

Abstract
The pharmacological properties of fatty acid amidohydrolase (FAAH) at different assay pH values were investigated using [3H]-anandamide ([3H]-AEA) as substrate in rat brain homogenates and in COS-7 cells transfected with wild type and mutant FAAH.
Rat brain hydrolysis of [3H]-AEA showed pH dependency with an optimum around pH 8-9. Between pH 6.3 and 8.2, the difference in activity was due to differences in the Vmax, rather than the KM values.
For inhibition of rat brain [3H]-AEA metabolism by a series of known FAAH inhibitors, the potencies of the enantiomers of ibuprofen and phenylmethylsulphonyl fluoride (PMSF) were higher at pH 5.28 than at pH 8.37, whereas the reverse was true for oleyl trifluoromethylketone (OTMK) and arachidonoylserotonin. At both pH values, (−)ibuprofen was a mixed-type inhibitor of FAAH. The Ki(slope) and Ki(intercept) values for (−)ibuprofen at pH 5.28 were 11 and 143 μM, respectively. At pH 8.37, the corresponding values were 185 and 3950 μM, respectively.
The pH dependency for the inhibition by OTMK and (−)ibuprofen was also seen in COS-7 cells transiently transfected with either wild type, S152A or C249A FAAH. No differences in potencies between the wild type and mutant enzymes were seen.
It is concluded that the pharmacological properties of FAAH are highly pH-dependent. The higher potency of ibuprofen at lower pH values raises the possibility that in certain types of inflamed tissue, the concentration of this compound following oral administration may be sufficient to inhibit FAAH.
Keywords: Anandamide, fatty acid amidohydrolase, ibuprofen, pH dependency
Introduction
The fatty acid amides are a class of endogenous compounds that have attracted considerable recent interest for their possible therapeutic usefulness. Two of these compounds are anandamide (AEA) and palmitoylethanolamide. AEA, which interacts with both cannabinoid and vanilloid receptors, has been shown to affect a number of processes including nociception (antinociceptive at low doses via effects on cannabinoid receptors, activation of peripheral nociceptors at high doses via effects on vanilloid receptors), cell proliferation and lung function (Calignano et al., 1998; 2000; Jaggar et al., 1998; Richardson et al., 1998; de petrocellis et al., 1998; Sarker et al., 2000; Maccarrone et al., 2000; Gauldie et al., 2001). Palmitoylethanolamide is inactive at cannabinoid receptors in vitro (Lambert et al., 1999), but has been shown to prevent substance P-induced mast cell degranulation and to have beneficial effects upon inflammatory pain in vivo (Aloe et al., 1993; Mazzari et al., 1996; Calignano et al., 1998; Jaggar et al., 1998). Given that the synthesis of fatty acid amides is increased under some conditions of cellular stress and inflammation (for review, see de petrocellis et al., 2000), the enzyme responsible for their metabolism may be a useful therapeutic target for the treatment of certain inflammatory states.
Fatty acid amide hydrolase, FAAH, is a membrane-associated enzyme capable of the metabolism of a large number of fatty acid amides (see Boger et al., 2000) and is expressed in a number of tissues, in particular the liver, brain, and testis (Deutsch & Chin, 1993). In the case of AEA, FAAH-catalyzed hydrolysis produces arachidonic acid and ethanolamine (Deutsch & Chin, 1993). A number of inhibitors of FAAH have been described, including substrate analogues such as oleyl trifluoromethylketone (OTMK) (Patterson et al., 1996) and arachidonyl serotonin (AA-5HT) (Bisogno et al., 1998), non-steroidal anti-inflammatory drugs such as ibuprofen (Fowler et al., 1997), and other substances such as the serine protease inhibitor phenylmethylsulphonyl fluoride (PMSF) (Deutsch & Chin, 1993). In 1996, the cDNA encoding the enzyme was cloned and shown to be a member of the amidase signature sequence group of enzymes (Cravatt et al., 1996). Subsequent mutation studies have identified some of the residues both within and outside the amidase signature sequence that are of importance for catalytic activity (Goparaju et al., 1999; Omeir et al., 1999; Patricelli et al., 1999; Patricelli & Cravatt, 2000), although information is lacking as to whether mutations retaining FAAH activity have altered pharmacological properties.
FAAH from a variety of sources, including both the naturally occurring enzyme (from the rat brain, for example) and the recombinant enzyme expressed in COS-7 cells, has been reported in several studies to have a pH optimum around pH 9 (Ueda et al., 1995; 1999; Maurelli et al., 1995; Hillard et al., 1995; Omeir et al., 1995; Bisogno et al., 1997; Patricelli et al., 1998); although one group found an apparently biphasic pH dependency for the metabolism of AEA by rat brain microsomes (Desarnaud et al., 1995). More recently, Ueda et al. (1999) reported the expression in a human megakaryoblastic cell line (CMK) of an AEA hydrolysing activity with a pH optimum of 5.0 and rather different pharmacological properties to FAAH. It was, however, not established whether the lack of sensitivity of this acid amidase to PMSF and the AEA substrate analogue methyl arachidonyl fluorophosphonate (MAFP) was a property of the enzyme per se or a reflection of the pH used. If the latter was the case, then similar anomalous sensitivities of inhibition of FAAH would be expected at acidic pH. In the present study, we have: (a) reinvestigated the pH dependency of the ability of rat brain homogenates to metabolise AEA, using two different buffer systems and (b) determined whether different assay pH values affect the pharmacological properties of FAAH. In the latter case, we have used both rat brain homogenates and COS-7 cells transfected with either wild type (wt) or FAAH with mutations within the amidase signature sequence that retain catalytic activity as enzyme sources.
