

Abstract
To elucidate mechanisms of hippocampal serotonin release and possible mechanisms of clinical action of carbamazepine (CBZ), we determined interaction between antagonists of N-type (ω-conotoxin GVIA:GVIA), P-type (ω-agatoxin IVA:IVA) Ca2+ channels, Na+ channel (tetrodotoxin: TTX) and CBZ on hippocampal basal, Ca2+- and K+-evoked serotonin releases, using microdialysis in freely moving rats.
Basal release was reduced by TTX, GVIA and IVA (GVIA>IVA). Ca2+-evoked release was reduced by GVIA but unaffected by TTX and IVA. K+-evoked release was reduced by TTX, GVIA and IVA (GVIA<IVA).
TTX inhibited actions of IVA and GVIA on respective basal and K+-evoked releases, without affecting Ca2+-evoked release.
Perfusion with 100 μM CBZ (estimated-concentration in hippocampal tissue: 19±2 μM) enhanced basal and Ca2+-evoked releases, but reduced K+-evoked release, whereas 1000 μM CBZ (estimated-concentration in hippocampal tissue: 188±16 μM) reduced three types of releases.
Under condition of pretreatment with 100 and 1000 μM CBZ, TTX unaffected basal and K+-evoked releases. Under condition of pretreatment with 100 μM CBZ, IVA and GVIA unaffected basal and K+-evoked releases, respectively, but GVIA reduced basal, Ca2+-evoked releases and IVA also reduced K+-evoked release. Under condition of pretreatment with 1000 μM CBZ, GVIA unaffected three types of releases, and IVA unaffected basal release but reduced K+-evoked release.
These findings contribute towards the possible mechanisms of concentration-dependent antiepileptic action of CBZ, which possibly inhibits Na+ channel related neurotransmitter release mechanisms during K+-evoked stage, and simultaneously enhances N-type Ca2+ channel related basal serotonin release at the resting stage.
Keywords: Ca2+ channel, Na+ channel, carbamazepine, serotonin, epilepsy, hippocampus
Introduction
The antiepileptic drug carbamazepine (CBZ), 5-carbamoyl-5H-dibenz[b,f]azepine was synthesized about 40 years ago (Schindler, 1960). CBZ has a wide clinical spectrum of use in epileptic (Scheffer et al., 1994; Loiseau & Duche, 1995), bipolar (Okuma et al., 1990), anxiety disorders (Keck et al., 1992) and schizophrenia (Neppe, 1982). The major mechanisms of antiepileptic action of CBZ were considered to be its inhibitory effects on Na+ channel activity (Mclean & Macdonald, 1986; Mattson, 1997). Numerous investigators have already studied the effects of CBZ on monoaminergic transmission (Kaneko et al., 1980; Pratt et al., 1985; Elphick et al., 1990; Okada et al., 1997a, 1997b, 1998a, 1998b; Ichikawa & Meltzer, 1999), because monoaminergic function has been related to the aetiology of several psychiatric and neurological disorders. It has been established that inhibition of Na+ channels reduces basal release of monoamine and acetylcholine (Westerink et al., 1988, 1989). However, we have already demonstrated concentration-dependent biphasic effects of CBZ on various neurotransmitter releases, including acetylcholine, dopamine, serotonin and glutamate, in which plasma-concentration of CBZ associated with its antiepileptic action, enhanced the synthesis and basal release of monoamine and acetylcholine, and inhibited the depolarization-induced glutamate release, whereas more than plasma-concentration of CBZ associated with its antiepileptic action reduced them (Kaneko et al., 1980; Okada et al., 1997a, 1997b; 1998a, 1998b; Mizuno et al., 2000). Our previous studies, however, could not rule out the possibility that the inhibitory effects of CBZ on basal release observed at more than plasma-concentration of CBZ associated with its antiepileptic action may also be present at plasma-concentration of CBZ associated with its antiepileptic action in the event of neuronal excitation leading to high-frequency activity (i.e. during seizures).
Synaptic neurotransmitter release is mediated by the entry of Ca2+ into presynaptic nerve terminals through voltage-sensitive Ca2+ channels (Dunlap et al., 1995; Randall & Tsien, 1995; Harvey et al., 1996; Wu & Saggau, 1997; Bergquist et al., 1998; Okada et al., 1998c; 2001) of which different types have been shown to exist (Dunlap et al., 1995; Randall & Tsien, 1995; Wu & Saggau, 1997). Electrophysiological experiments have demonstrated at least seven subtypes of voltage-sensitive Ca2+ channels, including L-, N-, O-, P-, Q-, R- and T-type Ca2+ channel subtypes (Olivera et al., 1994; Randall & Tsien, 1995; Jones, 1998). Pharmacological experiments have demonstrated that monoamine release is regulated by N-, P- and Q-type Ca2+ channels (Kato et al., 1992; Harvey et al., 1996; Bergquist et al., 1998; Okada et al., 1998c; 2001). Recently, we have indicated that neurotransmitter release is composed of Ca2+ channel sensitive multiple formations of exocytosis (Okada et al., 1996, 1998b, 1998c; 2001). However, neither effects of CBZ on Ca2+ channels activities nor on multiple neurotransmitter release formations have been determined.
Hence, using the in vivo microdialysis method, the present study was designed to determine the effects of CBZ on three types of hippocampal serotonin release in the presence of antagonists of Ca2+ and Na+ channels in order to clarify the following: (1) to characterize the mechanisms of three types of hippocampal serotonin release, including basal, Ca2+- and K+-evoked releases, (2) to determine the mechanism of biphasic concentration-dependent effect of CBZ on basal serotonin release, (3) to determine whether or not the effects of plasma-concentration of CBZ associated with its antiepileptic action on the activities of Ca2+ and Na+ channels are dependent upon neuronal excitability?
Methods
Materials
All experiments described in this study were performed in accordance with the specifications of the Ethical Committee of Hirosaki University and met the guidelines of the responsible governmental agency. Male Wistar rats (Clea, Tokyo, Japan), weighing 250 – 300 g, were housed under conditions of constant temperature 22±2°C with a 12 h light-dark cycle.
Microdialysis system preparation
Each rat was placed in a stereotaxic frame and kept under halothane anaesthesia (1.5% mixture of halothane and O2 with N2O). A concentric I-type dialysis probe (0.22 mm diameter; 2 mm exposed membrane; Eicom Co., Japan) was implanted in the hippocampus (A=−5.8 mm, L=4.8 mm, V=−4.0 mm relative to bregma) and the perfusion experiments were started 18 – 24 h after the rats had recovered from anaesthesia (Okada et al., 1999; 2001).
The perfusion rate was always 1 μl min−1, using modified Ringer's solution (MRS) composed of (in mM): Na+ 145, K+ 2.7, Ca2+ 1.2, Mg2+ 1.0, and buffered with 1 mM phosphate buffer and 1.1 mM Tris buffer to adjust the pH to 7.40. To study Ca2+-dependent release, we used Ca2+-free MRS (Okada et al., 1996; 1998b; 2001), in which the Ca2+ and the appropriate amount of Na+ were replaced by 40 mM Mg2+ (Westerink et al., 1988; Okada et al., 1996; 1998a, 1998b; 2001). To study the effects of an increase in the extracellular Ca2+ or K+ level (Ca2+- or K+-evoked stimulation) on hippocampal serotonin release, MRS containing 3.4 mM Ca2+ or 50 mM K+ was infused for 20 min. The ionic composition was modified and isotonicity was maintained by an equimolar decrease of sodium ion (Okada et al., 1996; 1998b).
