

Abstract
YM471, (Z)-4′-{4,4-difluoro-5-[2-(4-dimethylaminopiperidino)-2-oxoethylidene]-2,3,4,5-tetrahydro-1H-1-benzoazepine-1-carbonyl}-2-phenylbenzanilide monohydrochloride, is a newly synthesized potent vasopressin (AVP) receptor antagonist. Its effects on binding to and signal transduction by cloned human AVP receptors (V1A, V1B and V2) stably expressed in Chinese hamster ovary (CHO) cells, and oxytocin receptors in human uterine smooth muscle cells (USMC) were studied.
YM471 potently inhibited specific [3H]-AVP binding to V1A and V2 receptors with Ki values of 0.62 nM and 1.19 nM, respectively. In contrast, YM471 exhibited much lower affinity for V1B and oxytocin receptors with Ki values of 16.4 μM and 31.6 nM, respectively.
In CHO cells expressing V1A receptors, YM471 potently inhibited AVP-induced intracellular Ca2+ concentration ([Ca2+]i) increase, exhibiting an IC50 value of 0.56 nM. However, in human USMC expressing oxytocin receptors, YM471 exhibited much lower potency in inhibiting oxytocin-induced [Ca2+]i increase (IC50=193 nM), and did not affect AVP-induced [Ca2+]i increase in CHO cells expressing V1B receptors. Furthermore, in CHO cells expressing V2 receptors, YM471 potently inhibited the production of cyclic AMP stimulated by AVP with an IC50 value of 1.88 nM. In all assays, YM471 showed no agonistic activity.
These results demonstrate that YM471 is a potent, nonpeptide human V1A and V2 receptor antagonist which will be a valuable tool in defining the physiologic and pharmacologic actions of AVP.
Keywords: Human vasopressin receptors, antagonist, YM471, SR 49059, SR 121463A
Introduction
Arginine vasopressin (AVP), a peptide hormone secreted by the posterior pituitary, is an important regulator of fluid and cardiovascular homeostasis. The actions of AVP are mediated by specific receptors located in a variety of tissues and organs including blood vessels, liver, brain, lung, spleen, testis, urinary bladder and kidneys (Serradeil-Le Gal et al., 1995; Howl et al., 1991; Jard et al., 1986; Butlen et al., 1978; Tahara et al., 1997a,1997b; 1998c). Due to this heterogeneous tissue distribution, the possibility of multiple AVP receptor subtypes has been recognized for some time. The data supporting these proposals were based largely upon the relative affinities or potencies of agonist and antagonist peptide analogues of AVP in various tissues and organs. These AVP receptor subtypes have been classified according to the second messenger system to which they are coupled, and at least three AVP receptor subtypes (V1A, V1B and V2) have been identified. AVP activates phospholipase A2, C and D through the V1A and V1B receptors (Thibonnier, 1992). This results in the production of inositol 1,4,5-triphosphate and 1,2-diacylglycerol, the mobilization of intracellular calcium, and the activation of protein kinase C resulting in protein phosphorylation (Michell et al., 1979). In contrast, V2 receptors stimulate adenylate cyclase resulting in the production of cyclic AMP (Butlen et al., 1978), which stimulates phosphorylation of the collecting duct-specific water channel, aquaporin-2, via activation of protein kinase A (Nishimoto et al., 1999; Yasui et al., 1997). The three AVP receptor subtypes have recently been cloned and stably expressed, and found to belong to the family of seven membrane-spanning receptors that transmits signals through G proteins (Thibonnier et al., 1994; Sugimoto et al., 1994; Birnbaumer et al., 1992; Tahara et al., 1998b).
AVP is thought to play a role in several diseases, including heart failure, hypertension, renal diseases, hyponatremia and the syndrome of inappropriate antidiuretic hormone secretion (SIADH). Consequently, AVP receptor antagonists may be useful in treating these diseases. Recently, several orally effective and receptor subtype-selective nonpeptide AVP receptor antagonists have been discovered, namely the V1A receptor selective antagonists OPC-21268 and SR 49059 (Yamamura et al., 1991; Serradeil-Le Gal et al., 1993), the V2 receptor selective antagonists OPC-31260, OPC-41061 and SR 121463A (Yamamura et al., 1992; 1998; Serradeil-Le Gal et al., 1996) and the V1A/V2 receptor antagonist conivaptan (YM087; Tahara et al., 1997b).
We have previously reported on the discovery and characterization of a high-affinity mixed V1A/V2 receptor antagonist, conivaptan (Tahara et al., 1997b; Matsuhisa et al., 2000). Although the recent identification of nonpeptide AVP receptor antagonists represents an important milestone in AVP research, it is likely that elucidating the role of AVP in the pathophysiology of diseases in various systems will require potent compounds for both animals and human. This study set out to determine the effects of YM471 ((Z)-4′-{4,4-difluoro-5-[2-(4-dimethylaminopiperidino)-2-oxoethylidene]-2,3,4,5-tetrahydro-1H-1-benzoazepine-1-carbonyl}-2-phenylbenzanilide monohydrochloride; Figure 1), the lead compound from a new chemical series of potent nonpeptide AVP receptor antagonist (Shimada et al., 2000), on ligand binding to and signal transduction by human AVP and oxytocin receptors, using receptor binding and second messenger assays. In addition, the effects of YM471 were compared with those of the nonpeptide V1A receptor selective antagonist, SR 49059 and the V2 receptor selective antagonist, SR 121463A.
Figure 1.
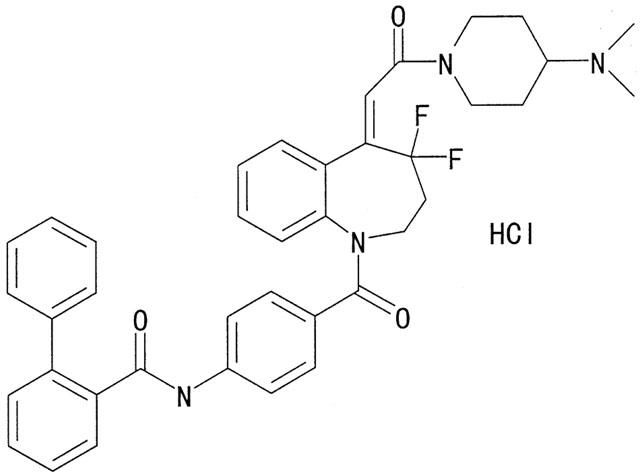
Chemical structure of YM471, (Z)-4′-{4,4-difluoro-5-[2-(4-dimethylaminopiperidino)-2-oxoethylidene]-2,3,4,5-tetrahydro-1H-1-benzoazepine-1-carbonyl}-2-phenylbenzanilide monohydrochloride.