Methods
Materials
Arachidonyl-ethanolamide-[1-3H] ([3H]-AEA) (specific activity 60 Ci mmol−1) was obtained from American Radiolabelled Chemicals Inc., St. Louis, MO, U.S.A. Non-radioactive AEA and the enantiomers of ibuprofen (α-methyl-4-(2-methylpropyl) benzeneacetic acid) were obtained from Research Biochemicals International, Natick, MA, U.S.A. Oleyl trifluoromethylketone (OTMK) and arachidonoyl-serotonin (AA-5HT) were obtained from the Cayman Chemical Company, Ann Arbor, MI, U.S.A. Phenylmethylsulphonyl fluoride (PMSF) and fatty acid free bovine serum albumin were obtained from the Sigma Chemical Co. (St Louis, MO, U.S.A.). AEA, ibuprofen enantiomers, OTMK, AA-5HT and PMSF were dissolved in ethanol. COS-7 cells were a kind gift of Dr Göran Bucht, Swedish Defence Research Agency, NBC Defence Division, Department of Microbiology, Umeå, Sweden.
Preparation of rat brain homogenates
Adult (9 month old) male Wistar and Sprague Dawley rats were used in the study. The animals were killed by carbon dioxide exposure followed by decapitation, and whole brains (minus the cerebellum) were dissected and frozen. For preparation of homogenates, the tissue was thawed, weighed and homogenized in 10 mM Tris-HCL buffer pH 7.6 containing 1 mM EDTA, in a volume of 2.5 ml/g wet weight. After determination of protein concentration, the homogenates were stored in aliquots of 250 μl at −80°C prior to assay of FAAH activity.
Preparation of homogenates of COS-7 cells transfected with wild-type and mutant FAAH
The pcDNA3.1 vectors containing wild-type or mutated FAAH cDNAs have been described previously (Omeir et al., 1999). The vectors were transported to Umeå and recovered as described by Rosman & Miller (1990) before being transformed into competent E. coli strain JM 109. Colonies were picked and propagated in Luria-Bertani medium. After pelleting the bacteria, the plasmids were purified using Qiagen Plasmid Midi kit (Qiagen, Hilden, Germany). Aliquots of 1.5×105 COS-7 cells were seeded into 6-well plates and grown for 18 – 24 h prior to transfection. Transfections were performed using the Lipofectamine™ or Lipofectamine2000™ protocol (Life Technologies, Tåstrup, Denmark). A 1.5 μg aliquot of plasmid (pFAAH-wt, pFAAH-S152A, pFAAH-C249A or pFAAH-S218A) was used for each transfection. As a control, cells were treated with the Lipofectamine™ or Lipofectamine2000™ alone (‘mock transfection'). After growing the transfected cells overnight, the cells were supplemented with fresh Dulbecco's minimal essential medium containing 10% FBS, and were grown for an additional 20 h. The cells were then trypsinized, pelleted by centrifugation, and stored at −70°C. Upon thawing, the samples were suspended in 10 mM Tris-HCl buffer with 1 mM EDTA, pH 8.0 and frozen in aliquots at −80°C prior to assay of FAAH activity.
Assay of FAAH activity
FAAH was assayed essentially as described by Omeir et al. (1995) who used carbon-14 labelled AEA as substrate (for full description using [3H]-AEA, see Fowler et al., 1997). Briefly, assay mixtures contained homogenate or cell lysate, test compound (when appropriate), and radiolabelled AEA (containing 10 mg ml−1 fatty acid-free bovine serum albumin) in an assay volume of 200 μl. Test compounds were diluted with ethanol and compared with controls containing the same concentration of ethanol (25 μl in a 200 μl assay volume). Blanks contained homogenization buffer instead of homogenate or cell lysate. The mixtures were incubated at 37°C for the times stated in the figure legends. The reactions were stopped by placing the samples on ice, adding 400 μl methanol-chloroform (1 : 1), and vortexing and centrifuging the samples. The unmetabolized [3H]-AEA is retained in the chloroform whereas the [3H]-ethanolamine formed by the action of FAAH upon AEA is soluble in the methanol/buffer phase. Aliquots (200 μl) of the methanol/buffer phase were removed for analyses of radioactivity by liquid scintillation spectroscopy with quench correction. To obtain the actual pH during the experiments, the assays were scaled up (albeit using homogenization buffer in place of the homogenate or cell lysate) and the pH was measured after incubation at 37°C.
Protein determination
Protein contents were measured using the method of Harrington (1990) using bovine serum albumin as standard.
Determination of pI50, KM and Vmax values
For the inhibition curves, the specific activity at each inhibitor concentration in each experiment was expressed as per cent of control and pIC50 values [−log10(IC50 value)] were analysed using the built-in equation ‘sigmoid dose-response (variable slope)' of the GraphPad Prism computer programme (GraphPad Software Inc., San Diego, CA, U.S.A.), with ‘top' (i.e. control) and ‘bottom' (i.e. blank) values fixed at 100 and 0, respectively. This method was used to mimize the impact of inter-experimental variations in enzyme specific activity.
