

Abstract
Authentic nitric oxide (NO; 0.1–10 μmoles) caused transient, dose-dependent relaxation of phenylephrine-induced tone without changing membrane potential in mesenteric arteries. Larger doses, above 10 μmoles, did not evoke more relaxation (maximal relaxation to 150 μmoles NO in denuded arteries, 69±7%, n=8) but stimulated muscle hyperpolarization (maximum 19±3 mV, n=5).
The soluble guanylyl cyclase inhibitor, 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ; 10 μM), abolished relaxation to low doses of NO (n=4), but did not modify hyperpolarization with higher doses of NO (n=4). The potassium channel blocker charybdotoxin (ChTX; 50 nM) abolished hyperpolarization to high doses of NO and significantly reduced the maximal relaxation (to 43±6%, n=4; P<0.01). ODQ and ChTX together abolished tension and membrane potential change to all doses of NO (n=4).
All relaxations to 3-morpholino-sydnonimine (SIN-1; 0.01–10 μM) were associated with hyperpolarization. When the endothelium was intact, ChTX inhibited hyperpolarization and relaxation to SIN-1 (n=5), while iberiotoxin (IbTX; 50 nM) or 4-aminopyridine (4-AP; 500 μM) reduced relaxation by 40% and 20%, respectively and by 80% in combination (n=6 in each case).
In denuded arteries, relaxation to SIN-1 was unaffected by either ChTX or ODQ alone, but abolished by the inhibitors together (n=6). Alone, 4-AP did not alter relaxation, but in the presence of ODQ it reduced the maximal response by around 45% (n=6; P<0.01). 4-AP, ODQ and IbTX together inhibited relaxation to SIN-1 by 75% (n=6; P<0.01).
Therefore, cyclic guanosine 3′,5′-monophosphate (cyclic GMP)-independent smooth muscle hyperpolarization, possibly involving direct activation of calcium-activated and voltage-sensitive potassium channels, contributes to relaxation evoked by authentic NO and SIN-1. However, the importance of each pathway depends on the source of NO and with SIN-1 the relative contribution from each pathway is modified by the endothelium.
Keywords: Nitric oxide, potassium channels, soluble guanylyl cyclase, vascular smooth muscle
Introduction
Smooth muscle relaxation to nitric oxide (NO) is thought to be explained by the activation of soluble guanylyl cyclase leading to an increase in cytoplasmic concentrations of cyclic guanosine-3′,5′-monophosphate (cyclic GMP; Ignarro, 1991). cyclic GMP then activates specific cyclic GMP-dependent protein kinases, which act in a number of ways to decrease the cytoplasmic calcium concentration (Cornwell et al., 1991) and reduce the sensitivity of the contractile myofilaments (Tran et al., 1998).
One effect of cyclic GMP is to activate charybdotoxin (ChTX)-sensitive potassium channels, leading to hyperpolarization of the smooth muscle membrane potential and reducing calcium influx through voltage-sensitive channels (Robertson et al., 1993). In addition, authentic NO has been shown to activate directly ChTX-sensitive potassium channels in isolated smooth muscle cells from a number of vessels, including rabbit aorta and rat mesenteric arteries (Bolotina et al., 1994; Mistry & Garland, 1998). Therefore, it is not surprising that NO can evoke smooth muscle hyperpolarization in arterial smooth muscle (Tare et al., 1990; Garland & Mcpherson, 1992; Murphy & Brayden, 1995), and that in a number of preparations, relaxation to NO can be inhibited with ChTX (Khan et al., 1993; Bolotina et al., 1994; Archer et al., 1995, Plane et al., 1996; Cohen et al., 1997).
The extent to which smooth muscle relaxation follows the activation of potassium channels by NO, and the involvement of cyclic GMP, is not clear. It may in fact vary both with the arterial preparation and the source of the NO. In the rabbit isolated carotid artery, authentic NO and the NO donors, 3-morpholino-sydnonimine (SIN-1) and S-nitroso-N-acetylpenicillamine, evoked smooth muscle relaxation and hyperpolarization with a similar potency. In each case, inhibition of membrane hyperpolarization significantly reduced relaxation. However, the contribution of cyclic GMP to the changes in membrane potential and tension differed, such that inhibiting the generation of cyclic GMP effectively abolished both the hyperpolarization and relaxation to the NO donors, while the equivalent responses to authentic NO were only reduced by about 40%. The persistent relaxation and hyperpolarization to NO was abolished with ChTX, suggesting it may directly activate the potassium channels (Cohen et al., 1997).