ECD-HPLC system conditions for measuring extracellular serotonin levels
For the measurement of extracellular serotonin levels, the hippocampal perfusate was collected into microtubes containing 10 μl of 0.02 M acetic acid with 0.27 mM EDTA-2Na. After collection, all samples were frozen at −80°C until analysis. The samples were subsequently thawed to an HPLC system equipped with electrochemical detection (ECD). The HPLC system used for determination of the extracellular serotonin level was equipped with an electrochemical detector (ECD-300, Eicom, Kyoto, Japan), pump (EP-300, Eicom, Kyoto, Japan) and a graphite carbon electrode set at +450 mV (versus an Ag/AgCl reference electrode). The analytical column (100×1.5 mm internal diameter) was packed with mightysil RP-18 (particle size 3 μm), which was purchased from Kanto Chemicals (Tokyo, Japan), by Masis Inc. (Hirosaki, Japan). The mobile phase consisted of 0.1 M phosphate buffer, containing 20% (v v−1) methanol, 800 mg l−1 octansulphonic sodium and 0.14 mM EDTA-2Na; the final pH was 5.9 and the column temperature was maintained at 25°C with the flow rate set at 225 μl min−1 (Okada et al., 2001). The quantification limits for serotonin were 0.1 fmol sample−1 (10 μl).
Determination of diffusion rates of CBZ and antagonists of Ca2+ channels and Na+ channel
In order to accurately measure the concentration of the N-type Ca2+ channel antagonist, ω-conotoxin GVIA (GVIA), the P-type Ca2+ channel antagonist, ω-agatoxin IVA (IVA), the Q-type Ca2+ channel antagonist, ω-conotoxin MVIIC (MVIIC), the Na+ channel antagonist, tetrodotoxin (TTX) and CBZ in the extracellular fluid perfused from hippocampus, in vivo probe diffusion was determined according to the ‘reverse dialysis' procedure (Le Quellec et al., 1995). Because solute diffusion occurs in both directions across the dialysis membrane, loss of solute from the perfusate occurs at the same rate as recovery of solute into the perfusate. During analysis the temperature was maintained at 37°C with a perfusion warmer.
The concentration of GVIA, MVIIC and IVA was determined by HPLC system mainly according to our previous study (Okada et al., 1998c). The concentration of TTX was measured by HPLC according to the methods of Onoue et al. (1983), and that of CBZ was determined by HPLC according to the method of Juergens (Juergens, 1987).
Drug administration
Perfusion medium was commenced with MRS alone. At least 6 h after the starting perfusion, the hippocampal extracellular serotonin level was measured. After confirming that the extracellular serotonin level had reached plateau (stabilization) according to method of Okada et al. (1999), control data were obtained over a further 60 min, then the perfusion medium was switched to MRS containing the required agent: GVIA (0.1, 1, or 10 μM), MVIIC (0.1, 1, or 10 μM), IVA (0.1, 1, or 10 μM), TTX (0.1, 1, or 10 μM) or CBZ (100 or 1000 μM) for determination of the effects of each agent on basal serotonin release for 120 min. After confirming stabilization, the perfusion medium was switched to MRS containing 3.4 mM Ca2+ with the same agents for 20 min (Ca2+-evoked release) (Okada et al., 1998c). After confirming stabilization, the perfusion medium was switched to MRS containing 50 mM K+ with the same agents for 20 min (K+-evoked release) (Okada et al., 1998c; 2001).
Chemical agents
The chemical agents used in this study were, the N-type Ca2+ channel antagonist, ω-conotoxin GVIA (Peptide Institute Inc., Japan), the P-type Ca2+ channel antagonist, ω-agatoxin IVA (Peptide Institute Inc.), the Q-type Ca2+ channel antagonist, ω-conotoxin MVIIC (Peptide Institute Inc.), the Na+ channel antagonist tetrodotoxin (Sankyo., Tokyo, Japan) and carbamazepine (Tokyo Kasei Kogyo Co., Tokyo, Japan).
Statistics
The concentration-dependent effects of Ca2+ channel antagonists, Na+ channel antagonist and CBZ on basal, Ca2+ -and K+-evoked hippocampal serotonin releases were analysed by one-way analysis of variance (ANOVA) with Tukey's multiple comparison. The interaction between Na+ channel antagonist and Ca2+ channel antagonists, Ca2+ channel antagonists and CBZ, or Na+ channel antagonist and CBZ on basal, Ca2+ -and K+-evoked hippocampal serotonin releases were analysed by two-way ANOVA with Tukey's multiple comparison. Differences of P<0.05 were considered significant.
Results
In in vitro experiments, the recovery rate of probes for serotonin (external to internal probes) ranged 12.3±1.1% (mean±s.d., n=36). The basal hippocampal extracellular serotonin level (basal release) was 5.6±0.6 fmol sample−1 (10 μl) (mean±s.d., n=6) in 10 min (Figure 1a,b). An increase in the extracellular Ca2+ level from 1.2 – 3.4 mM (Ca2+-evoked stimulation for 20 min) increased from 5.5 – 10.2 fmol sample−1 of the extracellular serotonin level (Ca2+-evoked release: 4.7±0.6 fmol sample−1, mean±s.d., n=6) (Figure 1a,b). An increase in the extracellular K+ level from 2.7 – 50 mM (K+-evoked stimulation for 20 min) increased from 5.5 – 25.5 fmol sample−1 of the extracellular serotonin level (K+-evoked release: 20.0±3.8 fmol sample−1, mean±s.d., n=6) (Figure 1a,b). Other in vitro experiments indicated that the rate at which CBZ diffused from the dialysis probe (internal to external probes) was 18.8±1.6% (mean±s.d., n=12). The rate at which GVIA, IVA, MVIIC and TTX diffused from the dialysis probe were 0.98±0.28%, 0.59±0.19%, 0.87±0.32% and 12.4±2.6% (mean±s.d., n=6), respectively.
Figure 1.
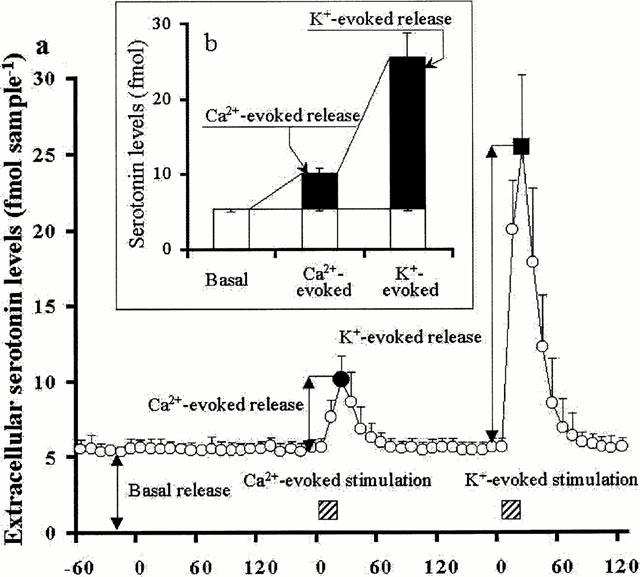
Criteria for the levels of basal, Ca2+- and K+-evoked hippocampal serotonin releases. (a) At least 6 h after starting the perfusion with MRS, the serotonin level in hippocampal perfusate was measured. After confirming the stabilization of extracellular serotonin level, the serotonin level was measured for 120 min as control data (basal release). After determination of basal release, perfusion medium was switched to 3.4 mM Ca2+ including MRS for 20 min (Ca2+-evoked stimulation). After confirming the stabilization of extracellular serotonin level, perfusion medium was switched to 50 mM K+ including MRS for 20 min (K+-evoked stimulation). (b) The value of Ca2+-evoked serotonin release was defined as the level of basal extracellular serotonin level subtracted from the maximal extracellular serotonin level during Ca2+-evoked stimulation. The level of K+-evoked release was defined as the level of basal extracellular level subtracted from the maximal extracellular level during K+-evoked stimulation.