However, when the inhibition curve for PMSF at pH 5.5 was analysed using the raw data at each inhibitor concentration (but excluding the control and blank values), the calculated ‘top' and ‘bottom' values agreed well with those found for the uninhibited and blank samples, respectively. In addition, the pIC50 value obtained (5.84±0.18) agreed well with that found using the data as percent of control (Table 1). KM and Vmax values (or apparent KM and Vmax values for experiments determining the mode of inhibition by (−)ibuprofen) for FAAH activities were calculated using the Direct Linear Plot analysis (Eisenthal & Cornish-Bowden, 1974) using the Enzyme Kinetics v 1.4 software package, Trinity Software, Campton, NH, U.S.A. Since this analysis is non-parametric in nature, KM and Vmax values have been given as medians and ranges. For the experiments with (−)ibuprofen, the apparent KM and Vmax values calculated from the mean data at each inhibitor concentration were used in secondary replots to determine Ki(slope) and Ki(intercept) values. Significance testing was undertaken using the Statview™ computer programme (SAS Institute Inc., Cary, NC, U.S.A.).
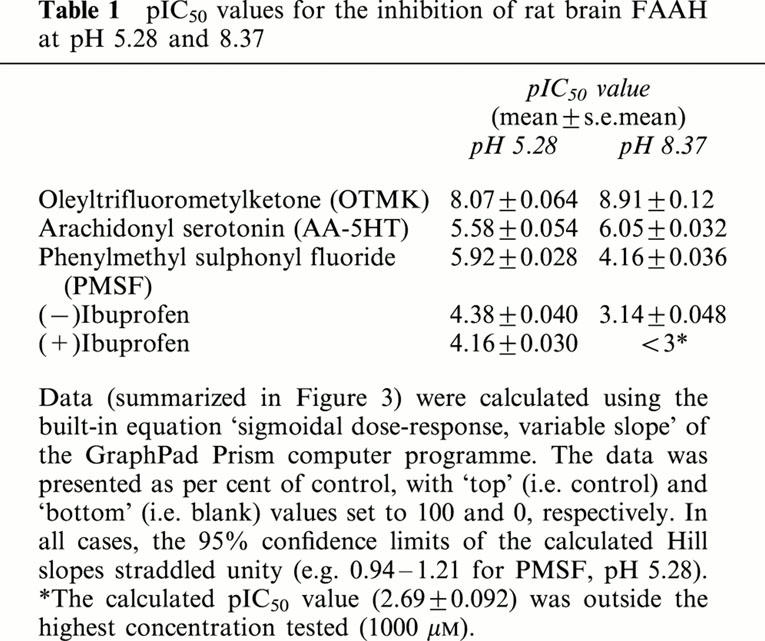
Table 1.
pIC50 values for the inhibition of rat brain FAAH at pH 5.28 and 8.37
Results
pH dependency of AEA hydrolysis by rat brain homogenates
Two different combinations of buffers were used. One consisted of sodium acetate, tris-HCl, and disodium tetraborate (Figure 1a), as was used in the study of Desarnaud et al. (1995) (where a biphasic pH curve for rat brain microsomal FAAH activity was seen). The other consisted of sodium citrate, tris-HCl, and sodium carbonate (Figure 1b) as was used in the study of Ueda et al. (1999) (where monphasic pH curves with pH optima of ∼9 and ∼5 were seen for AEA metabolising activity from RBL2H3 and CMK cells, respectively). An optimum around pH 8 – 9 was seen for both buffer systems, with no sign of any obvious top at acid pH values. The 100 mM disodium tetraborate buffer used to give the highest pH values in Figure 1a appeared to inhibit [3H]-AEA metabolism by the rat brain homogenates.
Figure 1.

pH dependency of the hydrolysis of 2 μM [3H]-AEA by rat brain homogenates. Two different buffer systems were used: (a) 100 mM sodium acetate buffer, 100 mM tris-HCl buffer, and 100 mM disodium tetraborate buffer (b) 50 mM sodium citrate, 50 mM tris-HCl and 50 mM sodium carbonate buffer. The pH values shown are the actual assay pH values, tested in scaled up assays, rather than the nominal pH of the buffers added to the homogenates. Data are means from two separate experiments using an incubation time of 20 min and of 4 μg protein/assay.
The effect of variations in the pH upon the KM and Vmax of FAAH was determined (Figure 2). For the three pH values tested using the same tris-HCl buffer system, the difference in activity was found to be due to differences in Vmax, rather than KM values. Thus, KM values (medians, n=3, with ranges in brackets) of 0.36 [0.35 – 0.37], 0.33 [0.30 – 0.38] and 0.41 [0.31 – 0.66] μM were found at assay pH values of 6.3, 7.27 and 8.2, respectively. The corresponding Vmax values were 645 [471 – 885], 828 [698 – 1124] and 1374 [1050 – 1572] pmol (mg protein)−1 min−1, respectively. The Friedman nonparametric test for analysis of variance by ranks of matched samples gave a significant effect of pH upon the Vmax values (P<0.05) but not on the KM values.
Figure 2.