A very different mechanistic profile appears to exist in the rat isolated mesenteric artery. In this small resistance artery, bolus doses of authentic NO (0.1–1 μmol) reversibly hyperpolarized the smooth muscle resting potential via a glibenclamide-sensitive mechanism, but relaxed cells pre-stimulated with noradrenaline without a change in membrane potential (Garland & Mcpherson, 1992). In endothelium-intact segments of the mesenteric artery, the NO-donor, SIN-1, evoked smooth muscle relaxation which was abolished with ChTX, but was unaffected by the guanylyl cyclase inhibitor, 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ; Garthwaite et al., 1995; Plane et al., 1996). However, in mesenteric arteries denuded of endothelium, relaxation to SIN-1 was only partially inhibited with either ChTX or ODQ, but blocked completely when these two agents were applied simultaneously (Plane et al., 1996).
The aim of the present study was to extend these observations to investigate further the relative importance of a change in membrane potential to the relaxation in rat isolated mesenteric arteries stimulated with either authentic NO or the NO donor, SIN-1.
Methods
Tension measurements
Male Wistar rats (250–300 g) were stunned and then killed by cervical dislocation. Segments of third order branches of the superior mesenteric artery (D100=290±15 μm; n=40) were mounted in a Mulvany-Halpern myograph under a normalized tension as described previously (Plane et al., 1996). Briefly, the segments of artery were mounted between two tungsten wires (25 μm in diameter) and maintained in a static bath at 37°C in oxygenated Krebs buffer, containing indomethacin (2.8 μM).
Cumulative concentration-response curves were constructed to SIN-1 in arterial segments depolarized and constricted with phenylephrine (1–3 μM). Authentic NO was applied as bolus doses. In some experiments, the endothelial cell layer was removed by gently rubbing the intimal surface with a hair and successful removal of the endothelium confirmed by the absence of relaxation to acetylcholine (1 μM). In all experiments, the concentration of phenylephrine was adjusted to give a similar level of tone in the presence and absence of inhibitors (mean depolarization and contraction: 28±5 mV (n=10 cells from six preparations) and 15±3 mN (n=14), respectively).
Electrophysiology
Measurement of smooth muscle membrane potential was made with a glass microelectrode advanced through the adventitial surface of the arterial segment. The electrodes were back filled with 2 M KCl and had resistances of 60–120 MΩ. Membrane electrical events were recorded through a high impedance d.c. pre-amplifier (Neurolog 102G) and together with data from the isometric force transducer, stored on disc (MacLab, AD Instruments, Hastings, U.K.).
Solutions and drugs
Tissues were maintained in Krebs buffer of the following composition (mM): NaCl 119.0, NaHCO3 25.0, KCl 4.7, MgSO4 1.2, KH2PO4 1.18, glucose 11, disodium EDTA 0.027 and CaCl2 2.5. All drugs were from Sigma except for SIN-1 (Tocris), ODQ (Tocris), Iberiotoxin (IbTX; Calbiochem) and ChTX (Calbiochem). All drugs were dissolved in Krebs buffer except for ODQ which was dissolved in DMSO, and indomethacin which was dissolved in 2% Na2CO3.
Preparation of NO solutions
Solutions of NO were prepared by injecting research grade NO gas (BDH) into de-gassed Krebs buffer as described previously (Plane et al., 1998). NO solutions were injected in to the myograph (bath volume 10 mls) close to the segments of artery and in volumes of less than 250 μl. Control injections of Krebs solution were made to assess the extent of any injection artefacts.
Analysis of data
Arterial relaxation is expressed as a percentage decrease in the phenylephrine-induced contraction and smooth muscle hyperpolarization either in mV or as a percentage reversal of phenylephrine-induced depolarisation. All data are expressed as mean±s.e.mean and the significance of differences between mean values calculated using the paired Students t-test.
Results
Membrane potential and tension responses to authentic NO solutions
Bolus additions of authentic NO (0.1–10 μmoles) caused transient, dose-dependent relaxation in arterial segments pre-stimulated with phenylephrine (1–3 μM), without altering significantly the smooth muscle membrane potential. In endothelium-denuded arteries, a maximum reversal of contraction (70±7%; n=8) was achieved with 10 μmoles of authentic NO. With higher doses of NO, up to 150 μmoles, the amplitude of NO-evoked relaxation did not increase but smooth muscle hyperpolarization was now evident. The maximal relaxation and hyperpolarization obtained with 150 μmoles of NO was 69±7% (n=8) and 19±3 mV (n=5), respectively. Representative traces showing simultaneous changes in membrane potential and tension to bolus doses of NO in an endothelium-denuded arterial segment are shown in Figure 1a.
Figure 1.