Concentration-dependent effects of antagonists of Ca2+, Na+ channels and carbamazepine on hippocampal serotonin release
The effects of concentration-dependent GVIA, IVA, MVIIC, TTX and CBZ on hippocampal basal, Ca2+- and K+-evoked serotonin releases are shown in Figures 2, 3, 4 and 5. The basal hippocampal serotonin release was reduced by GVIA (F=63.3, P<0.01), IVA (F=7.1, P<0.01), MVIIC (F=28.4, P<0.01), TTX (F=189.2, P<0.01), in a concentration-dependent manner (Figures 2a, 3a, 4a and 5a), whereas the basal hippocampal serotonin release was increased and decreased by 100 μM CBZ (estimated concentration in hippocampal tissue was 19±2 μM) and 1000 μM CBZ (estimated concentration in hippocampal tissue was 188±16 μM), respectively (F=53.6, P<0.01, n=6) (Figures 4a and 5a). The Ca2+-evoked hippocampal serotonin release was decreased by GVIA (F=26.69, P<0.01) and MVIIC (F=9.6, P<0.01), in a concentration-dependent manner, but were not affected by IVA (F=0.2) or TTX (F=1.7) (Figures 2b, 3b, 4b and 5b). The Ca2+-evoked serotonin release was increased and decreased by 100 μM and 1000 μM CBZ, respectively (F=21.4, P<0.01) (Figures 4b and 5b). The K-evoked hippocampal serotonin release was inhibited by GVIA (F=3.7, P<0.05, n=6), MVIIC (F=19.0, P<0.01), IVA (F=28.8, P<0.01), TTX (F=17.2, P<0.01) and CBZ (F=18.4, P<0.01), in a concentration-dependent manner (Figures 2c, 3c, 4c and 5c).
Figure 2.
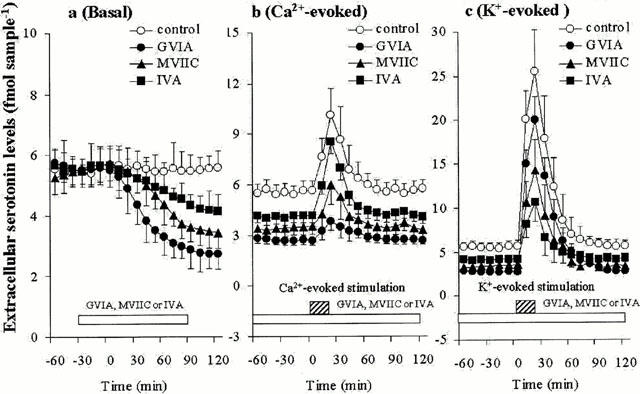
Effects of Ca2+ channel antagonists on serotonin release. The effects of 1 μM GVIA, IVA and MVIIC on basal, Ca2+- and K+-evoked hippocampal serotonin releases are represented in a, b and c, respectively. The ordinates indicate the mean±s.d. (n=6) of extracellular serotonin levels (fmol sample−1), and abscissas show the time in minutes (min). The mean values of serotonin release level obtained between perfusion with and without Ca2+ antagonists were compared by one-way ANOVA with Tukey's multiple comparison. Asterisk marks which indicate the statistical significance were excluded to avoid over complicating the figures.
Figure 3.
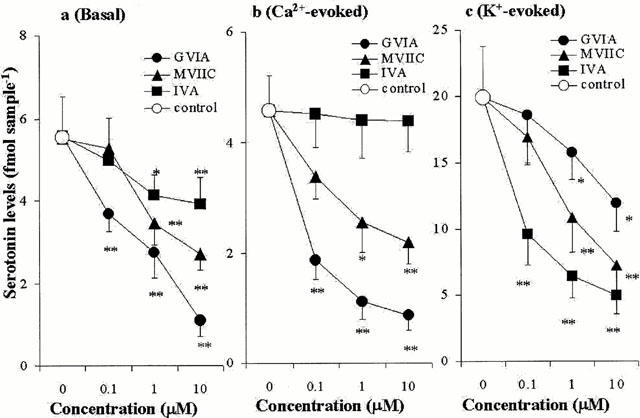
Concentration-dependent effects of Ca2+ channel antagonists on serotonin release. The concentration-dependent effects of Ca2+ channel antagonists, on basal, Ca2+- and K+-evoked hippocampal serotonin releases are represented in a, b and c, respectively. The ordinates indicate the mean±s.d. (n=6) of serotonin release level (fmol sample−1), and abscissas show the concentration of Ca2+ channel antagonists (μM). The mean values of serotonin release obtained between perfusion without (control) and with Ca2+ channel antagonists were compared by one-way ANOVA with Tukey's multiple comparison (*: P<0.05; **: P<0.01).
Figure 4.
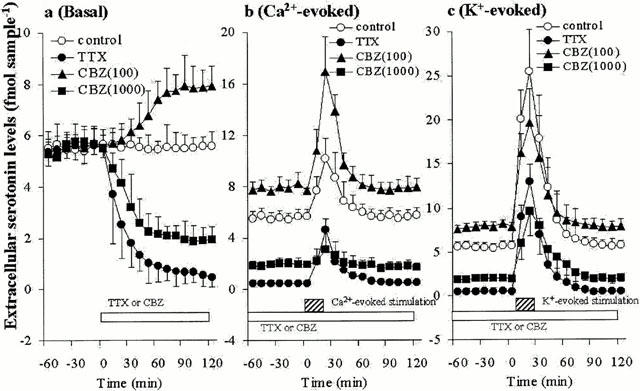
Effects of Na+ channel antagonist and carbamazepine on serotonin release. The effects of 1 μM TTX, 100 and 1000 μM CBZ on basal, Ca2+- and K+-evoked hippocampal serotonin releases are represented in a, b and c, respectively. The ordinates indicate the mean±s.d. (n=6) of extracellular serotonin levels (fmol sample−1), and abscissas show the time in minutes (min). The mean values of serotonin release level obtained between perfusion with and without TTX or CBZ were compared by one-way ANOVA with Tukey's multiple comparison. Asterisk marks which indicate the statistical significance were excluded to avoid over complicating the figures.
Figure 5.