Hydrolysis of a range of concentrations of [3H]-AEA by rat brain homogenates at different pH values. In each case, 50 mM tris-HCl was used as assay buffer. The pH values shown are the actual assay pH values, tested in scaled up assays, rather than the nominal pH of the buffers added to the homogenates. Shown are means±s.e.mean of three experiments using an incubation time of 10 min and 2.5 μg protein/assay.
Pharmacological properties of FAAH at different pH values.
To see if the sensitivity of FAAH to different known inhibitors changes with the pH, homogenates were incubated with different concentrations of the inhibitors at pH 5.28 and 8.37 (Figure 3). PMSF and the two ibuprofen enantiomers were found to be more potent inhibitors of [3H]-AEA metabolism at pH 5.28 than at pH 8.37, whereas the reverse was true for OTMK and AA-5HT. The pI50 values calculated from these experiments are summarized in Table 1.
Figure 3.

Inhibition of the hydrolysis of 2 μM [3H]AEA by rat brain homogenates by: (a) OTMK and AA-5HT; (b) (−)ibuprofen; (c) PMSF; and (d) (+)ibuprofen. Assays were performed either at pH 5.28 using 50 mM sodium citrate or at pH 8.37 using 50 mM tris-HCl as assay buffer. The pH values shown are the actual assay pH values, tested in scaled up assays, rather than the nominal pH of the buffers added to the homogenates. Shown are means±s.e. mean, n=3 using an incubation time of 20 min and either 8 (pH 5.28) or 4 (pHμ8.37) μg protein/assay. The two filled symbols shown in Panels (b) and (d) indicate data from separate experimental series at pHμ5.28.
The mode of inhibition of FAAH by (−)ibuprofen was determined at pH 5.28 and 8.37 (Figure 4). The inhibition was mixed-type at both pH values, although once again the potency of (−)ibuprofen was greater at the acidic pH.
Figure 4.

Mode of inhibition by (−)ibuprofen of the metabolism of [3H]-AEA by rat brain homogenates. The pH values (at assay) were: (a) pH 5.28 and (b) pH 8.37 using 50 mM sodium citrate and 50 mM tris-HCl, respectively, as assay buffers. Shown are means±s.e.mean of three experiments using an incubation time of 10μmin and 4μμg protein/assay. Inserts to the figures are secondary replots of the mean data to illustrate the mixed-type nature of the inhibition. The Ki(slope) and Ki(intercept) values were calculated as described in Methods.
Pharmacological properties of wt and mutant FAAH transiently expressed in COS-7 cells
One of the drawbacks associated with the use of rat brain homogenates is that they can represent a heterogeneous population of enzyme activities, although no evidence for pharmacological differences in FAAH in different brain regions or in different subcellular fractions has been presented (Fowler et al., 1999; Tiger et al., 2000). In order to circumvent this drawback, and to provide data on the pharmacological properties of catalytically active FAAH mutants, we investigated the sensitivity of wt, S152A and C249A enzymes expressed in COS-7 cells. The latter mutant is of particular interest since the rodent variant (used here) is catalytically active, whereas the porcine variant is not (Omeir et al., 1999; Goparaju et al., 1999).
Two transfection strategies were used, one with Lipofectamine™ reagent and one with Lipofectamine2000™ reagent. Both strategies gave essentially identical data in terms of relative enzyme activities and pharmacological properties. Initial time-course experiments indicated that good expression levels of the enzymes, ∼20 μg lysate/assay of cells expressing wt, S152A and C249A FAAH being able completely to metabolise essentially all available substrate within 120 min of incubation (Figure 5a). Mock transfected cells showed very low, but measureable, [3H]-AEA metabolizing activity. However, this potentially confounding factor was not seen when more diluted samples and shorter incubation times were used (Figure 5b). A fourth variant, S218A, was also tested, but the activity was rather low and was therefore not used in subsequent experiments (Figure 5a,b). The relative activities of the mutants are in agreement with the original study of Omeir et al. (1999), although these authors required longer incubation periods to assess activity, presumably reflecting lower transfection efficiencies.
Figure 5.

Properties of wt and mutant FAAH expressed in COS cells using 2 μM [3H]-AEA as substrate (a) and (b) show mean data from single experiments using lysates from cells transfected using the Lipofectamine™ reagent; where the assay protein concentration of the samples in (a) was set to ≈20 and ≈1.5μμg in (b). The assay buffer used was 10μmM Tris HCl, pHμ8.0 containing 1μmM EDTA. In (c), the inhibition of 2 μM [3H]-AEA metabolism by OTMK and (−)ibuprofen is shown. The pH values (at assay) were pHμ6.1 and pHμ8.0 using 10μmM tris-HCl containing 1 mM EDTA as assay buffer in both cases. Shown are means of two experiments using one set of samples from cells transfected using the Lipofectamine™ reagent and one set using cells transfected using the Lipofectamine2000™ reagent using incubation times of 30μmin and the appropriate protein contents to ensure that initial velocities were measured.