Methods
Materials
The radioligands [3H]-AVP (specific activity, 80 Ci mmol−1), [3H]-oxytocin (specific activity, 50 Ci mmol−1) and [3H]-cyclic AMP (specific activity, 27 Ci mmol−1) were obtained from DuPont-New England Nuclear (Boston, MA, U.S.A.). AVP and oxytocin were obtained from the Peptide Institute Inc. (Osaka, Japan). YM471, SR 49059 ((2S) 1-[(2R 3S)-5-chloro-3-(2-chlorophenyl)-1-(3,4-dimethoxybenzene-sulphonyl)-3-hydroxy-2,3-dihydro-1H-indole-2-carbonyl]-pyrrolidine-2-carboxamide) and SR 121463A (1-[4-(N-tert-butylcarbamoyl)-2-methoxybenzene sulphonyl]-5-ethoxy-3-spiro-[4-(2-morpholinoethoxy)cyclohexane]indol-2-one;equatorial isomer) were synthesized at the Yamanouchi Pharmaceutical Co. (Ibaraki, Japan). These nonpeptide antagonists were initially dissolved in dimethyl sulphoxide (DMSO) at 10−2 M and diluted to the desired concentration with the buffer used in the receptor binding and second messenger assays. The final concentration of DMSO in the assay buffer did not exceed 1%, at which specific [3H]-AVP and [3H]-oxytocin binding were not affected. Fura 2-acetoxymethyl ester (AM) was obtained from Dojindo Laboratories (Kumamoto, Japan) and EGTA, ionomycin, 3-isobutyl-1-methylxanthine (IBMX) and bovine heart tissue were from Wako Pure Chemicals (Osaka, Japan). Minimum essential medium (MEM)-alpha, LipofectAMINE, foetal calf serum (FCS), antibiotics (penicillin and streptomycin) and trypsin-EDTA were from Gibco (Grand Island, NY, U.S.A.). Chinese hamster ovary (CHO) cells were from the American Tissue Culture Collection (Rockville, MD, U.S.A.). Bovine serum albumin (BSA) was purchased from Nacalai Tesque Inc. (Kyoto, Japan). Reagents for protein assay were purchased from Bio-Rad Laboratories (Richmond, CA, U.S.A.). All other chemicals were of the highest available reagent grade.
Membrane preparations
Human AVP receptor subtypes were stably expressed in CHO cells and membranes prepared as described in Tahara et al. (1998b). Briefly, CHO cells deficient in dihydrofolate reductase were stably transfected with the mammalian expression vector pEF-BOS (Mizushima & Nagata, 1990) which contains the dihydrofolate reductase gene, using LipofectAMINE. After 2 weeks of selection in MEM-alpha without nucleosides but supplemented with 10% FCS and 100 nM of the dihydrofolate reductase inhibitor amethopterin, surviving colonies of cells were isolated and grown in medium with 10% FCS and 1 μM amethopterin. Confluent cells were harvested in ice-cold 10 mM Tris-HCl, pH 7.4, containing 5 mM EDTA followed by homogenization and centrifugation at 35,000×g for 20 min at 4°C. The pellet was resuspended in 50 mM Tris-HCl, pH 7.4, containing 10 mM MgCl2 and stored at −80°C until use. Membrane preparations from human uterine smooth muscle cells (USMC) were prepared as described in Tahara et al. (1999). Human USMC imported from Clonetics (San Diego, CA, U.S.A.) were purchased from IWAKI (Tokyo, Japan). The cells were grown in SmBM culture medium (Clonetics) supplemented with 0.5 μg ml−1 human epidermal growth factor, 5 mg ml−1 insulin, 1 μg ml−1 human fibroblast growth factor, 5% FCS and antibiotics (GA-1000). Cells at the 5–10 passage stage were used and cells were identified histochemically by anti-α-actin and factor VIII antibodies.
Binding assays
For saturation binding studies, membrane preparations were incubated with increasing concentrations of [3H]-AVP or [3H]-oxytocin (0.05–3.0 nM). For competition studies, radioligand (0.5 nM) was added to each membrane preparation (V1A: 15 μg, V1B: 7.0 μg, V2: 6.0 μg, oxytocin: 100 μg), and incubated with increasing concentrations of compounds in 250 μl of assay buffer containing 50 mM Tris-HCl, pH 7.4, 10 mM MgCl2 and 0.1% BSA. The binding reactions were initiated by the addition of plasma membrane preparations and incubated for 60 min at 25°C (V1A, V1B and V2 receptors) or 30°C (oxytocin receptors), which allowed equilibrium to be established. After incubation, the reaction was terminated by addition of 3 ml of ice-cold Tris buffer (50 mM Tris-HCl, pH 7.4, and 10 mM MgCl2) followed immediately by rapid filtration through 96-well GF/C UniFilter Plates using a MicroMate Cell Harvester (Packard Instrument Company; Meriden, CT, U.S.A.). The filters were rinsed twice and the radioactivity retained on the filters was counted with TopCount Microplate Scintillation Counter (Packard Instrument Company). Nonspecific binding was determined using 1 μM unlabeled AVP or oxytocin in the reaction mixture. Specific binding was calculated as the total binding minus nonspecific binding. The concentration of test compound that caused 50% inhibition (IC50) of the specific binding of [3H]-AVP or [3H]-oxytocin was determined by regression analysis of displacement curves. The inhibition constant (Ki) was calculated from the following formula (Cheng & Prusoff, 1973): Ki=IC50/(1+[L]/KD), where [L] is the concentration of radioligand present in the tubes and KD is the dissociation constant of radioligand obtained from the saturation studies. The Hill coefficient (nH) was calculated from the following four parameter-logistic equation: Y = Bottom + (Top − Bottom)/(1+10(Log IC50−X)nH), where Y is the response and X is the concentration of compound. To investigate whether YM471 interacts competitively or noncompetitively, we examined saturation binding of [3H]-AVP with or without YM471 in CHO cells expressing human AVP receptors.
Measurement of [Ca2+]i
CHO cells expressing human V1A or V1B receptors and human USMC were grown on coverglasses and serum-starved for 24 h. Cell monolayers (2×105 cells cm−2) were loaded with fura 2-AM (2 μM/coverglass) in Krebs-Henseleit–HEPES buffer (containing (mM) NaCl 130, KCl 5, CaCl2 1.25, MgSO4 0.8, glucose 5.5, HEPES 20, and BSA 0.1%, pH 7.4) for 30 min at 37°C. The monolayers of cells were washed, then transferred to fura 2-free Krebs-Henseleit–HEPES buffer and incubated for an additional 30 min at 37°C. The coverglass was placed into a quartz cuvette containing 2 ml Krebs-Henseleit–HEPES buffer and maintained at 37°C with continuous stirring. When thermal equilibrium was reached, the fluorescence signal was recorded with a CAF-110 spectrofluorometer (Japan Spectrometer Co.; Tokyo, Japan) at both 340 and 380 nm excitation wavelengths, and 500 nm emission wavelength. After recording the baseline signal for 3 min, AVP or oxytocin was added to the cuvette to stimulate the mobilization of intracellular calcium in the presence or absence of antagonists. Fluorescence measurements were converted to [Ca2+]i by determining maximal fluorescence (Fmax) with the nonfluorescent Ca2+ ionophore, ionomycin (25 μM), after which minimal fluorescence (Fmin) was obtained by adding 3 mM EGTA. From the ratio (R) of fluorescence at 340 and 380 nm, the [Ca2+]i was determined using the following equation: [Ca2+]i (nM)=KD×[(R-Rmin) / (Rmax - R)]×b, where b is the ratio of fluorescence of fura 2 at 380 nm in zero and saturating Ca2+ concentrations, and KD is the dissociation constant of fura 2 for Ca2+, assumed to be 224 nM (Grynkiewicz et al., 1985).