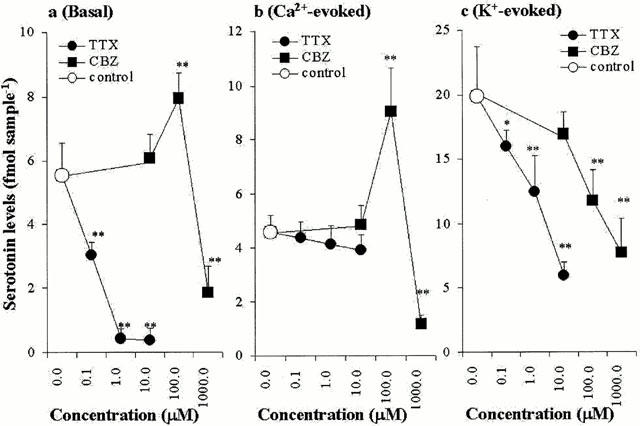
Concentration-dependent effects of Na+ channel antagonist and carbamazepine on serotonin release. The concentration-dependent effects of TTX and CBZ, on basal, Ca2+- and K+-evoked hippocampal serotonin releases are represented in a, b and c, respectively. The ordinates indicate the mean±s.d. (n=6) of serotonin release level (fmol sample−1), and abscissas show the concentration of TTX and CBZ (μM). The mean values of serotonin release obtained between the perfusion without (control) and with TTX or CBZ were compared by one-way ANOVA with Tukey's multiple comparison (*: P<0.05; **: P<0.01).
Interaction between Ca2+ channel antagonists and Na+ channel antagonist on hippocampal serotonin release
The interaction between antagonists of Ca2+ channels and Na+ channel on three types of hippocampal serotonin release is shown in Figure 6. Both under the conditions of perfusion with and without 0.1 μM TTX, 1 μM GVIA reduced basal serotonin release (P<0.01) (Figure 6a). Under the condition of functional Na+ channels, 1 μM IVA reduced basal serotonin release (P<0.05), however, under the condition of Na+ channel blockade by perfusion with 0.1 μM TTX, IVA had no effect (Figure 6a). Both under the condition of perfusion with and without 0.1 μM TTX, GVIA reduced Ca2+-evoked serotonin release (P<0.01), whereas IVA had no effect (Figure 6b). Both under the condition of perfusion with and without 0.1 μM TTX, IVA reduced K+-evoked serotonin release (P<0.01) (Figure 6c). Under the conditions of functional Na+ channels, GVIA reduced K+-evoked serotonin release (P<0.05), however, under the condition of Na+ channel blockade by 0.1 μM TTX, GVIA had no effect (Figure 6c).
Figure 6.
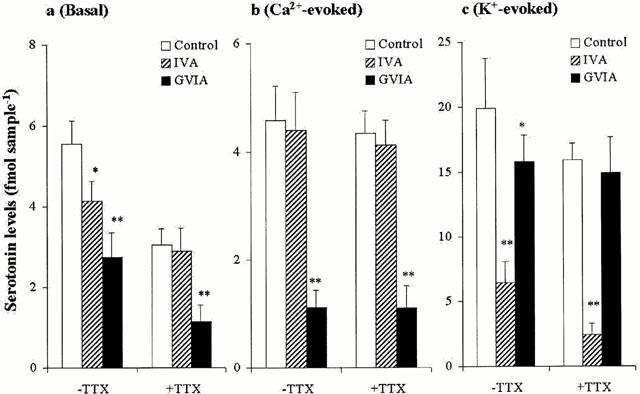
Interaction between Ca2+ channel antagonists and Na+ channel antagonist on serotonin release. The interaction between Ca2+ channel antagonists (1 μM GVIA and IVA) and 0.1 >μM TTX on basal, Ca2+- and K+-evoked hippocampal serotonin releases are represented in a, b and c, respectively. The ordinates indicate the mean±s.d. (n=6) of serotonin release level (fmol sample−1). The data was analysed by two-way ANOVA with Tukey's multiple comparison (*: P<0.05; **: P<0.01).
Interaction between CBZ and Na+ channel antagonist on hippocampal serotonin release
The interaction between CBZ and TTX on three types of hippocampal serotonin release is shown in Figure 7. The inhibitory effect of 0.1 μM TTX on basal release was abolished by perfusion with both 100 and 1000 μM CBZ (Figure 7a). TTX (0.1 μM) had no effect on Ca2+-evoked release, under the condition of pre-perfusion with and without CBZ (100 and 1000 μM) (Figure 7b). Both 100 and 1000 μM CBZ abolished the inhibitory effects of 0.1 μM TTX on K+-evoked release (Figure 7c).
Figure 7.
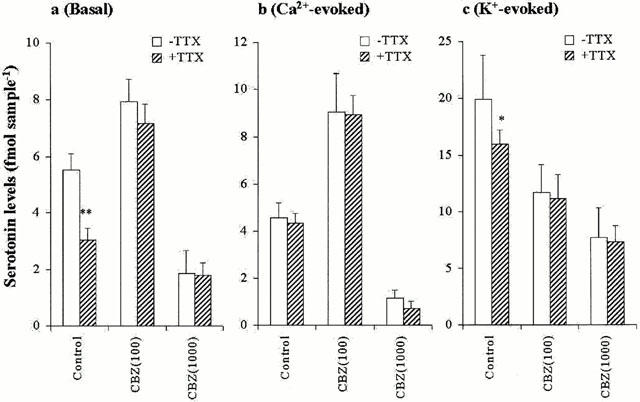
Interaction between Na+ channel antagonist and carbamazepine on serotonin release. The interaction between 0.1 μM TTX, 100 and 1000 μM CBZ on basal, Ca2+- and K+-evoked hippocampal serotonin releases are represented in a, b and c, respectively. The ordinates indicate the mean±s.d. (n=6) of serotonin release level (fmol sample−1). The data was analysed by two-way ANOVA with Tukey's multiple comparison (*: P<0.05; **: P<0.01).
Interaction between CBZ and Ca2+ channel antagonists on hippocampal serotonin release
The interaction between CBZ and Ca2+ channel antagonists on three types of hippocampal serotonin releases is shown in Figure 8. Under the condition of pre-perfusion with 100 μM CBZ, 1 μM GVIA reduced basal release (P<0.01), but IVA was unaffected (Figure 8a). Under the condition of perfusion with 1000 μM CBZ, neither 1 μM GVIA nor IVA affected basal release (Figure 8a). Both under the conditions of pre-perfusion with and without CBZ, 1 μM IVA did not affect Ca2+-evoked release (Figure 8b). Under the conditions of both without and with 100 μM CBZ, 1 μM GVIA reduced Ca2+-evoked release (P<0.01), whereas pre-perfusion with 1000 μM CBZ abolished the inhibitory effects of GVIA on Ca2+-evoked release (Figure 8b). Under the condition of perfusion with 100 and 1000 μM CBZ, 1 μM IVA reduced K+-evoked release (P<0.01), however, both pre-perfusion with 100 and 1000 μM CBZ abolished the inhibitory effect of GVIA on K+-evoked (Figure 8c).
Figure 8.
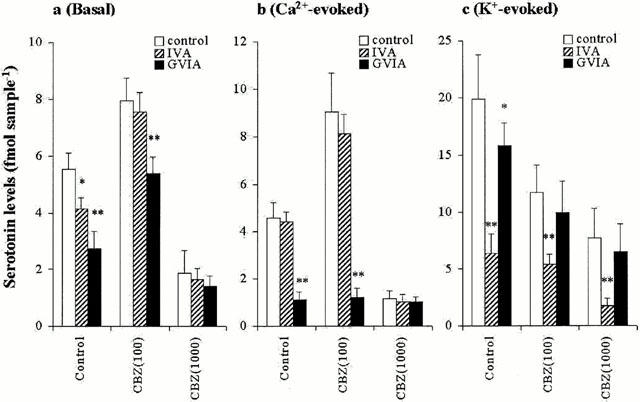
Interaction between Ca2+ channel antagonists and carbamazepine on serotonin release. The interaction between 1 μM GVIA, IVA and CBZ on basal, Ca2+- and K+-evoked hippocampal serotonin releases are represented in a, b and c, respectively. The ordinates indicate the mean±s.d. (n=6) of serotonin release level (fmol sample−1), under the condition of perfusion without or with 100 μM, 1000 μM CBZ. The data was analysed by two-way ANOVA with Tukey's multiple comparison (*: P<0.05; **: P<0.01).