Using assay conditions where no relevant metabolism of [3H]-AEA by mock transfected cells could be seen, the effects of OTMK and (−)ibuprofen upon wt, S152A and C249A FAAH were assessed at pH 6.1 and 8.0 (Figure 5c). The pattern seen for the rat brain was repeated, with OTMK being less potent and (−)ibuprofen more potent at the lower pH. There was no marked difference in the sensitivities to inhibition between the wt and mutant FAAH. Thus, the pIC50 values calculated from the mean data for wt, S152A and C249A FAAH, respectively, were OTMK pH 6.1 : 6.84, 6.79 and 6.84; OTMK pH 8.0 : 7.45, 7.55 and 7.50; (−)ibuprofen pH 6.1 : 4.19, 4.23 and 4.18; (−)ibuprofen, pH 8.0 : 3.15, 3.13 and 3.35.
Discussion
In the present study, the pharmacological properties of FAAH have been investigated at different pH values, since information is lacking in this respect. Consistent with previous studies using homogenates from cow brain (Omeir et al., 1995) and membranes from rat brain (Hillard et al., 1995), mouse N18TG2 neuroblastoma cells (Maurelli et al., 1995), rat RBL2H3 and RBL-1 basophilic leukaemia cells (Bisogno et al., 1997; Ueda et al., 1999), purified recombinant rat FAAH (Patricelli et al., 1998), and partially purified FAAH from pig brain microsomes (Ueda et al., 1995), a single optimum at pH 8-9 was seen. The variation in activity, at least between pH 6.3 and 8.2, was due to a variation in Vmax rather than KM, indicating that in this range the variation does not merely reflect pH-dependent changes in the ionization state of the substrate but rather a change in the catalytic activity of the enzyme. Although the present study does not shed light into the molecular mechanisms giving rise to the pH sensitivity of the enzyme, it has been reported that the pH profile found here has been seen in both wild type FAAH and mutant FAAH lacking the putative transmembrane region of the enzyme near the N-terminus (Patricelli et al., 1998). Thus, the large predicted cytoplasmic region of the molecule defines the pH sensitivity.
The lack of a peak at low pH values would suggest that in the rat brain homogenates used here, there is no sign of any significant expression of an acid amidase capable of the hydrolysis of AEA, such as that expressed in CMK cells (Ueda et al., 1999). It is of course possible that microsomal fractions contain appreciable acid amidase, and thereby give the biphasic pH curve seen by Desarnaud et al. (1995) who used a microsomal fraction from rat brain and the buffer system shown in Figure 1a. Appreciable expression of such activity, however, would be expected to affect the observed sensitivity of microsomal [3H]-AEA metabolism to inhibition by PMSF relative to other subcellular fractions, given that the acid amidase activity found in CMK cells has a very low sensitivity to this inhibitor (Ueda et al., 1999). Such an anomalous sensitivity of microsomal fractions has not been seen (Tiger et al., 2000).
The pharmacological properties of FAAH at pH values near the optimum and at more acidic pH values is consistent for the enzyme activity in rat brain homogenates to that expressed following transfection of COS-7 cells with wt and mutant plasmids. In all cases, the potency of OTMK is lower at the lower pH, whereas the potency of (−)ibuprofen is higher. At both pH values tested, (−)ibuprofen acted as a mixed-type inhibitor, a result consistent with previous studies at pH 7.6 using both this enantiomer and racemic ibuprofen (Fowler et al., 1997; 1999).
In the brain samples, the pattern seen with (−)ibuprofen was also seen with its (+)-enantiomer (albeit with a slightly lower potency, consistent with previous data, Fowler et al., 1999) and with PMSF, whereas the potency of AA-5HT behaved more like OTMK. The increased potency of PMSF at low pH may at least in part be a reflection of the better stability of this compound in acidic aqueous solutions than in alkaline solutions (James, 1978). In the case of ibuprofen, there is a report that basic solutions can promote the racemization of ibuprofen enantiomers (Xie et al., 2000). However, such racemization cannot account for the pH difference in potencies of the ibuprofen enantiomers, since the enantiomeric selectivity of ibuprofen for FAAH is rather small, and indeed would have been expected to result in an increase in the potency of the (+)enantiomer, since this is less potent than the racemate (Fowler et al., 1999).
At first sight, the increased potency of the ibuprofen enantiomers at acid pH is at odds with the decreased potency of OTMK and AA-5HT. However, if it is argued that only the uncharged form of ibuprofen inhibits FAAH, an increased potency at acid pH would be predicted by the Henderson-Hasselbach equation. Indeed, this equation predicts a greater difference in potency than is actually found, which might reflect a general decrease in sensitivity of FAAH to all inhibitors at acid pH. This would be consistent with the data for OTMK and AA-5HT. This interpretation would predict that a plot of pIC50 for (−)ibuprofen vs assay pH would be linear. Simple regression analysis of the mean pIC50 values found here for the rat brain and wt FAAH inhibition by (−)ibuprofen at the four pH values tested indeed appeared linear (r2=0.979) and predicted a pI50 value at pH 7.6 of 3.42. This is in reasonable agreement with our previously published data at this pH using rat brain samples and 2 μM [3H]-AEA as substrate where pI50 values in the range 3.54 – 3.80 were found (Fowler et al., 1999; 2000).