Measurement of cyclic AMP production
CHO cells expressing human V2 receptors were grown in 24-well culture plates to 90–95% confluence and serum-starved for 24 h. Cell monolayers were incubated in MEM-alpha supplemented with 0.5 mM IBMX and 0.1% BSA containing vehicle or various concentrations of AVP and/or antagonists for 10 min at 37°C. At the end of incubation, the cell monolayers were washed three times with phosphate-buffered saline (PBS) followed by lysis in boiling 50 mM sodium acetate, pH 6.2, containing 2 mM IBMX. Extracts were then boiled for 3 min and kept at −40°C before determination of cyclic AMP. The amount of cyclic AMP was measured as previously described (Takeda et al., 1989) although some minor modifications were introduced. Briefly, crude binding protein was prepared from bovine heart tissue. Approximately, 100 g of bovine heart tissue was homogenized in 4 volumes of 20 mM sodium phosphate buffer containing 2 mM EDTA and 25 mM 2-mercaptoethanol (PEM buffer), pH 7.4, followed by centrifugation at 11,000×g for 30 min. The supernatant was precipitated with ammonium sulphate (400 g l−1) for 60 min with stirring. The suspension was centrifuged at 12,000×g for 20 min and the resulting supernatant discarded. The pellet was resuspended in a minimum volume of PEM buffer, followed by dialysis for 2 h against the PEM buffer. The dialysate was centrifuged again to remove insoluble proteins. The resulting supernatant was used as the crude binding protein. For competitive protein binding assays, [3H]-cyclic AMP (2 nM) was added to crude binding protein (0.05 mg). This mixture was incubated with extract samples or standard cyclic AMP solutions (0–80 pmol) in 250 μl of PEM buffer (pH 7.4), containing 0.5 mg ml−1 BSA and 1.5 mM IBMX. After incubation for 60 min at 25°C, the reaction was terminated by addition of 3 ml of ice-cold 20 mM sodium phosphate buffer (pH 7.4), containing 2 mM EDTA, followed immediately by rapid filtration through glass fiber filters (GF/B; Whatman, Maidstone, U.K.) pre-soaked in 0.5% polyethylenimine. The filters were rinsed twice and the radioactivity retained on the filters was counted with a liquid scintillation counter.
Data analysis
Experimental results are expressed as the mean±standard error of the mean (s.e.mean) or the mean with 95% confidence limits. The KD and Bmax were calculated by GraphPad PRISM (GraphPAD Software, Inc.; San Diego, CA, U.S.A.) in a linear regression analysis of the transformed data. The EC50 and IC50 values were estimated from the concentration-response curves by GraphPad PRISM. All experiments were repeated at least three times, and comparable results were obtained.
Results
Radioligand binding studies
Saturation experiments employing increasing concentrations of [3H]-AVP and human V1A, V1B and V2 receptors on CHO cell membranes preparations and [3H]-oxytocin and human uterine smooth muscle cell membrane preparations showed that specific binding was saturable. Scatchard analysis of these data gave linear plots consistent with the presence of a single class of high affinity binding site for each receptor. The apparent dissociation constant (KD) was 0.39±0.13 nM for V1A, 0.25±0.02 nM for V1B, 1.21±0.37 nM for V2 and 0.76±0.04 nM for oxytocin receptors. The calculated maximum binding capacity (Bmax) was 1580±150 fmol mg−1 protein for V1A, 5230±690 fmol mg−1 protein for V1B, 7020±450 fmol mg−1 protein for V2 and 153±4 fmol mg−1 protein for oxytocin receptors. YM471, SR 49059 and SR 121463A were tested for their ability to compete against specific [3H]-AVP or [3H]-oxytocin binding (Figure 2). The inhibition constants (Ki) of the tested compounds are shown in Table 1. YM471 potently inhibited specific binding of [3H]-AVP to human V1A and V2 receptors, exhibiting Ki values of 0.62 nM and 1.19 nM, respectively. In contrast, SR 49059 and SR 121463A exhibited high affinity and selectivity for V1A (Ki=0.53 nM) and V2 receptors (Ki=2.75 nM), respectively. The slopes of the inhibition curves (Hill coefficient: nH) of all nonpeptide antagonists were close to unity, which suggest a single-site competitive model. The comparison with the V1A receptor selective antagonist, SR 49059, and the V2 receptor selective antagonist, SR 121463A showed that these compound displayed high affinity for human V1A and V2 receptors, respectively, exhibiting Ki values comparable to those of YM471 and in agreement with the original published affinities for human adrenal V1A and kidney V2 receptors (Serradeil-Le Gal et al., 1993; 1996). The selectivity of YM471 for human V1A and V2 receptors was evaluated by measuring the ability of YM471 to inhibit the binding to human V1B receptors and to the related human oxytocin receptors. YM471 exhibited low affinity for V1B and oxytocin receptors with Ki values of 16.4 μM and 31.6 nM, respectively, showing an approximate 100–10 000-fold lower potency against oxytocin and V1B receptors, respectively, than against V1A and V2 receptors (Table 2).
Figure 2.
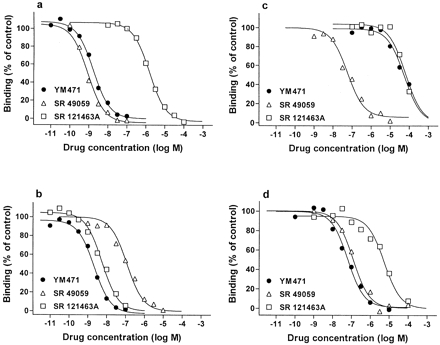
Inhibition of specific [3H]-AVP or [3H]-oxytocin bound to plasma membranes prepared from CHO cells expressing human (a) V1A, (b) V2, (c)V1B, and (d) oxytocin receptors by AVP receptor antagonists. Results are representative data from three to five independent experiments performed in duplicate. The combined results of all experiments are summarized in Table 1.
Table 1.