Discussion
In the present study, both perfusion with Ca2+-free MRS (data not shown) and MRS containing 1 μM TTX decreased the basal extracellular serotonin level to lower than 0.5 fmol sample−1 (10 μl). Furthermore, 50 mM K+-evoked stimulation increased the extracellular serotonin levels. Therefore, these experiments demonstrate that under microdialysis conditions presently employed, the serotonin level (basal serotonin release) in hippocampal perfusate was primarily of neuronal origin, because the extracellular serotonin level was TTX-sensitive, Ca2+-dependent and K+-sensitive (Westerink et al., 1989, Okada et al., 1998a; 2001). The present study clearly demonstrated that the three types of hippocampal serotonin releases, including basal, Ca2+- and K+-evoked releases, required specific mechanisms of both Na+ and Ca2+ inflow through each specific Na+ and Ca2+ channels. The basal release was reduced by the Na+ channel antagonist, TTX, the N-type Ca2+ channel antagonist, GVIA, the Q-type Ca2+ channel antagonist, MVIIC and the P-type Ca2+ channel antagonist, IVA. The relative sensitivity of basal release to Ca2+ channel antagonists was as follows: GVIA>MVIIC>IVA. The Ca2+-evoked serotonin release was inhibited by GVIA and MVIIC (GVIA>MVIIC), but was unaffected by IVA or TTX. The K+-evoked serotonin release was inhibited by TTX, GVIA, MVIIC and IVA. The relative sensitivity of K+-evoked release to Ca2+ channel antagonists was as follows: IVA>MVIIC>GVIA. MVIIC has been shown to block both P- and Q-type Ca2+ channels at concentrations of greater than 500 nM (Randall & Tsien, 1995). In addition, this toxin also weakly inhibits the N-type Ca2+ channel (Hillyard et al., 1992). Therefore, to clarify whether MVIIC is a specific antagonist of Q-type Ca2+ channels as well as the precise effect of Q-type Ca2+ channels on three types of hippocampal serotonin release, further studies are needed.
Yoshihara et al. (1999) have suggested that different mechanisms are required for depolarization induced and spontaneous neurotransmitter releases. The mechanism for depolarization-induced neurotransmitter release has been considered to occur as follows: after arrival of an action potential at the presynaptic terminal and resulting entry of Ca2+ through Ca2+ channels, the local levels in intraneuronal presynaptic terminal active-zone of Ca2+ rises abruptly to submillimolar level (Roberts et al., 1990). On the other hand, spontaneous fusion of the synaptic vesicle at the resting state generates miniature synaptic potentials. The frequency of these events changes when the cytosolic Ca2+ level is altered in into the micromolar range (Delaney & Tank, 1994; Xu et al., 1998). Our recent demonstration indicated that hippocampal serotonin release was regulated by the two functional complexes, N-type Ca2+ channel/calcium-phospholipid-dependent protein kinase (PKC)/syntaxin and P-type Ca2+ channel/cyclic AMP-dependent protein kinase (PKA)/ synaptobrevin (Okada et al., 2001). Furthermore, basal serotonin release was regulated by N-type Ca2+ channel/ PKC/syntaxin predominantly and P-type Ca2+ channel/ PKA/synaptobrevin weakly, whereas K+-evoked release was regulated by P-type Ca2+ channel/PKA/synaptobrevin predominantly and N-type Ca2+ channel/PKC/syntaxin weakly (Okada et al., 2001). Therefore, taken together with these hypotheses, the present results indicate that the hippocampal basal, Ca2+ - and K+-evoked serotonin releases require different specific Ca2+ inflow pathways.
To characterize the mechanisms of these three formations of serotonin release, the interaction between TTX and Ca2+ channel antagonists on basal, Ca2+- and K+-evoked serotonin releases were analysed. The K+-evoked release was inhibited by GVIA weakly, TTX and IVA predominantly. The inhibitory effect of GVIA on the K+-evoked release was abolished by blockade of Na+ channel activity by TTX, whereas that of IVA was not affected by TTX. This evidence indicates that the possible mechanisms of K+-evoked serotonin release may be organized by activation of both functional pathways, N-type Ca2+ channels/Na+ channel and P/Q-type Ca2+ channel. On the other hand, the basal release was inhibited by IVA weakly, TTX and GVIA predominantly. TTX abolished the inhibitory effect of IVA on basal release, whereas that of GVIA was not affected by TTX. Therefore, these evidences suggest that the basal serotonin release may be also organized by the activation of the two functional pathways N-type Ca2+ channel and P-type Ca2+ channel/Na+ channel, however, contrary to K+-evoked release, basal release was regulated by N-type Ca2+ channel activity predominantly, and P-type Ca2+ channel/ Na+ channel weakly.
The Ca2+-evoked release was inhibited by GVIA, but insensitive to IVA or TTX. We have already demonstrated that an increase in extracellular Ca2+ level from 1.2 – 3.4 mM (Ca2+-evoked stimulation) stimulated dopamine release in hippocampus (Okada et al., 1998c). Taken together with these previous results, thus, the present demonstration can advocate that the mechanisms of Ca2+-evoked release may be a new type of exocytotic release of serotonin, which was organized by N-type Ca2+ channel activity without being affected by activities of either Na+ channels or P-type Ca2+ channel. To clarify the mechanisms of Ca2+-evoked serotonin release including the interaction among activities of N-type Ca2+ channel (Jones, 1998), protein kinases (Turner et al., 1999) and synprint proteins (Sheng et al., 1998), we shall report further studies.
It has been well established that the plasma-concentration of CBZ associated with its antiepileptic action inhibits Na+ channel activity (Mclean & Macdonald, 1986; Mattson, 1997). However, our previous studies demonstrated that the biphasic concentration-dependent effects of CBZ on neurotransmitter release (Okada et al., 1997a, 1997b; 1998a, 1998b; Mizuno et al., 2000). The plasma-concentration of CBZ associated with its antiepileptic action (17 – 42 μM: Masuda et al., 1979) increased basal monoamine release without affecting monoamine oxidase activity (Okada et al., 1997a; 1998a), or basal glutamate release (Okada et al., 1998b), while more than the plasma-concentration of CBZ associated with its antiepileptic action reduced basal monoamine release (Okada et al., 1997a; 1998a) and K+-evoked glutamate release (Okada et al., 1998b). In the present study, 1000 μM CBZ (estimated CBZ concentration in hippocampal tissue was 188±16 μM) inhibited three types of serotonin releases. Under the condition of perfusion with 1000 μM CBZ, the inhibitory effects of TTX and GVIA on the three types of releases were abolished. Although under the same condition, the inhibitory effects of IVA on basal release was abolished, but that of IVA on K+-evoked release was not affected. Therefore, more than the plasma-concentration of CBZ associated with its antiepileptic action inhibits both activities of N-type, P-type Ca2+ channels and Na+ channel in reduction of basal, Ca2+ - and K+-evoked releases.