In conclusion, the present data would indicate (a) that there is no significant expression in brain homogenates (relative to FAAH) of an acid amidase such as is found in CMK cells; (b) that the sensitivities to inhibition by OTMK and (−)ibuprofen for two active FAAH mutants, S152A and C249A FAAH are similar to the wild type enzyme; (c) that the uncharged form of ibuprofen is presumed to account for its inhibitory potency towards FAAH. This latter observation raises a possibility previously discussed (Fowler et al., 1997; 1999) that FAAH inhibition may contribute to the antiinflammatory properties of this compound in vivo, given (a) that certain inflammatory stimuli produce an increased synthesis of AEA and palmitoylethanolamide, both of which have beneficial effects upon inflammatory pain (see Introduction) and (b) that the peak plasma concentration of ibuprofen following oral administration of 2×200 mg ibuprofen tablets is in the range 110 – 150 μM (Karttunen et al., 1990). This compares with the IC50 values for (−) and (+)ibuprofen of 42 and 70 μM (calculated from the mean pI50 values in Table 1) at pH 5.28. Although this pH value is admittedly low (the IC50 value for racemic ibuprofen at pH 7.6 is 270 μM (Fowler et al., 1997)), there is some data suggestive of a reduced intracellular pH in certain inflammatory conditions in addition to the well-documented changes in extracellular pH. Andersson et al. (1999) reported a reduction in both tissue and intracellular pH in periarticular soft tissue from arthritic knee joints of rats relative to the contralateral control joints following antigen-induced arthritis. It is tempting to speculate that under such conditions, the reduced pHi may result not only in an impaired ability of FAAH to metabolise AEA per se (on the basis of the pH optimum curve), but also in an increased sensitivity of FAAH to inhibition by ibuprofen so that significant inhibition can be produced in vivo following administration of this compound at the high doses generally used by patients with rheumatoid arthritis.
Acknowledgments
We thank Britt Jacobsson for excellent technical assistance for the experiments using wt and mutant FAAH, and Göran Bucht for his kindness in providing us with COS-7 cells. The help and cooperation of Dale Deutsch and Judy Howell with the FAAH plasmids is greatly appreciated. This work was supported by grants from the Swedish Medical Research Foundation (Grant no. 12158), the Swedish Asthma- and Allergy Association's Research Foundation, and the Research Funds of the Medical Odontological Faculty, Umeå University. Part of the present study was undertaken as a rotation project in the biomedical graduate school programme run at Umeå University.
Abbreviations
- AA-5HT
arachidonoyl-serotonin
- AEA
anandamide
- FAAH
fatty acid amidohydrolase, NSAID, non-steroidal anti-inflammatory drug
- OTMK
oleyl trifluoromethylketone
- PMSF
phenylmethylsulphonyl fluoride
References
- ALOE L., LEON A., LEVI-MOLTALCINI R. A proposed autacoid mechanism controlling mastocyte behaviour. Agents Actions. 1993;39:C145–C147. doi: 10.1007/BF01972748. [DOI] [PubMed] [Google Scholar]
- ANDERSSON S.E., LEXMÜLLER K., JOHANSSON A., EKSTRÖM G.M. Tissue and intracellular pH in normal periarticular soft tissue and during different phases of antigen induced arthritis in the rat. J. Rheumatol. 1999;26:2018–2024. [PubMed] [Google Scholar]
- BISOGNO T., MAURELLI S., MELCK D., DE PETROCELLIS L., DI MARZO V. Biosynthesis, uptake, and degradation of anandamide and palmitoylethanolamide in leukocytes. J. Biol. Chem. 1997;272:3315–3323. doi: 10.1074/jbc.272.6.3315. [DOI] [PubMed] [Google Scholar]
- BISOGNO T., MELCK D., DE PETROCELLIS L., BOBROV M.Y., GRETSKAYA N.M., BEZUGLOV V.V., SITACHITTA N., GERWICK W.H., DI MARZO V. Arachidonoylserotonin and other novel inhibitors of fatty acid amide hydrolase. Biochem. Biophys. Res. Commun. 1998;248:515–522. doi: 10.1006/bbrc.1998.8874. [DOI] [PubMed] [Google Scholar]
- BOGER D.L., FECIK R.A., PATTERSON J.E., MIYAUCHI H., PATRICELLI M.P., CRAVATT B.F. Fatty acid amide hydrolase substrate specificity. Bioorg. Med. Chem. Lett. 2000;10:2613–2616. doi: 10.1016/s0960-894x(00)00528-x. [DOI] [PubMed] [Google Scholar]
- CALIGNANO A., KÁTONA I., DÉSARNAUD F., GIUFFRIDA A., LA RANA G., MACKIE K., FREUND T.F., PIOMELLI D. Bidirectional control of airway responsiveness by endogenous cannabinoids. Nature. 2000;408:96–101. doi: 10.1038/35040576. [DOI] [PubMed] [Google Scholar]