Ki values of nonpeptide AVP receptor antagonists for human AVP and oxytocin receptors

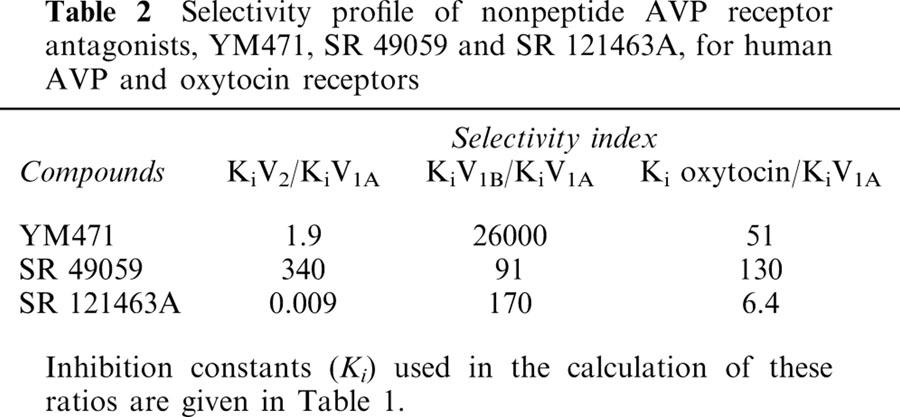
Table 2.
Selectivity profile of nonpeptide AVP receptor antagonists, YM471, SR 49059 and SR 121463A, for human AVP and oxytocin receptors
In the absence or presence of YM471 (0.1, 0.3 and 1.0 nM), [3H]-AVP saturation binding experiments were performed using CHO cell membranes expressing either human V1A or V2 receptors. In both preparations, increasing concentrations of YM471 caused successive decreases in the slopes of the curves, consistent with an increase in equilibrium dissociation constant (KD) without a reduction in receptor density (Bmax) (Figure 3a). From these saturation experiments, the calculated Ki values for YM471 were 0.93±0.33 nM (V1A) and 2.08±1.68 nM (V2) using the equation KDApp=KD×(1+C / Ki), where KDApp is the apparent KD in the presence of different concentrations of YM471 (0.1, 0.3 or 1.0 nM) and C is the concentration of YM471. These results were consistent with the directly measured Ki values obtained from competition experiments with [3H]-AVP (Table 1).
Figure 3.
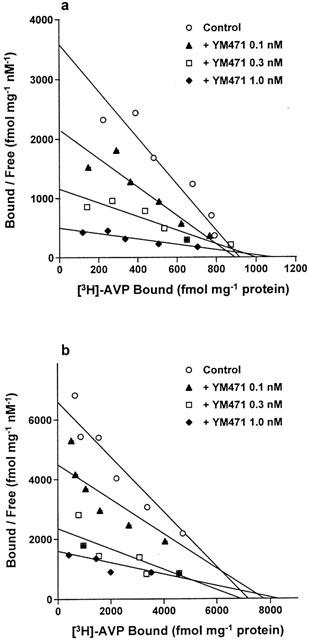
Scatchard plots of [3H]-AVP binding to plasma membranes prepared from CHO cells expressing human (a) V1A and (b) V2 receptors in the absence or presence of YM471. Results are representative data from four independent experiments performed in duplicate.
Measurement of [Ca2+]i
Addition of AVP to fura 2-loaded CHO cells expressing human V1A or V1B receptors increased [Ca2+]i concentration-dependently. (Figures 4a, 5a). In CHO cells expressing human V1A receptors, YM471 and SR 49059 strongly and concentration-dependently inhibited the increase in [Ca2+]i stimulated by AVP (10−8 M), exhibiting IC50 values of 0.56 (0.42–0.76) nM and 0.34 (0.24–0.48) nM, respectively (Figure 4b). However, SR 121463A did not potently inhibit [Ca2+]i increase (IC50=223 (167–298) nM). In contrast, in CHO cells expressing human V1B receptors, SR 49059 inhibited the increase in [Ca2+]i stimulated by AVP (10−8 M) with an IC50 value of 65.3 (39.8–107) nM, however, YM471 and SR 121463A did not inhibit the increase in [Ca2+]i (IC50>10 μM) (Figure 5b). Oxytocin added to fura 2-loaded human USMC expressing oxytocin receptors resulted in an increase in [Ca2+]i dependent upon oxytocin concentration (Figure 6a). YM471 inhibited the increase in [Ca2+]i stimulated by oxytocin (10−7 M) on human USMC, exhibiting an IC50 value of 193 (115–323) nM (Figure 6b). Under the same experimental conditions, SR 49059 also inhibited the increase in [Ca2+]i with an IC50 value of 653 (398–1070) nM, but SR 121463A did not potently inhibit [Ca2+]i increase (IC50=19.5 (13.0–29.1) μM). These IC50 values correspond well with the Ki values obtained from the binding studies. In all assays used, YM471 had no any agonistic activity (up to 10 μM).
Figure 4.
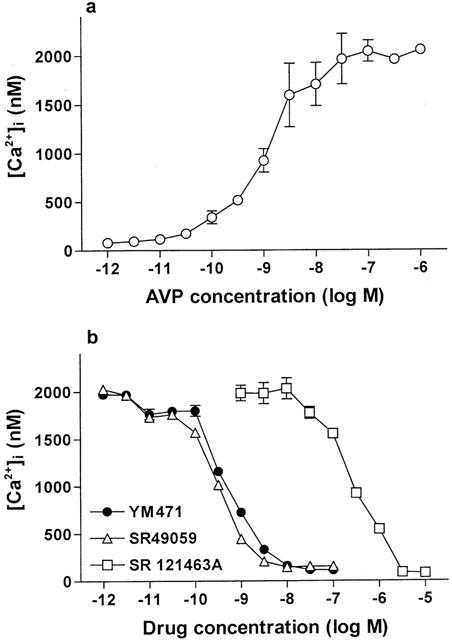
(a) Effect of AVP on [Ca2+]i increase and (b) effect of AVP receptor antagonists on 10 nM AVP-induced [Ca2+]i increases in CHO cells expressing human V1A receptors. Values are mean±s.e.mean from three to eight independent determinations.
Figure 5.
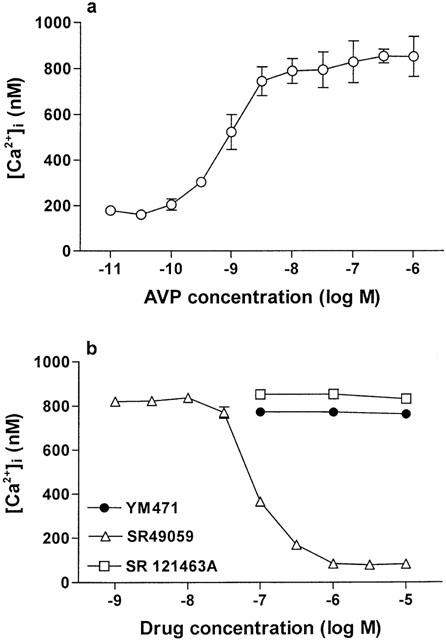
(a) Effect of AVP on [Ca2+]i increase and (b) effect of AVP receptor antagonists on 10 nM AVP-induced [Ca2+]i increases in CHO cells expressing human V1B receptors. Values are mean±s.e.mean from three to eight independent determinations.
Figure 6.