On the other hand, 100 μM CBZ reduced K+-evoked serotonin release but enhanced basal and Ca2+-evoked serotonin releases. Pre-treatment of 100 μM CBZ abolished the inhibitory effects of TTX on basal and K+-evoked releases. Under the condition of perfusion with 100 μM CBZ, the inhibitory effects of GVIA on K+-evoked release was abolished, however, under the same condition, GVIA and IVA could reduced basal and K+-evoked releases, respectively. In addition, under the condition of pre-perfusion with 100 μM CBZ, GVIA also could reduced Ca2+-evoked release. Thus, these results suggest that, 100 μM CBZ enhances and inhibits weakly N-type Ca2+ channel and Na+ channel activities, respectively, resulting in 100 μM CBZ increased both basal and Ca2+-evoked release but reduced K+-evoked release. Contrary to N-type Ca2+-channel, the inhibitory effects of IVA on basal and K+-evoked release were prevented and unaffected by pre-perfusion with 100 μM CBZ, respectively. This contradiction suggests that 100 μM CBZ inhibits P-type Ca2+-channel activity.
CBZ reduces Ca2+ influx (Kito et al., 1994; Yoshimura et al., 1995), but the effects of CBZ on voltage-sensitive Ca2+ channel subtypes have not been clarified yet. Kito et al. (1994) demonstrated, using a patch-clamp technique, the lack of effect of CBZ on various subtypes of Ca2+ channels in the human neuroblastoma NB1 cell, however, Yoshimura et al. (1995) demonstrated that CBZ (4 – 120 μM) inhibited N-type Ca2+ channel function in cultured bovine adrenal medullary cells. Together with these previous demonstrations, the present study indicates, at least partially, that 100 and 1000 μM CBZ enhanced and inhibited Ca2+-evoked release, respectively (biphasic concentration-dependently). The present evidences explain the possible mechanisms of concentration-dependent biphasic action of CBZ on both basal and Ca2+-evoked hippocampal serotonin releases. 100 μM CBZ inhibits Na+ channel activity during K+-evoked stimulation, and enhances N-type Ca2+ channel function at the resting stage. On the other hand, 1000 μM CBZ inhibits activities of N-type Ca2+ and Na+ channels. However, the possibility that the enhancement of basal serotonin release induced by 100 μM CBZ might be modulated by the stimulation of synthesis of serotonin cannot be readily ruled out, since the plasma-concentration of CBZ associated with its antiepileptic action enhances hippocampal monoamine synthesis without affecting either its degradation or re-uptake activities (Okada et al., 1997a; 1998a).
The occurrence of an epileptic seizure may be generated by the relative imbalance between excitatory and inhibitory neurotransmission, resulting in neurones showing increased excitability and abnormally frequent patterns of discharge (Gumnit et al., 1997; Hirose et al., 2000). Glutamate is the principal excitatory neurotransmitter, and excessive release of glutamate may produce seizures in epileptic patients (During et al., 1995), and epileptic animal models (Ueda & Tsuru, 1994). We have already demonstrated that clinically relevant concentration of CBZ reduced both hippocampal K+-evoked glutamate release and spreading depression induced glutamate release without affecting basal glutamate release (Okada et al., 1998b). In addition, enhancement of monoaminergic transmission reduced seizure threshold (Wahnschaffe & Loscher, 1991; Wada et al., 1993). Taken together with our previous results, the present study indicates that the mechanisms of antiepileptic action of 100 μM CBZ may be produced by the combination of effects resulting from enhancement of basal release of inhibitory neurotransmitter without affecting basal glutamate release, and reduction of neurotransmitter release at the event of neuronal excitation (i.e. during seizures).
We determined the different effects between 100 and 1000 μM CBZ in this study. In conclusion, 1000 μM CBZ inhibited the activities of N-type, P-type Ca2+ channels and TTX-sensitive Na+ channel, which resulted in inhibition of exocytosis. By contrast, the 100 μM CBZ enhanced N-type Ca2+ channel activity at the resting stage, and inhibited activities of TTX-sensitive Na+ channel and P-type Ca2+-channel activity, at both the resting and neuronal depolarizing stages, i.e. during K+-evoked stimulation. During resting stage, CBZ possibly exerts its antiepileptic action, at least partially, by combined effects of a blockade of both P-type Ca2+-channel and Na+ channel activities (inhibition of basal release) and an enhancement of N-type Ca2+ channel activity (enhancement of basal release). These evidences thus suggest that the 100 μM CBZ exerts its antiepileptic action by combined effects on the Na+ channel blockade together with an enhancement of N-type Ca2+ channel activity at resting stage and the blockade of Na+ channel and P-type Ca2+-channel activities at neuronal depolarizing stage.
Acknowledgments
This study was supported by a Grant-in-Aid for Scientific Research from Japanese Ministry of Education, Science and Culture (05454309, 12770515 and 11770532), a Grant from Hirosaki Research Institute for Neurosciences, a Grant from Pharmacopsychiatry Research Foundation and a Grant from Japan Epilepsy Research Foundation.
Abbreviations
- ANOVA
analysis of variance
- CBZ
carbamazepine
- ECD-HPLC
high performance liquid chromatography equipped with electrochemical detector
- EDTA
ethylenediamine-tetraacetic acid
- GVIA
ω-conotoxin GVIA
- IVA
ω-agatoxin IVA
- MRS
modified Ringer's solution
- MVIIC
ω-conotoxin MVIIC
- TTX
tetrodotoxin
References
- BERGQUIST F., JONASON J., PILEBLAD E., NISSBRANDT H. Effects of local administration of L-, N-, and P/Q-type calcium channel blockers on spontaneous dopamine release in the striatum and the substantia nigra: a microdialysis study in rat. J. Neurochem. 1998;70:1532–1540. doi: 10.1046/j.1471-4159.1998.70041532.x. [DOI] [PubMed] [Google Scholar]
- DELANEY K.R., TANK D.W. A quantitative measurement of the dependence of short-term synaptic enhancement on synaptic residual calcium. J. Neurosci. 1994;14:5885–5902. doi: 10.1523/JNEUROSCI.14-10-05885.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUNLAP K., LUEBKE J.I., TURNER T.J. Exocytotic Ca2+ channels in mammalian central neurons. Trends. Neurosci. 1995;18:89–98. [PubMed] [Google Scholar]