- CALIGNANO A., LA RANA G., GIUFFRIDA A., PIOMELLI D. Control of pain initiation by endogenous cannabinoids. Nature. 1998;394:277–281. doi: 10.1038/28393. [DOI] [PubMed] [Google Scholar]
- CRAVATT B.F., GIANG D.K., MAYFIELD S.P., BOGER D.L., LERNER R.A., GILULA N.B. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- DE PETROCELLIS L., MELCK D., BISOGNO T., DI MARZO V. Endocannabinoids and fatty acid amides in cancer, inflammation and related disorders. Chem. Phys. Lipids. 2000;108:191–207. doi: 10.1016/s0009-3084(00)00196-1. [DOI] [PubMed] [Google Scholar]
- DE PETROCELLIS L., MELCK D., PALMISANO A., BISOGNO T., LAEZZA C., BIFULCO M., DI MARZO V. The endogenous cannabinoid anandamide inhibits human breast cancer cell proliferation. Proc Natl Acad Sci U.S.A. 1998;95:8375–8380. doi: 10.1073/pnas.95.14.8375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DESARNAUD F., CADAS H., PIOMELLI D. Anandamide amidohydrolase activity in rat brain microsomes. Identification and partial characterization. J. Biol. Chem. 1995;270:6030–6035. doi: 10.1074/jbc.270.11.6030. [DOI] [PubMed] [Google Scholar]
- DEUTSCH D.G., CHIN S.A. Enzymatic synthesis and degradation of anandamide, a cannabinoid receptor agonist. Biochem. Pharmacol. 1993;46:791–796. doi: 10.1016/0006-2952(93)90486-g. [DOI] [PubMed] [Google Scholar]
- EISENTHAL R., CORNISH-BOWDEN A. The direct linear plot. A new graphical procedure for estimating enzyme kinetic parameters. Biochem. J. 1974;139:715–720. doi: 10.1042/bj1390715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FOWLER C.J., BÖRJESSON M., TIGER G. Differences in the pharmacological properties of rat and chicken brain fatty acid amidohydrolase. Br. J. Pharmacol. 2000;131:498–504. doi: 10.1038/sj.bjp.0703569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FOWLER C.J., JANSSON U., JOHNSON R.M., WAHLSTRÖM G., STENSTRÖM A., NORSTRÖM Å. &, TIGER G. Inhibition of anandamide hydrolysis by the enantiomers of ibuprofen, ketorolac and flurbiprofen. Arch. Biochem. Biophys. 1999;362:191–196. doi: 10.1006/abbi.1998.1025. [DOI] [PubMed] [Google Scholar]
- FOWLER C.J., TIGER G., STENSTRÖM A. Ibuprofen inhibits rat brain deamidation of anandamide at pharmacologically relevant concentrations. Mode of inhibition and structure-activity relationship. J. Pharmacol. Exp. Ther. 1997;283:729–734. [PubMed] [Google Scholar]
- GAULDIE S.D., MCQUEEN D.S., PERTWEE R., CHESSELL I.P. Anandamide activates peripheral nociceptors in normal and arthritic knee joints. Br. J. Pharmacol. 2001;132:617–621. doi: 10.1038/sj.bjp.0703890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOPARAJU S.K., KURAHASHI Y., SUZUKI H., UEDA N., YAMAMOTO S. Anandamide amidohydrolase of porcine brain: cDNA cloning, functional expression and site-directed mutagenesis. Biochim. Biophys. Acta. 1999;1441:77–84. doi: 10.1016/s1388-1981(99)00143-2. [DOI] [PubMed] [Google Scholar]
- HARRINGTON C.R. Lowry protein assay containing sodium dodecyl sulfate in microtiter plates for protein determination on fractions from brain tissue. Analyt. Biochem. 1990;186:285–287. doi: 10.1016/0003-2697(90)90081-j. [DOI] [PubMed] [Google Scholar]
- HILLARD C., WILKISON D., EDGEMOND W., CAMPBELL W. Characterization of the kinetics and distribution of N-arachidonylethanolamine (anandamide) hydrolysis by rat brain. Biochim. Biophys. Acta. 1995;1257:249–256. doi: 10.1016/0005-2760(95)00087-s. [DOI] [PubMed] [Google Scholar]
- JAGGAR S.I., HASNIE F.S., SELLATURAY S., RICE A.S.C. The anti-hyperalgesic actions of the cannabinoid anandamide and the putative CB2 receptor agonist palmitoylethanolamide in visceral and somatic inflammatory pain. Pain. 1998;76:189–199. doi: 10.1016/s0304-3959(98)00041-4. [DOI] [PubMed] [Google Scholar]
- JAMES G.T. Inactivation of the protease inhibitor phenylmethylsulfonyl fluoride in buffers. Anal. Biochem. 1978;86:574–579. doi: 10.1016/0003-2697(78)90784-4. [DOI] [PubMed] [Google Scholar]
- KARTTUNEN P., SAANO V., PARONEN P., PEURA P., VIDGREN M. Pharmacokinetics of ibuprofen in man: a single-dose comparison of two over-the-counter, 200 mg preparations. Int. J. Clin. Pharmacol. Ther. Toxicol. 1990;28:251–255. [PubMed] [Google Scholar]