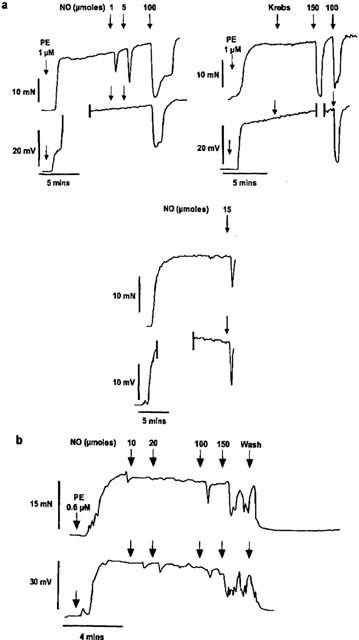
Representative traces showing simultaneous recording of changes in smooth muscle tension (mN) and membrane potential (mV) to bolus doses of authentic NO (1–150 μmoles) in an endothelium-denuded artery segments pre-stimulated with phenylephrine (1 μM) in the absence (a) and presence (b) of ODQ (10 μM). In the presence of ODQ, the concentration of phenylephrine was reduced to 0.6 μM to ensure that a comparable level of pre-contraction and depolarization was achieved. Vertical lines denote loss of electrode impalement.
Application of ODQ (10 μM; 10 min), an inhibitor of soluble guanylyl cyclase, abolished the relaxation to lower doses of NO (0.1–5 μmoles; n=4), and significantly reduced relaxation to higher doses (maximal relaxation to 150 μmoles 50±5%; n=5; P<0.01). However, the hyperpolarizations which accompanied relaxations to 10–150 μmoles of NO were not altered by this inhibitor (14±2 mV; n=3; P>0.05). Using radioimmunoassay, this concentration of ODQ has been shown to abolish completely SIN-1-evoked formation of cyclic GMP in this artery (Plane et al., 1996). Representative traces showing simultaneous changes in membrane potential and tension to NO in an endothelium-denuded arterial segment in the presence of ODQ are shown in Figure 1b and mean dose-response curves showing the effect of ODQ on relaxation and hyperpolarization to authentic NO are shown in Figure 2 a and b, respectively. In the presence of ODQ, the contraction to phenylephrine increased somewhat, although the variability in this effect meant the change was not significant (n=8, data not shown). However, the concentration of phenylephrine was varied to ensure the contraction matched that obtained in the absence of ODQ
Figure 2.
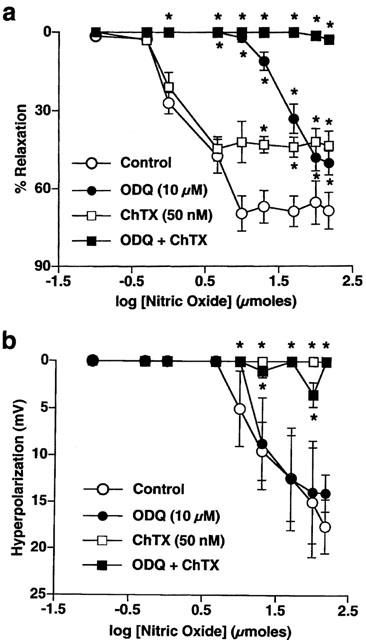
Mean dose-response curves for NO (0.1–150 μmoles)-evoked relaxation (a) and hyperpolarization (b) in the absence and presence of ODQ (10 μM) and ChTX (50 nM) alone and in combination. All points are the mean of 3–8 observations with s.e.means shown by vertical lines. *P<0.01 compared to control values.
The potassium channel inhibitor ChTX (50 nM; 10 min) abolished hyperpolarization to higher doses of NO and significantly blunted the accompanying relaxations such that the maximum relaxation to 150 μmoles of NO was only 43±6% (n=4; P<0.01). In contrast, relaxations to lower doses were largely unaffected (n=4). In the presence of ODQ and ChTX together, tension and membrane potential responses to all doses of NO were abolished (n=4). Mean dose-response curves showing the effect of ChTX, alone and with ODQ, on relaxation and hyperpolarization to authentic NO are shown in Figure 2a and b, respectively.
Bolus additions of NO also evoked transient, dose-dependent relaxations of phenylephrine-induced tone in endothelium-intact arterial segments, although the threshold dose was higher (1 μmole compared to 0.1 μmole) than in denuded arteries (n=7). As in denuded vessels, relaxation of intact tissues to higher doses of NO (above 10 μmoles) was accompanied by membrane hyperpolarization, the maximal changes in tension and membrane potential being similar to those observed in the absence of a functional endothelial cell layer (75±8%, n=5 and 18±3 mV, n=3, respectively).
The effect of ODQ (10 μM) and ChTX (50 nM) on responses to authentic NO in endothelium-intact tissues was identical to denuded arteries. ODQ abolished relaxation to lower doses of NO and significantly reduced the maximal relaxation to 45±6% (n=5). Pre-incubation with ChTX abolished both the hyperpolarization to doses of NO above 10 μmoles (n=4) and reduced the maximal relaxation to 150 μmoles to 23±7% (n=4). Application of ChTX and ODQ together abolished responses to all doses of NO (n=5).
