, the polydesoxyribonucleotide defibrotide, which exerts an antithrombotic effect in animal experiments, inhibits platelet activation by activated neutrophils at concentrations between 1 and 100 μg ml−1 (Evangelista et al., 1992a). At 3 μg ml−1, defibrotide nearly completely inhibited cathepsin G-induced relaxation. Suramin, a hexasulphonated naphthylurea, which binds to cathepsin G with a Ki value of 80 nM, inhibits the cathepsin G (0.25 μM)-induced aggregation of washed human platelets with an IC50 of 4 μM (Cadene et al., 1997) which again is in the same range of concentrations as that used in the present vascular studies. The thrombin inhibitor hirudin neutralizes the effect of thrombin but not that of cathepsin G or trypsin.
Thrombin causes a homologous desensitisation of its relaxant response but this is not the case with cathepsin G. After exposure to cathepsin G the response to a second challenge was not significantly reduced. Also after washout for 45 min the second relaxant effect of cathepsin G was unchanged. In addition, there was no heterologous desensitization between cathepsin G and thrombin or trypsin, i.e., cathepsin G did not reduce the subsequent response to thrombin or trypsin and vice versa. These findings lead to the assumption that the vascular cathepsin G receptor seems to be distinct from the thrombin receptor (Molino et al., 1995; Renesto et al., 1997). Similarly, the specific receptor of platelets to which cathepsin G binds has been distinguished from the thrombin receptor, PAR-1 (Selak, 1994; Selak & Smith, 1990). Cathepsin G neither activated PAR-2 nor prevented its subsequent activation by trypsin (Molino et al., 1997). At this time it is not clarified whether the effects of cathepsin G on some cell types are due to preferential cleavage of PAR-1 or due to activation of a not yet identified receptor that might belong to the family of PARs. Recent studies indicate that cathepsin G activates platelets via stimulation of PAR-4 (Sambrano et al., 2000). In contrast, there is little information about PAR-4 in blood vessels (Xu et al., 1998). In precontracted rat aorta, very high concentrations of the PAR-4 receptor activating peptide GYPGQV-NH2 (EC50 300 μM) were required to induce an endothelium-dependent relaxation (Hollenberg et al., 1999) suggesting that at least PAR-4 is not involved in the vascular response to cathepsin G.
In summary, the present study demonstrates that cathepsin G may induce the release of endothelial NO thereby eliciting endothelium-dependent vascular relaxation. The cathepsin G concentrations used in the present study correspond to those concentrations which might be reached locally in the vascular space at sites of injury or inflammation. Leukocytes contain relatively large amounts (approximately 1 – 4 μg per 106 cells) of cathepsin G (Weksler et al., 1989; Evangelista et al., 1991; Renesto & Chignard, 1993; Owen et al., 1995). This implies that at sites of injury or inflammation activated neutrophils can express up to 160 ng of catalytically active cathepsin G per 106 cells (Owen et al., 1995), a concentration sufficiently high to induce a local endothelium-dependent vasodilatation and hyperaemia thereby contributing to an inflammatory process.
Acknowledgments
The skilful technical assistance of Mrs I. Weiss is gratefully acknowledged. This study was supported by a grant of the Deutsche Forschungsgemeinschaft.
Abbreviations
- L-NAME
NG-nitro-L-arginine methyl ester
- NO
nitric oxide
- PAR
proteinase-activated receptor
- PGF2α
prostaglandin F2α
- SBTI
soybean trypsin inhibitor
References
- ADAMS S.A., KELLY S.L., KIRSCH R.E., ROBSON S.C., SHEPARD E.G. Role of neutrophil membrane proteases in fibrin degradation. Blood Coagul. Fibrinol. 1995;6:693–702. doi: 10.1097/00001721-199512000-00001. [DOI] [PubMed] [Google Scholar]
- ALLEN D.H., TRACY P.B. Human coagulation factor V is activated to the functional cofactor by elastase and cathepsin G expressed at the monocyte surface. J. Biol. Chem. 1995;270:1408–1415. doi: 10.1074/jbc.270.3.1408. [DOI] [PubMed] [Google Scholar]