- DURING M.J., RYDER K.M., SPENCER D.D. Hippocampal GABA transporter function in temporal-lobe epilepsy. Nature. 1995;376:174–177. doi: 10.1038/376174a0. [DOI] [PubMed] [Google Scholar]
- ELPHICK M., ANDERSON S.M., HALLIS K.F., GRAHAME-SMITH D.G. Effects of carbamazepine on 5-hydroxytryptamine function in rodents. Psychopharmacology. 1990;100:49–53. doi: 10.1007/BF02245789. [DOI] [PubMed] [Google Scholar]
- GUMNIT R.J., RISINGER M., LEPPIK I.E., MAISTER B., GIL-NAGEL A.The epilepsy and convulsive disorders Clinical Neurology 1997Vol 3New York: Lippincott-Raven publishers; 1–95.ed. Joint, R.J. ppChapter 31 [Google Scholar]
- HARVEY J., WEDLEY S., FINDLAY J.D., SIDELL M.R., PULLAR I.A. ω-Agatoxin IVA identifies a single calcium channel subtype which contributes to the potassium-induced release of acetylcholine, 5-hydroxytryptamine, dopamine, γ-aminobutylic acid and glutamate from rat brain slices. Neuropharmacology. 1996;35:385–392. doi: 10.1016/0028-3908(96)00010-x. [DOI] [PubMed] [Google Scholar]
- HILLYARD D.R., MONJE V.D., MINTZ I.M., BEAN B.P., NADASDI L., RAMACHANDRAN J., MILJANICH G., AZIMI-ZOONOOZ A., MCINTOSH J.M., CRUZ L.J., IMPERIAL J.S., OLIVERA B.M. A new Conus peptide ligand for mammalian presynaptic Ca2+ channels. Neuron. 1992;9:69–77. doi: 10.1016/0896-6273(92)90221-x. [DOI] [PubMed] [Google Scholar]
- HIROSE S., OKADA M., KANEKO S., MITSUDOME A. Are some idiopathic epilepsies disorders of the ion channels?: A working hypothesis. Epilepsy Res. 2000;41:191–204. doi: 10.1016/s0920-1211(00)00141-8. [DOI] [PubMed] [Google Scholar]
- ICHIKAWA J., MELTZER H.Y. Valproate and carbamazepine increase prefrontal dopamine release by 5-HT1A receptor activation. Eur. J. Pharmacol. 1999;380:R1–R3. doi: 10.1016/s0014-2999(99)00517-8. [DOI] [PubMed] [Google Scholar]
- JONES S.W. Overview of voltage-dependent calcium. J. Bioenerg. Biomembr. 1998;30:299–312. doi: 10.1023/a:1021977304001. [DOI] [PubMed] [Google Scholar]
- JUERGENS U. Simultaneous determination of zonisamide and nine other anti-epileptic drugs and metabolites in serum. J. Chromatogr. 1987;385:233–240. doi: 10.1016/s0021-9673(01)94635-7. [DOI] [PubMed] [Google Scholar]
- KANEKO S., FUKUSHIMA Y., SATO T., HIRAMATSU M., MORI A. Effects of carbamazepine on catecholamine level in mouse brain. IRCS Med. Sci. 1980;9:80–81. [Google Scholar]
- KATO T., OTSU Y., FURUNE Y., YAMAMOTO T. Different effects of L-, N- and T-type calcium channel blockers on striatal dopamine release measured by microdialysis in freely moving rats. Neurochem. Int. 1992;21:99–107. doi: 10.1016/0197-0186(92)90072-y. [DOI] [PubMed] [Google Scholar]
- KECK P.E.J., MCELROY S.L., FRIEDMAN L.M. Valproate and carbamazepine in the treatment of panic and posttraumatic stress disorders, withdrawal states, and behavioural dyscontrol syndromes. J. Clin. Psychopharmacol. 1992;12:36S–41S. doi: 10.1097/00004714-199202001-00006. [DOI] [PubMed] [Google Scholar]
- KITO M., MAEHARA M., WATANABE K. Antiepileptic drugs-calcium current interaction in cultured human neuroblastoma cells. Seizure. 1994;3:141–149. doi: 10.1016/s1059-1311(05)80205-5. [DOI] [PubMed] [Google Scholar]
- LE QUELLEC A., DUPIN S., GENISSEL P., SAIVIN S., MARCHAND B., HOUIN G. Microdialysis probes calibration: gradient and tissue dependent changes in no net flux and reverse dialysis methods. J. Pharmacol. Toxicol. Methods. 1995;33:11–16. doi: 10.1016/1056-8719(94)00049-a. [DOI] [PubMed] [Google Scholar]
- LOISEAU P., DUCHE B.Carbamazepine Clinical Use Antiepileptic drugs, Fourth edition 1995New York: Raven Press; 555–566.ed. Levy, R.H., Mattson, R.H. & Meldrum, B.S. pp [Google Scholar]
- MASUDA Y., UTSUI Y., SHIRAISHI Y., KARASAWA T., YOSHIDA K., SHIMIZU M. Relationships between plasma concentrations of diphenylhydantoin, phenobarbital, carbamazepine, and 3-sulfamoylmethyl-1,2-benzisoxazole(AD810), a new anticonvulsant agent, and their anticonvulsant or neurotoxic effects in experimental animals. Epilepsia. 1979;20:623–633. doi: 10.1111/j.1528-1157.1979.tb04846.x. [DOI] [PubMed] [Google Scholar]
- MATTSON R.H.Carbamazepine Epilepsy: a comprehensive textbook 1997Philadelphia: Lippincott-Raven Publishers; 1491–1502.ed. Engel, Jr. J. & Pedley, T.A. pp [Google Scholar]
- MCLEAN M.J., MACDONALD R.L. Carbamazepine and 10,11-epoxycarbamazepine produce use- and voltage-dependent limitation of rapidly firing action potentials of mouse central neurons in cell culture. J. Pharmacol. Exp. Ther. 1986;238:727–738. [PubMed] [Google Scholar]
- MIZUNO K., OKADA M., MURAKAMI T., KAMATA A., ZHU G., KAWATA Y., WADA K., KANEKO S. Effects of carbamazepine on acetylcholine release and metabolism. Epilepsy. Res. 2000;40:187–195. doi: 10.1016/s0920-1211(00)00129-7. [DOI] [PubMed] [Google Scholar]
- NEPPE V.M. Carbamazepine in the psychiatric patient. Lancet. 1982;2:334. doi: 10.1016/s0140-6736(82)90305-1. [DOI] [PubMed] [Google Scholar]
- OKADA M., HIRANO T., MIZUNO K., CHIBA T., KAWATA Y., KIRYU K., WADA K., TASAKI H., KANEKO S. Biphasic effects of carbamazepine on the dopaminergic system in rat striatum and hippocampus. Epilepsy. Res. 1997a;28:143–153. doi: 10.1016/s0920-1211(97)00042-9. [DOI] [PubMed] [Google Scholar]
- OKADA M., HIRANO T., MIZUNO K., KAWATA Y., WADA K., MURAKAMI T., TASAKI H., KANEKO S. Effects of carbamazepine on hippocampal serotonergic system. Epilepsy Res. 1998a;31:187–198. doi: 10.1016/s0920-1211(98)00025-4. [DOI] [PubMed] [Google Scholar]
- OKADA M., KAWATA Y., MIZUNO K., WADA K., KONDO T., KANEKO S. Interaction between Ca2+, K−1, carbamazepine and zonisamide on hippocampal extracellular glutamate monitored with a microdialysis electrode. Br. J. Pharmacol. 1998b;124:1277–1285. doi: 10.1038/sj.bjp.0701941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OKADA M., KAWATA Y., MURAKAMI T., WADA K., MIZUNO K., KANEKO S. Interaction between purinoceptor subtypes on hippocampal serotonergic transmission using in vivo microdialysis. Neuropharmacology. 1999;38:707–715. doi: 10.1016/s0028-3908(98)00226-3. [DOI] [PubMed] [Google Scholar]