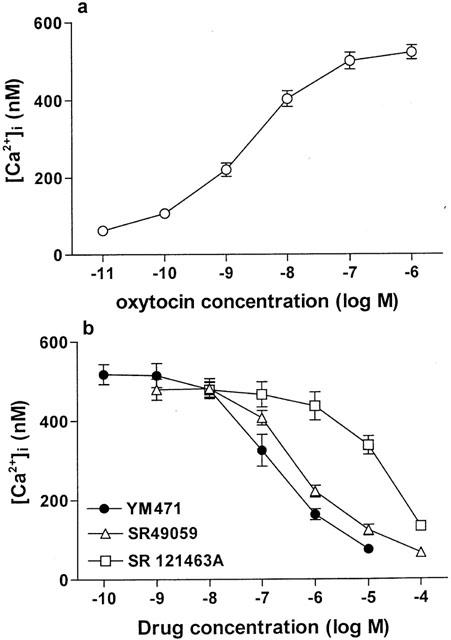
(a) Effect of oxytocin on [Ca2+]i increase and (b) effect of AVP receptor antagonists on 100 nM oxytocin-induced [Ca2+]i increases in human USMC. Values are mean±s.e.mean from three to eight independent determinations.
Measurement of cyclic AMP production
Addition of AVP to CHO cells expressing human V2 receptors resulted in a concentration-dependent increase in cellular cyclic AMP production (Figure 7a). YM471 strongly and concentration-dependently inhibited 10 nM AVP-induced increase in cyclic AMP production in CHO cells expressing human V2 receptors, exhibiting an IC50 value of 1.88 (1.14–3.09) nM (Figure 7b). No change in basal cyclic AMP production occurred when YM471 was tested alone (up to 10 μM). Under the same experimental conditions, SR 121463A also potently inhibited AVP-induced increase in cyclic AMP production, exhibiting an IC50 value of 1.66 (1.18–2.33) nM, but SR 49059 did not potently inhibit the increase in cyclic AMP production (IC50=186 (104–330) nM).
Figure 7.
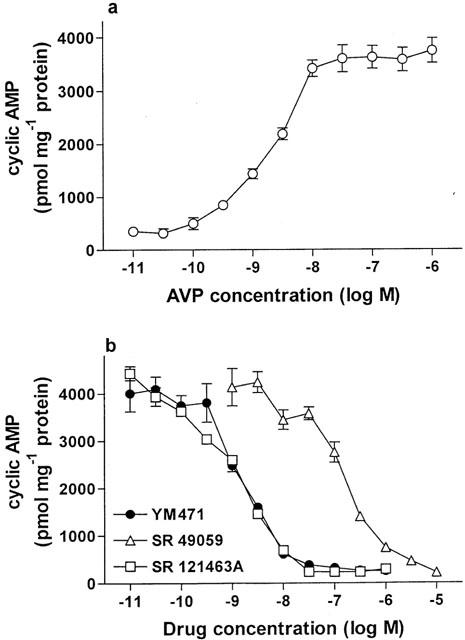
(a) Effect of AVP on production of cellular cyclic AMP and (b) effect of AVP receptor antagonists on 10 nM AVP-induced cellular cyclic AMP production in CHO cells expressing human V2 receptors. Values are mean±s.e.mean from eight independent determinations.
Discussion
The pharmacologic properties of YM471, a potent, new non-peptide AVP receptor antagonist, were investigated using radioligand binding and second messenger assays. The affinity of YM471 for human AVP and oxytocin receptors was examined in a radioligand binding assay using plasma membranes from CHO cells stably expressing human V1A, V1B and V2 receptors, and human USMC expressing oxytocin receptors. Nanomolar concentrations of YM471 effectively inhibited [3H]-AVP binding to human V1A and V2 receptors. To determine whether YM471 interacts reversibly or irreversibly with V1A and V2 receptors, [3H]-AVP saturation binding was determined in the presence or absence of YM471 and analysed using Scatchard's method (1949). In CHO cells expressing human V1A or V2 receptors, YM471 concentration-dependently reduced the slope without affecting the intercept of the Scatchard plot, indicating a change in the KD values of the radioligand for its receptors without a change in the Bmax values. The data show YM471 interacts reversibly and competitively with V1A and V2 receptors. The Ki values for YM471 in these experiments were estimated to be 0.62 nM (V1A) and 1.19 nM (V2). In contrast, YM471 showed much lower affinity for human V1B and oxytocin receptors. These results suggest that YM471 possesses potent affinity and selectivity for human V1A and V2 receptors, exhibiting Ki values similar to those of the V1A receptor selective antagonist, SR 49059, and the V2 receptor selective antagonist, SR 121463A. Experiments employing human AVP receptors are necessary since previously reports demonstrated marked species differences in the in vitro affinity and potency of several nonpeptide AVP receptor antagonists (Guillon et al., 1982; Pettibone et al., 1992; Serradeil-Le Gal et al., 1995; Tahara et al., 1998a, 1998b).
Another important question was whether the affinity observed in these binding studies translated into a corresponding potency to inhibit signal transduction; consequently several in vitro functional studies were performed to characterize the nature of the interaction of YM471 with human V1A, V1B, V2 and oxytocin receptors. AVP and oxytocin activate phospholipase C-mediated hydrolysis of polyphosphoinositides via the V1A, V1B and oxytocin receptors to generate two second messengers, inositol-1,4,5-triphosphate, which induces an increase in free intracellular calcium from the endoplasmic reticulum, and 1,2-diacylglycerol, which activates protein kinase C (Michell et al., 1979; Tasaka et al., 1991; Jasper et al., 1995; Holda et al., 1996). In CHO cells expressing either human V1A or V1B receptors, AVP treatment concentration-dependently increased [Ca2+]i. YM471 potently antagonized this AVP-induced increase in [Ca2+]i mediated by V1A receptors, but did not inhibit the increase in [Ca2+]i mediated by V1B receptors. Furthermore, in human USMC, which express only oxytocin receptors (Tahara et al., 2000), oxytocin treatment increased [Ca2+]i in a concentration-dependent manner. Although YM471 inhibited oxytocin-induced increase in [Ca2+]i, its inhibitory potency on oxytocin receptors was 300 times lower than that on V1A receptors. These results are consistent with the Ki values of YM471 obtained from [3H]-AVP and [3H]-oxytocin binding studies. With regard to V2 receptors, in contrast, AVP stimulates adenylate cyclase resulting in the production of cyclic AMP (Jans et al., 1989). In the present study using CHO cells which express human V2 receptors, AVP concentration-dependently stimulated intracellular cyclic AMP production and YM471 potently and concentration-dependently inhibited the production of cyclic AMP induced by AVP. Furthermore, in the absence of AVP, YM471 did not stimulate [Ca2+]i increase or cyclic AMP production, indicating that YM471 possesses no agonistic activity for AVP and oxytocin receptors. These results suggest that YM471 is a potent human AVP receptor antagonist which possesses dual V1A and V2 receptor antagonistic activities with no agonistic activity.