- ANDERSSEN T., HALVORSEN H., BAJAK S.P., OSTERUD B. Human leukocyte elastase and cathepsin G inactivate factor VII by limited proteolysis. Thromb. Haemost. 1993;70:414–417. [PubMed] [Google Scholar]
- CADENE M., DURANTON J., NORTH A., SI-TAHAR M., CHIGNARD M., BIETH J.G. Inhibition of neutrophil serine proteinases by suramin. J. Biol. Chem. 1997;272:9950–9955. doi: 10.1074/jbc.272.15.9950. [DOI] [PubMed] [Google Scholar]
- CERLETTI C., EVANGELISTA V., MOLINO M., DE GAETANO G. Platelet activation by polymorphonuclear leukocytes: role of cathepsin G and P-selectin. Thromb. Haemost. 1995;74:218–223. [PubMed] [Google Scholar]
- DERY O., CORVERA C.U., STEINHOFF M., BUNNETT N.W. Proteinase-activated receptors: novel mechanisms of signaling by serine proteases. Am. J. Physiol. 1998;274:C1429–C1452. doi: 10.1152/ajpcell.1998.274.6.C1429. [DOI] [PubMed] [Google Scholar]
- ERMOLIEFF J., BOUDIER C.B., LAINE A., MEYER B., BIETH J.G. Heparin protects cathepsin G against inhibition by protein proteinase inhibitors. J. Biol. Chem. 1994;269:29502–29508. [PubMed] [Google Scholar]
- EVANGELISTA V., PICCARDONI P., DE GAETANO G., CERLETTI C. Defibrotide inhibits platelet activation by cathepsin G released from stimulated polymorphonuclear leukocytes. Thromb. Haemost. 1992a;67:660–664. [PubMed] [Google Scholar]
- EVANGELISTA V., PICCARDONI P., MAUGERI N., DE GAETANO G., CERLETTI C. Inhibition by heparin of platelet activation induced by neutrophil-derived cathepsin G. Eur. J. Pharmacol. 1992b;216:401–405. doi: 10.1016/0014-2999(92)90437-9. [DOI] [PubMed] [Google Scholar]
- EVANGELISTA V., RAJTAR G., DE GAETANO G., WHITE J.G., CERLETTI C. Platelet activation by fMLP-stimulated polymorphonuclear leukocytes: the activity of cathepsin G is not prevented by antiproteinases. Blood. 1991;77:2379–2388. [PubMed] [Google Scholar]
- FERRER-LOPEZ P., RENESTO P., PREVOST M.C., GOUNON P., CHIGNARD M. Heparin inhibits neutrophil-induced platelet activation via cathepsin G. J. Lab. Clin. Med. 1992;119:231–239. [PubMed] [Google Scholar]
- GLUSA E., BRETSCHNEIDER E., PAINTZ M. Contractile effects of thrombin in porcine pulmonary arteries and influence of thrombin inhibitors. Naunyn-Schmiedeberg's Arch. Pharmacol. 1994;349:101–106. doi: 10.1007/BF00178213. [DOI] [PubMed] [Google Scholar]
- GLUSA E., PAINTZ M. Relexant and contractile responses of porcine pulmonary arteries to a thrombin receptor activating peptide (TRAP) Naunyn-Schmiedeberg's Arch. Pharmacol. 1994;349:431–436. doi: 10.1007/BF00170891. [DOI] [PubMed] [Google Scholar]
- GLUSA E., SAFT A., PRASA D., STÜRZEBECHER J. Trypsin- and SLIGRL-induced vascular relaxation and the inhibition by benzamidine derivatives. Thromb. Haemost. 1997;78:1399–1403. [PubMed] [Google Scholar]
- HASE-YAMAZAKI T., AOKI Y. Stimulation of human lymphocytes by cathepsin G. Cell. Immunol. 1995;160:24–32. doi: 10.1016/0008-8749(95)80005-4. [DOI] [PubMed] [Google Scholar]
- HOF P., MAYR I., HUBER R., KORZUS E., POTEMPA J., TRAVIS J., POWERS J.C., BODE W. The 1.8 A crystal structure of human cathepsin G in complex with Suc-Val-Pro-PheP-(OPh)2: a Janus-faced proteinase with two opposite specificities. EMBO J. 1996;15:5481–5491. [PMC free article] [PubMed] [Google Scholar]
- HOLLENBERG M.D., SAIFEDDINE M., AL ANI B., GUI Y. Proteinase-activated receptor 4 (PAR (4)): action of PAR(4)-activating peptides in vascular and gastric tissue and lack of cross-reactivity with PAR(1) and PAR(2) Can. J. Physiol. Pharmacol. 1999;77:458–464. [PubMed] [Google Scholar]
- KINLOUGH-RATHBONE R.L., PERRY D.W., RAND M.L., PACKHAM M.A. Effects of cathepsin G pretreatment of platelets on their subsequent responses to aggregating agents. Thromb. Res. 1999;95:315–323. doi: 10.1016/s0049-3848(99)00051-1. [DOI] [PubMed] [Google Scholar]
- LA ROSA C.A., ROHRER M.J., BENOIT S.E., RODINO L.J., BARNARD M.R., MICHELSON A.D. Human neutrophil cathepsin G is a potent platelet activator. J. Vasc. Surg. 1994;19:306–319. doi: 10.1016/s0741-5214(94)70106-7. [DOI] [PubMed] [Google Scholar]
- MACIVOR D.M., SHAPIRO S.D., PHAM C.T.N., BELAAOUAJ A., ABRAHAM S.N., LEY T.J. Normal neutrophil function in cathepsin G-deficient mice. Blood. 1999;94:4282–4293. [PubMed] [Google Scholar]