- OKADA M., KIRYU K., KAWATA Y., MIZUNO K., WADA K., TASAKI H., KANEKO S. Determination of the effects of caffeine and carbamazepine on striatal dopamine release by in vivo microdialysis. Eur. J. Pharmacol. 1997b;32:81–188. doi: 10.1016/s0014-2999(96)00938-7. [DOI] [PubMed] [Google Scholar]
- OKADA M., MIZUNO K., OKUYAMA M., KANEKO S. Magnesium ion augmentation of inhibitory effects of adenosine on dopamine release in the rat striatum. Psychiatry and Clin Neurosci. 1996;50:147–156. doi: 10.1111/j.1440-1819.1996.tb01680.x. [DOI] [PubMed] [Google Scholar]
- OKADA M., NUTT D.J., MURAKAMI T., ZHU G., KAMATA A., KAWATA Y., KANEKO S. Adenosine receptor subtypes modulate two major functional pathways for hippocampal serotonin release. J. Neurosci. 2001;21:628–640. doi: 10.1523/JNEUROSCI.21-02-00628.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OKADA M., WADA K., KIRYU K., KAWATA Y., MIZUNO K., KONDO T., TASAKI H., KANEKO S. Effects of Ca2+ channel antagonists on striatal dopamine and DOPA release, studied by in vivo microdialysis. Br. J. Pharmacol. 1998c;123:805–814. doi: 10.1038/sj.bjp.0701675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OKUMA T., YAMASHITA I., TAKAHASHI R., ITOH H., OTUKI S., WATANABE S., HAZAMA H., INAGAWA K. Comparison of the antimanic efficacy of carbamazepine and lithium carbonate by double-blind controlled study. Parmacopsychiatry. 1990;23:143–150. doi: 10.1055/s-2007-1014497. [DOI] [PubMed] [Google Scholar]
- OLIVERA B.M., MILJANICH G.P., RAMACHANDRAN J., ADAMS M.E. Calcium channel deversity and neurotransmitter release: the omega-conotoxin and omega-agatoxin. Annu. Rev. Biochem. 1994;63:823–867. doi: 10.1146/annurev.bi.63.070194.004135. [DOI] [PubMed] [Google Scholar]
- ONOUE Y., NOGUCHI T., NAGASHIMA Y., HASHIMOTO K., KANOH S., ITO M., TSUKADA K. Separation of tetrodotoxin and paralytic shellfish poisons by high-performance liquid chromatography with a fluorometric detection using o-phthalaldehyde. J. Chromatogr. 1983;257:373–379. doi: 10.1016/s0021-9673(01)88193-0. [DOI] [PubMed] [Google Scholar]
- PRATT J.A., JENNER P., MARSDEN C.D. Comparison of the effects of benzodiazepines and other anticonvulsant drugs on synthesis and utilization of 5-HT in mouse brain. Neuropharmacology. 1985;24:59–68. doi: 10.1016/0028-3908(85)90096-6. [DOI] [PubMed] [Google Scholar]
- RANDALL A., TSIEN R.W. Pharmacological dissection of multiple types of Ca2+ channel currents in rat cerebellar granule neurons. J. Neurosci. 1995;15:2995–3012. doi: 10.1523/JNEUROSCI.15-04-02995.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROBERTS W.M., JACOBS R.A., HUDSPETH A.J. Colocalization of ion channels involved in frequency selective and synaptic transmission at presynaptic active zone of hair cells. J. Neurosci. 1990;10:3664–3684. doi: 10.1523/JNEUROSCI.10-11-03664.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHEFFER I.E., BHATIA K.P., LOPES-CENDES I., FISH D.R., MARSDEN C.D., ANDERMANN F., ANDERMANN E., DESBIENS R., CENDES F., MANSON J.I. Autosomal dominant frontal epilepsy misdiagnosed as sleep disorder. Lancet. 1994;343:515–517. doi: 10.1016/s0140-6736(94)91463-x. [DOI] [PubMed] [Google Scholar]
- SCHINDLER W.New N-heterocyclic compounds U.S.Patent 2,948,718 1960. August 9, 1960
- SHENG Z.H., WESTENBROEK R.E., CATTERALL W.A. Physical link and functional coupling of presynaptic calcium channels and the synaptic vesicle docking/fusion machinery. J. Bioenerg. Biomembr. 1998;30:335–345. doi: 10.1023/a:1021985521748. [DOI] [PubMed] [Google Scholar]
- TURNER K.M., BURGOYNE R.D., MORGAN A. Protein phosphorylation and the regulation of synaptic membrane traffic. Trends. Neurosci. 1999;22:459–464. doi: 10.1016/s0166-2236(99)01436-8. [DOI] [PubMed] [Google Scholar]
- UEDA Y., TSURU N. Bilateral seizure-related changes of extracellular glutamate concentration in hippocampi during development of amygdaloid kindling. Epilepsy. Res. 1994;18:85–88. doi: 10.1016/0920-1211(94)90036-1. [DOI] [PubMed] [Google Scholar]
- WADA Y., NAKAMURA M., HASEGAWA H., YAMAGUCHI N. Intra-hippocampal injection of 8-hydroxy-2-(di-n-propylamino)tetralin (8-OH-DPAT) inhibits partial and generalized seizures induced by kindling stimulation in cats. Neurosci. Lett. 1993;159:179–182. doi: 10.1016/0304-3940(93)90828-9. [DOI] [PubMed] [Google Scholar]
- WAHNSCHAFFE U., LOSCHER W. Anticonvulsant effects of ipsilateral but not contralateral microinjections of the dopamine D2 agonist LY 171555 into the nucleus accumbens of amygdala - kindled rats. Brain. Res. 1991;553:181–187. doi: 10.1016/0006-8993(91)90822-d. [DOI] [PubMed] [Google Scholar]
- WESTERINK B.H.C., HOFSTEEDE H.M., DAMSMA G., DEVRIES J.B. The significance of extracellular calcium for the release of dopamine, acetylcholine and amino acids in conscious rats, evaluated by brain microdialysis. Naunyn-Schmiedeberg's Arch. Pharmacol. 1988;337:373–378. doi: 10.1007/BF00169526. [DOI] [PubMed] [Google Scholar]
- WESTERINK B.H.C., HOFSTEEDE R.M., TUNTLER J., DE VRIES J.B. Use of calcium antagonism for the characterization of drug-evoked dopamine release from the brain of conscious rats determined by microdialysis. J. Neurochem. 1989;52:722–729. doi: 10.1111/j.1471-4159.1989.tb02514.x. [DOI] [PubMed] [Google Scholar]
- WU L.G., SAGGAU P. Presynaptic inhibition of elicited neurotransmitter release. Trends Neurosci. 1997;20:204–212. doi: 10.1016/s0166-2236(96)01015-6. [DOI] [PubMed] [Google Scholar]
- XU T., BINZ T., NIEMANN H., NEHER E. Multiple kinetic components of exocytosis distinguished by neurotoxin sensitivity. Nat. Neurosci. 1998;1:192–200. doi: 10.1038/642. [DOI] [PubMed] [Google Scholar]
- YOSHIHARA M., UEDA A., ZHANG D., DEITCHER D.L., SCHWARZ T.L., KIDOKORO Y. Selective effects of neuronal-synaptobrevin mutations on transmitter release evoked by sustained versus transient Ca2+ increases and by cAMP. J. Neurosci. 1999;19:2432–2441. doi: 10.1523/JNEUROSCI.19-07-02432.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YOSHIMURA R., YANAGIHARA N., TERAO T., MINAMI K., ABE K., IZUMI F. Inhibition by carbamazepine of various ion channels-mediated catecholamine secretion in cultured bovine adrenal medullary cells. Naunyn Schmiedebergs Arch. Pharmacol. 1995;352:297–303. doi: 10.1007/BF00168560. [DOI] [PubMed] [Google Scholar]