AVP exerts a variety of biological effects such as vasoconstriction via the V1A receptors and regulation of water excretion via the V2 receptors. It is generally assumed that AVP might be involved in several circulatory disorders and diseases including congestive heart failure, renal diseases, hyponatremia, SIADH, vasospasm and hypertension through the V1A and V2 receptors (Fujisawa et al., 1993a, 1993b; Naitoh et al., 1994; Laszlo et al., 1991). The development of AVP receptor antagonists appears essential for assessing the pathophysiologic roles of AVP and could lead to new therapeutic agents for many circulatory and hypertensive disorders. Recently, several potent and orally effective nonpeptide AVP receptor antagonists have been reported, namely the V1A receptor selective antagonist SR 49059, the V2 receptor selective antagonist SR 121463A, and the V1A/V2 receptor antagonist conivaptan. Among them, several experimental and clinical studies have demonstrated that conivaptan improves cardiohemodynamics and exerts aquauretic effects in SIADH or congestive heart failure; the V1A/V2 receptor antagonist is a valuable therapeutic agent in the treatment of heart failure (Yatsu et al., 1999; Abraham et al., 1999; 2000; Udelson et al., 2000; Painchaud et al., 2000; Suresh et al., 1999). The results of this study indicate that YM471 is the most potent V1A/V2 receptor antagonist with Ki values comparable to those to SR 49059 and SR 121463A and, thus, may serve as a useful pharmacologic tool to examine the role of these receptor subtypes and offer a effective approach to treatment of several circulatory diseases.
In summary, the data from this study demonstrate that YM471 has the highest affinity for and is the most potent nonpeptide antagonist of human V1A and V2 receptors as assayed by in vitro radioligand binding and functional assays. To the extent that AVP is involved in the etiology and maintenance of various diseases such as heart failure, YM471 may prove to be a valuable therapeutic drug for the treatment of these chronic disorders.
Acknowledgments
The authors acknowledge Drs Toichi Takenaka, Takeshi Fujikura, Noboru Satoh, Isao Yanagisawa, Gensei Kon, Osamu Inagaki, Hisataka Shikama, Nobuyuki Yamamoto and Kazuo Honda (Yamanouchi Pharmaceutical Co., Ltd.) for their valuable comments and continuing encouragement.
Abbreviations
- AM
acetoxymethyl ester
- AVP
arginine vasopressin
- BSA
bovine serum albumin
- CHO
Chinese hamster ovary
- DMSO
dimethyl sulphoxide
- EDTA
ethylenediaminetetraacetic acid
- EGTA
ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′,-tetraacetic acid
- FCS
foetal calf serum
- HEPES
N-2-hydroxyethylpiperazine-N′-2-ethanesulphonic acid
- IBMX
3-isobutyl-1-methylxanthine
- MEM
minimum essential medium
- PBS
phosphate-buffered saline
- SIADH
syndrome of inappropriate antidiuretic hormone secretion
- SR 121463A
1-[4-(N-tert-butylcarbamoyl)-2-methoxybenzene sulphonyl]-5-ethoxy-3-spiro-[4-(2-morpholinoethoxy)cyclohexane]indol-2-one equatorial isomer
- SR 49059
(2S) 1-[(2R 3S)-5-chloro-3-(2-chlorophenyl)-1-(3,4-dimethoxybenzene-sulfonyl)-3-hydroxy-2,3-dihydro-1H-indole-2-carbonyl]-pyrrolidine-2-carboxamide
- USMC
uterine smooth muscle cells
- YM471
(Z)-4′-{4,4-difluoro-5-[2-(4-dimethylaminopiperidino)-2-oxoethylidene]-2,3,4,5-tetrahydro-1H-1-benzoazepine-1-carbonyl}-2-phenylbenzanilide monohydrochloride
References
- ABRAHAM W., KOREN M., BICHET D.G., VERBALIS J.G., KLAPHOLZ M., SELARU P., BAKKER-ARKEMA R.G., RUMMEL S.A. Treatment of hyponatremia in patients with SIADH or CHF with intravenous conivaptan (YM087), a new combined vasopressin V1A/V2 receptor antagonist. Eur. Heart J. 2000;21:345. [Google Scholar]
- ABRAHAM W.T., SURESH D.P., WAGONER L.E., HAAS G.J., MCCORD J., RYDZINSKI S., NELSON C.B., BAKKER-ARKEMA R.G. Pharmacotherapy for hyponatremia in heart failure: effects of a new dual V1A/V2 vasopressin antagonist YM087. Circulation. 1999;100:I299. [Google Scholar]
- BIRNBAUMER M., SEIBOLD A., GILBERT S., ISHIDO M., BARBERIS C., ANTARAMIAN A., BRABET P., ROSENTHAL W. Molecular cloning of the receptor for human antidiuretic hormone. Nature. 1992;357:333–335. doi: 10.1038/357333a0. [DOI] [PubMed] [Google Scholar]
- BUTLEN D., GUILLON G., RAJERISON R.M., JARD S., SAWYER W.H., MANNING M. Structural requirements for activation of vasopressin-sensitive adenylate cyclase, hormone binding, and antidiuretic actions. Mol. Pharmacol. 1978;14:1006–1017. [PubMed] [Google Scholar]
- CHENG Y., PRUSOFF W.H. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (IC50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- FUJISAWA G., ISHIKAWA S., OKADA K., SAKUMA N., TSUBOI Y., SAITO T. Improvement by a non-peptide vasopressin antagonist OPC-31260 of water retention in experimental rats with myocardial infarction. J. Am. Soc. Nephrol. 1993a;4:852. [Google Scholar]
- FUJISAWA G., ISHIKAWA S., TSUBOI Y., OKADA K., SAITO T. Therapeutic efficacy of non-peptide ADH antagonist OPC-31260 in SIADH rats. Kidney Int. 1993b;44:19–23. doi: 10.1038/ki.1993.207. [DOI] [PubMed] [Google Scholar]
- GRYNKIEWICZ G., POENIE M., TSIEN R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- GUILLON G., BUTLEN D., CANTAU B., BARTH T., JARD S. Kinetic and pharmacological characterization of vasopressin membrane receptors from human kidney medulla: relation of adenylate cyclase activation. Eur. J. Pharmacol. 1982;85:291–304. doi: 10.1016/0014-2999(82)90216-3. [DOI] [PubMed] [Google Scholar]