- LAMBERT D.M., DIPAOLO F.G., SONVEAUX P., KANYONYO M., GOVAERTS S.J., HERMANS E., BUEB J., DELZENNE N.M., TSCHIRHART E.J. Analogues and homologues of N-palmitoylethanolamide, a putative endogenous CB2 cannabinoid, as potential ligands for the cannabinoid receptors. Biochim. Biophys. Acta. 1999;1440:266–274. doi: 10.1016/s1388-1981(99)00132-8. [DOI] [PubMed] [Google Scholar]
- MACCARRONE M., LORENZON T., BARI M., MELINO G., FINAZZI-AGRÒ A. Anandamide induces apoptosis in human cells via vanilloid receptors. Evidence for a protective role of cannabinoid receptors. J. Biol. Chem. 2000;275:31938–31945. doi: 10.1074/jbc.M005722200. [DOI] [PubMed] [Google Scholar]
- MAURELLI S., BISOGNO T., DE PETROCELLIS L., LUCCIA A.D., MARINO G., DI MARZO V. Two novel classes of neuroactive fatty acid amides are substrates for mouse neuroblastoma ‘anandamide amidohydrolase'. FEBS Letts. 1995;377:82–86. doi: 10.1016/0014-5793(95)01311-3. [DOI] [PubMed] [Google Scholar]
- MAZZARI S., CANELLA R., PETRELLI L., MARCOLONGO G., LEON A. N-(2-hydroxyethyl)hexadecamide is orally active in reducing edema formation and inflammatory hyperalgesia by down-modulating mast cell activation. Eur. J. Pharmacol. 1996;300:227–236. doi: 10.1016/0014-2999(96)00015-5. [DOI] [PubMed] [Google Scholar]
- OMEIR R.L., ARREAZA G., DEUTSCH D.G. Identification of two serine residues involved in catalysis by fatty acid amide hydrolase. Biochem. Biophys. Res. Commun. 1999;264:316–320. doi: 10.1006/bbrc.1999.1524. [DOI] [PubMed] [Google Scholar]
- OMEIR R.L., CHIN S., HONG Y., AHERN D.G., DEUTSCH D.G. Arachidonoyl ethanolamide-[1,2-14C] as a substrate for anandamide amidase. Life Sci. 1995;56:1999–2005. doi: 10.1016/0024-3205(95)00181-5. [DOI] [PubMed] [Google Scholar]
- PATRICELLI M.P., CRAVATT B.F. Clarifying the catalytic roles of conserved residues in the amidase signature family. J. Biol. Chem. 2000;275:19177–19184. doi: 10.1074/jbc.M001607200. [DOI] [PubMed] [Google Scholar]
- PATRICELLI M.P., LASHUEL H.A., GIANG D.K., KELLY J.W., CRAVATT B.F. Comparative characterization of a wild type and transmembrane domain-deleted fatty acid amide hydrolase: identification of the transmembrane domain as a site for oligomerization. Biochemistry. 1998;37:15177–15187. doi: 10.1021/bi981733n. [DOI] [PubMed] [Google Scholar]
- PATRICELLI M.P., LOVATO M.A., CRAVATT B.F. Chemical and mutagenic investigations of fatty acid amide hydrolase: evidence for a family of serine hydrolases with distinct catalytic properties. Biochemistry. 1999;38:9804–9812. doi: 10.1021/bi990637z. [DOI] [PubMed] [Google Scholar]
- PATTERSON J.E., OLLMAN I.R., CRAVATT B.F., BOGER D.L., WONG C.-H., LERNER R.A. Inhibition of oleamide hydrolase catalyzed hydrolysis of the endogenous sleep-inducing lipid cis-9-octadecenamide. J. Am. Chem. Soc. 1996;118:5938–5945. [Google Scholar]
- RICHARDSON J.D., KILO S., HARGREAVES K.M. Cannabinoids reduce hyperalgesia and inflammation via interaction with peripheral CB1 receptors. Pain. 1998;75:111–119. doi: 10.1016/S0304-3959(97)00213-3. [DOI] [PubMed] [Google Scholar]
- ROSMAN G.J., MILLER A.D. Improved method for plasmid shipment. Biotechniques. 1990;8:509. [PubMed] [Google Scholar]
- SARKER K.P, , OBARA S., NAKATA M., KITAJIMA I., MARUYAMA I. Anandamide induces apoptosis of PC-12 cells: involvement of superoxide and caspase-3. FEBS Letts. 2000;472:39–44. doi: 10.1016/s0014-5793(00)01425-3. [DOI] [PubMed] [Google Scholar]
- TIGER G., STENSTRÖM A., FOWLER C.J. Pharmacological properties of rat brain fatty acid amidohydrolase in different subcellular fractions using palmitoylethanolamide as substrate. Biochem. Pharmacol. 2000;59:647–653. doi: 10.1016/s0006-2952(99)00373-1. [DOI] [PubMed] [Google Scholar]
- UEDA N., KURAHASHI Y., YAMAMOTO S., TOKUNAGA T. Partial purification and characterization of the porcine brain enzyme hydrolyzing and synthesizing anandamide. J. Biol. Chem. 1995;270:23823–23827. doi: 10.1074/jbc.270.40.23823. [DOI] [PubMed] [Google Scholar]
- UEDA N., YAMANAKA K., TERASAWA Y., YAMAMOTO S. An acid amidase hydrolyzing anandamide as an endogenous ligand for cannabinoid receptors. FEBS Letts. 1999;454:267–270. doi: 10.1016/s0014-5793(99)00820-0. [DOI] [PubMed] [Google Scholar]
- XIE Y., LIU H., JIAYONG C. Kinetics of base catalyzed racemization of ibuprofen enantiomers. Int. J. Pharmaceut. 2000;196:21–26. doi: 10.1016/s0378-5173(99)00438-x. [DOI] [PubMed] [Google Scholar]