- MOLINO M., BLANCHARD N., BELMONTE E., TARVER A.P., ABRAMS C., HOXIE J.A., CERLETTI C., BRASS L.F. Proteolysis of the human platelet and endothelial cell thrombin receptor by neutrophil-derived cathepsin G. J. Biol. Chem. 1995;270:11168–11175. doi: 10.1074/jbc.270.19.11168. [DOI] [PubMed] [Google Scholar]
- MOLINO M., WOOLKALIS M.J., REAVEY-CANTWELL J., PRATICO D., ANDRADE-GORDON P., BARNATHAN E.S., BRASS L.F. Endothelial cell thrombin receptors and PAR-2. J. Biol. Chem. 1997;272:11133–11141. doi: 10.1074/jbc.272.17.11133. [DOI] [PubMed] [Google Scholar]
- OWEN C.A., CAMPBELL M.A., BOUKEDES S.S., CAMPBELL E.J. Inducible binding of bioactive cathepsin G to the cell surface of neutrophils. J. Immunol. 1995;155:5803–5810. [PubMed] [Google Scholar]
- RENESTO P., CHIGNARD M. Enhancement of cathepsin G-induced platelet activation by leukocyte elastase: consequence for the neutrophil-mediated platelet activation. Blood. 1993;82:139–144. [PubMed] [Google Scholar]
- RENESTO P., SI-TAHAR M., MONIATTE M., BALLOY V., VAN DORSSELAER A., PIDARD D., CHIGNARD M. Specific inhibition of thrombin-induced cell activation by the neutrophil proteinases elastase, cathepsin G, and proteinase 3: evidence for distinct cleavage sites within the aminoterminal domain of the thrombin receptor. Blood. 1997;89:1944–1953. [PubMed] [Google Scholar]
- RÖBENACK C., PAINTZ M., GLUSA E. Cathepsin G induces endothelium-dependent relaxation in isolated porcine coronary and pulmonary arteries. Naunyn-Schmiedeberg's Arch. Pharmacol. 1998;357 Suppl:R48. [Google Scholar]
- ROTONDO S., EVANGELISTA V., MANARINI S., DE GAETANO G., CERLETTI C. Different requirement of intracellular calcium and protein kinase C for arachidonic acid release and serotonin secretion in cathepsin G-activated platelets. Thromb. Haemost. 1997;78:919–925. [PubMed] [Google Scholar]
- SAMBRANO G.R., HUANG W., FARUQI T., MAHRUS S., CRAIK C., COUGHLIN S.R. Cathepsin G activates protease-activated receptor-4 in human platelets. J. Biol. Chem. 2000;275:6819–6823. doi: 10.1074/jbc.275.10.6819. [DOI] [PubMed] [Google Scholar]
- SEEMÜLLER U., FRITZ H., EULITZ M. Eglin: elastase-cathepsin G inhibitor from leeches. Methods Enzymol. 1981;80:804–816. [Google Scholar]
- SELAK M.A. Cathepsin G and thrombin: evidence for two different platelet receptors. Biochem. J. 1994;297:269–275. doi: 10.1042/bj2970269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SELAK M.A., CHIGNARD M., SMITH J.B. Cathepsin G is a strong platelet agonist released by neutrophils. Biochem. J. 1988;251:293–299. doi: 10.1042/bj2510293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SELAK M.A., SMITH J.B. Cathepsin G binding to human platelets. Evidence for a specific receptor. Biochem. J. 1990;266:55–62. doi: 10.1042/bj2660055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SI-TAHAR M., RENESTO P., FALET M., RENDU F., CHIGNARD M. The phospholipase C/protein kinase C pathway is involved in cathepsin G-induced human platelet activation: comparison with thrombin. Biochem. J. 1996;313:401–408. doi: 10.1042/bj3130401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TOTANI L., PICCOLI A., PELLEGRINI G., DI SANTO A., LORENZET R. Polymorphonuclear leukocytes enhance release of growth factors by cultured endothelial cells. Arterioscler. Thromb. 1994;14:125–132. doi: 10.1161/01.atv.14.1.125. [DOI] [PubMed] [Google Scholar]
- TURKINGTON P.T. Cathepsin G activates human factor V in vitro. Thromb. Res. 1993;72:333–337. doi: 10.1016/0049-3848(93)90142-b. [DOI] [PubMed] [Google Scholar]
- WALSMANN P. Über die Reinigung von Thrombinpräparaten. Pharmazie. 1968;23:401–402. [PubMed] [Google Scholar]
- WEKSLER B., JAFFE E.A., BROWER M.S., COLE O.F. Human leukocyte cathepsin G and elastase specifically suppress thrombin-induced prostacyclin production in human endothelial cells. Blood. 1989;74:1627–1634. [PubMed] [Google Scholar]
- XU W.-F., ANDERSEN H., WHITMORE T.E., PRESNELL S.R., YEE D.P., CHING A., GILBERT T., DAVIE E.W., FOSTER D.C. Cloning and characterization of human protease-activated receptor 4. Proc. Natl. Acad. Sci. U.S.A. 1998;95:6642–6646. doi: 10.1073/pnas.95.12.6642. [DOI] [PMC free article] [PubMed] [Google Scholar]