- HOLDA J.R., OBERTI C., PEREZ-REYES E., BLATTER L.A. Characterization of an oxytocin-induced rise in [Ca2+]i in single human myometrium smooth muscle cells. Cell Calcium. 1996;20:43–51. doi: 10.1016/s0143-4160(96)90049-4. [DOI] [PubMed] [Google Scholar]
- HOWL J., ISMAIL T., STRAIN A.J., KIRK C.J., ANDERSON D., WHEATLEY M. Characterization of the human liver vasopressin receptor. Biochem. J. 1991;276:189–195. doi: 10.1042/bj2760189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JANS D.A., PETERS R., ZSIGO J., FAHRENHOLZ F. The adenylate cyclase-coupled vasopressin V2-receptor is highly laterally mobile in membranes in LLC-PK1 renal epithelial cells at physiological temperature. EMBO J. 1989;8:2481–2488. doi: 10.1002/j.1460-2075.1989.tb08384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JARD S., GAILLARD R.C., GUILLON G., MARIE J., SCHOENENBERG P., MULLER A.F., MANNING M., SAWYER W.H. Vasopressin antagonists allow demonstration of a novel type of vasopressin receptor in the rat adenohypophysis. Mol. Pharmacol. 1986;30:171–177. [PubMed] [Google Scholar]
- JASPER J.R., HARRELL C.M., O'BRIEN J.A., PETTIBONE D.J. Characterization of the human oxytocin receptor stably expressed in 293 human embryonic kidney cells. Life Sci. 1995;57:2253–2261. doi: 10.1016/0024-3205(95)02218-8. [DOI] [PubMed] [Google Scholar]
- LASZLO F.A., LASZLO F., Jr, DE WIED D. Pharmacology and clinical perspectives of vasopressin antagonists. Pharmacol. Rev. 1991;43:73–108. [PubMed] [Google Scholar]
- MATSUHISA A., TANIGUCHI N., KOSHIO H., YATSU T., TANAKA A. Nonpeptide arginine vasopressin antagonists for both V1A and V2 receptors: synthesis and pharmacological properties of 4-(1,4,5,6-tetrahydroimidazo[4,5-d][1]benzoazepine-6-carbonyl)benzanili de derivatives and 4′-(5,6-dihydro-4H-thiazolo[5,4-d][1]benzoazepine-6-carbonyl)benzanilid e derivatives. Chem. Pharm. Bull. 2000;48:21–31. doi: 10.1248/cpb.48.21. [DOI] [PubMed] [Google Scholar]
- MICHELL R.H., KIRK C.J., BILLAH M.M. Hormonal stimulation of phosphatidylinositol breakdown with particular reference to the hepatic effects of vasopressin. Biochem. Soc. Trans. 1979;7:861–865. doi: 10.1042/bst0070861. [DOI] [PubMed] [Google Scholar]
- MIZUSHIMA S., NAGATA S. pEF-BOS, a powerful mammalian expression vector. Nucleic Acids Res. 1990;18:5322. doi: 10.1093/nar/18.17.5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAITOH M., SUZUKI H., MURAKAMI M., MATSUMOTO A., ARAKAWA K., ICHIHARA A., NAKAMOTO H., OKA K., YAMAMURA Y., SARUTA T. Effects of oral AVP receptor antagonists OPC-21268 and OPC-31260 on congestive heart failure in conscious dogs. Am. J. Physiol. 1994;267:H2245–H2254. doi: 10.1152/ajpheart.1994.267.6.H2245. [DOI] [PubMed] [Google Scholar]
- NISHIMOTO G., ZELENINA M., LI D., YASUI M., APERIA A., NIELSEN S., NAIRN A.C. Arginine vasopressin stimulates phosphorylation of aquaporin-2 in rat renal tissue. Am. J. Physiol. 1999;276:F254–F259. doi: 10.1152/ajprenal.1999.276.2.F254. [DOI] [PubMed] [Google Scholar]
- PAINCHAUD C.A., GHAZZI M.M., SELARU P., BICHET D.G., CHARTIER K.K., UDELSON J.E. Urinary water clearance after infusion of conivaptan, a combined vasopressin V1a and V2 receptor antagonist, in patients with NYHA class III/IV heart failure. Circulation. 2000;102:II535. [Google Scholar]
- PETTIBONE D.J., KISHEL M.T., WOYDEN C.J., CLINESCHMIDT B.V., BOCK M.G., FREIDINGER R.M., VEBER D.F., WILLIAMS P.D. Radioligand binding studies reveal marked species differences in the vasopressin V1 receptor of rat, rhesus and human tissues. Life Sci. 1992;50:1953–1958. doi: 10.1016/0024-3205(92)90524-s. [DOI] [PubMed] [Google Scholar]
- SCATCHARD G. The attraction of proteins for small molecules and ions. Ann. N. Y. Acad. Sci. 1949;51:660–672. [Google Scholar]
- SERRADEIL-LE GAL C., HERBERT J.M., DELISEE C., SCHAEFFER P., RAUFASTE D., GARCIA C., DOL F., MARTY E., MAFFRAND J.P., LE FUR G. Effect of SR-49059, a vasopressin V1a antagonist, on human vascular smooth muscle cells. Am. J. Physiol. 1995;268:H404–H410. doi: 10.1152/ajpheart.1995.268.1.H404. [DOI] [PubMed] [Google Scholar]
- SERRADEIL-LE GAL C., LACOUR C., VALETTE G., GARCIA G., FOULON L., GALINDO G., BANKIR L., POUZET B., GUILLON G., BARBERIS C., CHICOT D., JARD S., VILAIN P., GARCIA C., MARTY E., RAUFASTE D., BROSSARD G., NISATO D., MAFFRAND J.P., LE FUR G. Characterization of SR 121463A, a highly potent and selective, orally active vasopressin V2 receptor antagonist. J. Clin. Invest. 1996;98:2729–2738. doi: 10.1172/JCI119098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SERRADEIL-LE GAL C., WAGNON J., GARCIA C., LACOUR C., GUIRAUDOU P., CHRISTOPHE B., VILLANOVA G., NISATO D., MAFFRAND J.P., LE FUR G., GUILLON G., CANTAU B., BARBERIS C., TRUEBA M., ALA Y., JARD S. Biochemical and pharmacological properties of SR 49059, a new, potent, nonpeptide antagonist of rat and human vasopressin V1a receptors. J. Clin. Invest. 1993;92:224–231. doi: 10.1172/JCI116554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIMADA Y., TANIGUCHI N., MATSUHISA A., SAKAMOTO K., YATSU T., TANAKA A. Highly potent and orally active non-peptide arginine vasopressin antagonists for both V1A and V2 receptors: synthesis and pharmacological properties of 4′-(4,4-difluoro-5-methylidene-2,3,4,5-tetrahydro-1H-1-benzoazepin-1-yl)carbonyl)-2-phenylbenzanilide derivatives. Chem. Pharm. Bull. 2000;48:1644–1651. doi: 10.1248/cpb.48.1644. [DOI] [PubMed] [Google Scholar]
- SUGIMOTO T., SAITO M., MOCHIZUKI S., WATANABE Y., HASHIMOTO S., KAWASHIMA H. Molecular cloning and functional expression of a cDNA encoding the human V1b vasopressin receptor. J. Biol. Chem. 1994;269:27088–27092. [PubMed] [Google Scholar]
- SURESH D.P., WAGONER L.E., HAAS G.J., McCORD J., RYDZINSKI S., NELSON C.B., BAKKER-ARKEMA R.G. Pharmacotherapy for hyponatremia in heart failure: effects of a new dual V1A/V2 vasopressin antagonist YM087. Circulation. 1999;100:1–299. [Google Scholar]
- TAHARA A., SAITO M., SUGIMOTO T., TOMURA Y., WADA K., KUSAYAMA T., TSUKADA J., ISHII N., YATSU T., UCHIDA W., TANAKA A. Pharmacological characterization of YM087, a potent, nonpeptide human vasopressin V1A and V2 receptor antagonist. Naunyn-Schmiedebergs Arch. Pharmacol. 1998a;357:63–69. doi: 10.1007/pl00005139. [DOI] [PubMed] [Google Scholar]
- TAHARA A., SAITO M., SUGIMOTO T., TOMURA Y., WADA K., KUSAYAMA T., TSUKADA J., ISHII N., YATSU T., UCHIDA W., TANAKA A. Pharmacological characterization of the human vasopressin receptor subtypes stably expressed in Chinese hamster ovary cells. Br. J. Pharmacol. 1998b;125:1463–1470. doi: 10.1038/sj.bjp.0702220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAHARA A., TOMURA Y., WADA K., KUSAYAMA T., TSUKADA J., ISHII N., YATSU T., UCHIDA W., TANAKA A. Effect of YM087, a potent nonpeptide vasopressin antagonist, on vasopressin-induced hyperplasia and hypertrophy of cultured vascular smooth-muscle cells. J. Cardiovasc. Pharmacol. 1997a;30:759–766. doi: 10.1097/00005344-199712000-00010. [DOI] [PubMed] [Google Scholar]
- TAHARA A., TOMURA Y., WADA K., KUSAYAMA T., TSUKADA J., TAKANASHI M., YATSU T., UCHIDA W., TANAKA A. Pharmacologic profile of YM087, a novel potent nonpeptide vasopressin V1A and V2 receptor antagonist, in vitro and in vivo. J. Pharmacol. Exp. Ther. 1997b;282:301–308. [PubMed] [Google Scholar]
- TAHARA A., TOMURA Y., WADA K., KUSAYAMA T., TSUKADA J., TAKANASHI M., YATSU T., UCHIDA W., TANAKA A. Characterization of vasopressin receptor in rat lung. Neuropeptides. 1998c;32:281–286. doi: 10.1016/s0143-4179(98)90049-x. [DOI] [PubMed] [Google Scholar]
- TAHARA A., TSUKADA J., ISHII N., TOMURA Y., WADA K., KUSAYAMA T., YATSU T., UCHIDA W., TANAKA A. Comparison of vasopressin binding sites in human uterine and vascular smooth muscle cells. Eur. J. Pharmacol. 1999;378:137–142. doi: 10.1016/s0014-2999(99)00403-3. [DOI] [PubMed] [Google Scholar]
- TAHARA A., TSUKADA J., TOMURA Y., WADA K., KUSAYAMA T., ISHII N., YATSU T., UCHIDA W., TANAKA A. Pharmacologic characterization of the oxytocin receptor in human uterine smooth muscle cells. Br. J. Pharmacol. 2000;129:131–139. doi: 10.1038/sj.bjp.0702996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAKEDA T., KUNO T., SHUNTOH H., TANAKA C. A rapid filtration assay for cAMP. J. Biochem. 1989;105:327–329. doi: 10.1093/oxfordjournals.jbchem.a122662. [DOI] [PubMed] [Google Scholar]
- TASAKA K., MASUMOTO N., MIYAKE A., TANIZAWA O. Direct measurement of intracellular free calcium in cultured human puerperal myometrial cells stimulated by oxytocin: effects of extracellular calcium and calcium channel blockers. Obstet. Gynecol. 1991;77:101–106. [PubMed] [Google Scholar]
- THIBONNIER M. Signal transduction of V1-vascular vasopressin receptors. Regul. Pep. 1992;38:1–11. doi: 10.1016/0167-0115(92)90067-5. [DOI] [PubMed] [Google Scholar]
- THIBONNIER M., AUZAN C., MADHUN Z., WILKINS P., BERTI-MATTERA L., CLAUSER E. Molecular cloning, sequencing, and functional expression of a cDNA encoding the human V1a vasopressin receptor. J. Biol. Chem. 1994;269:3304–3310. [PubMed] [Google Scholar]
- UDELSON J.E., SMITH W.B., HENDRIX G.H., PAINCHAUD C.A., GHAZZI M.M., THOMAS I., GHALI J.K., SELARU P., PRESSLER M.L., KONSTAM M.A. Hemodynamic effects of conivaptan hydrochloride (YM087, CI-1025) a combined vasopressin V1a and V2 receptor antagonist in patients with advanced heart failure. Circulation. 2000;102:II593. doi: 10.1161/hc4501.099313. [DOI] [PubMed] [Google Scholar]
- YAMAMURA Y., NAKAMURA S., ITOH S., HIRANO T., ONOGAWA T., YAMASHITA T., YAMADA Y., TSUJIMAE K., AOYAMA M., KOTOSAI K., OGAWA H., YAMASHITA H., KONDO K., TOMINAGA M., TSUJIMOTO G., MORI T. OPC-41061, a highly potent human vasopressin V2-receptor antagonist: pharmacological profile and aquaretic effect by single and multiple oral dosing in rats. J. Pharmacol. Exp. Ther. 1998;287:860–867. [PubMed] [Google Scholar]
- YAMAMURA Y., OGAWA H., CHIHARA T., KONDO K., ONOGAWA T., NAKAMURA S., MORI T., TOMINAGA M., YABUUCHI Y. OPC-21268, an orally effective, nonpeptide vasopressin V1 receptor antagonist. Science. 1991;252:572–574. doi: 10.1126/science.1850553. [DOI] [PubMed] [Google Scholar]
- YAMAMURA Y., OGAWA H., YAMASHITA H., CHIHARA T., MIYAMOTO H., NAKAMURA S., ONOGAWA T., YAMASHITA T., HOSOKAWA T., MORI T., TOMINAGA M., YABUUCHI Y. Characterization of a novel aquaretic agent, OPC-31260, as an orally effective, nonpeptide vasopressin V2 receptor antagonist. Br. J. Pharmacol. 1992;105:787–791. doi: 10.1111/j.1476-5381.1992.tb09058.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YASUI M., ZELEIN S.M., CELSI G., APERIA A. Adenylate cyclase-coupled vasopressin receptor activates AQP2 promoter via a dual effect on CRE and AP1 elements. Am. J. Physiol. 1997;272:F443–F450. doi: 10.1152/ajprenal.1997.272.4.F443. [DOI] [PubMed] [Google Scholar]
- YATSU T., TOMURA Y., TAHARA A., WADA K., KUSAYAMA T., TSUKADA J., TOKIOKA T., UCHIDA W., INAGAKI O., IIZUMI Y., TANAKA A., HONDA K. Cardiovascular and renal effects of conivaptan hydrochloride (YM087), a vasopressin V1A and V2 receptor antagonist, in dogs with pacing-induced congestive heart failure. Eur. J. Pharmacol. 1999;376:239–246. doi: 10.1016/s0014-2999(99)00379-9. [DOI] [PubMed] [Google Scholar]