Membrane potential and tension responses to SIN-1
The NO donor, SIN-1 (0.01–10 μM), elicited concentration-dependent, slow hyperpolarization and relaxation in both endothelium-intact and -denuded arteries pre-stimulated with phenylephrine (1–3 μM). There was no significant difference in the threshold concentration (0.03 μM) or EC50 values for SIN-1-evoked changes in membrane potential and tension between intact (EC50 values; 0.11±0.05 μM, n=5 and 0.13±0.05 μM, n=3, respectively), and denuded arteries (EC50 values; 0.10±0.04 μM, n=7 (P>0.05) and 0.11±0.05 μM, n=3, (P>0.05), respectively). Representative traces illustrating simultaneous changes in membrane potential and tension to cumulative additions of SIN-1 (0.01–10 μM) in an endothelium-intact arterial segment are shown in Figure 3a.
Figure 3.
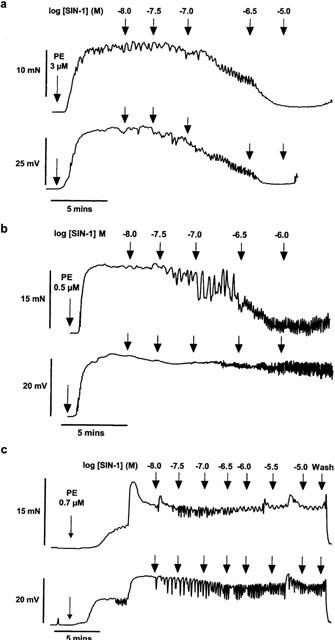
(a) Representative traces illustrating simultaneous changes in tension (mN) and membrane potential (mV) to cumulative additions of SIN-1 (0.01–10 μM) in an endothelium-intact arterial segment. (b) Representative traces illustrating simultaneous recordings of changes in tension and membrane potential to cumulative additions of SIN-1 (0.01–10 μM) in an endothelium-denuded arterial segment in the presence of ChTX alone and (c) in combination with ODQ.
In intact arterial segments, hyperpolarization and relaxation to SIN-1 was abolished by prior exposure to ChTX (50 nM; 10 min; n=4). In contrast, in endothelium-denuded tissues, although hyperpolarization evoked by SIN-1 was abolished by pre-incubation with ChTX (n=3), the relaxation was unaffected either by ChTX (n=6) or by ODQ (10 μM; 10 min; n=4) alone. However, following pre-incubation with ChTX and ODQ together, both the sustained hyperpolarization (n=4) and relaxation (n=7) of endothelium-denuded arterial segments in response to SIN-1 were abolished although small oscillations in both membrane potential and tension were still observed. Representative traces illustrating simultaneous recordings of changes in membrane potential and tension to cumulative additions of SIN-1 (0.01–10 μM) in an endothelium-denuded arterial segment in the presence of ChTX, alone and in combination with ODQ, are shown in Figure 3b and c. Mean concentration-response curves showing the effect of ChTX and ODQ, alone and in combination, on relaxation and hyperpolarization to SIN-1 in denuded vessels are shown in Figure 4a and b, respectively.
Figure 4.
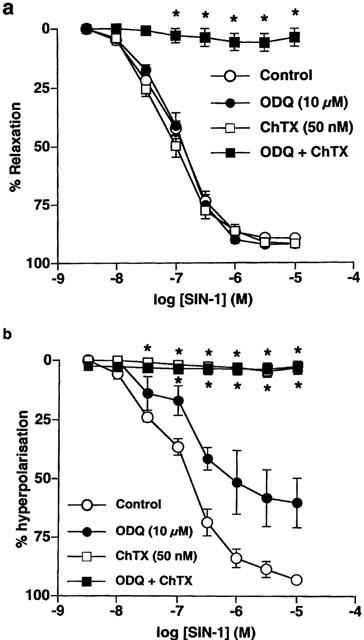
Mean concentration-response curves for SIN-1-evoked relaxation (a) and hyperpolarization (b) in endothelium-denuded arteries in the absence and presence of ODQ (10 μM) and ChTX (50 nM) alone and in combination. All points are the mean of 4–5 observations with s.e.means shown by vertical lines. *P<0.01 compared to control values.
Application of either IbTX (50 nM) or 4-aminopyridine (4-AP; 500 μM) alone reduced the maximum SIN-1-evoked relaxation in endothelium-intact strips by around 40% and 20% (n=6; P<0.01 in each case), respectively. Combination of the two inhibitors reduced the maximal response by around 80% to 15±5% (n=6; P<0.01). Mean concentration-response curves showing the effect of IbTX and 4-AP, alone and in combination, on relaxation to SIN-1 in endothelium-intact vessels are shown in Figure 5a.
Figure 5.
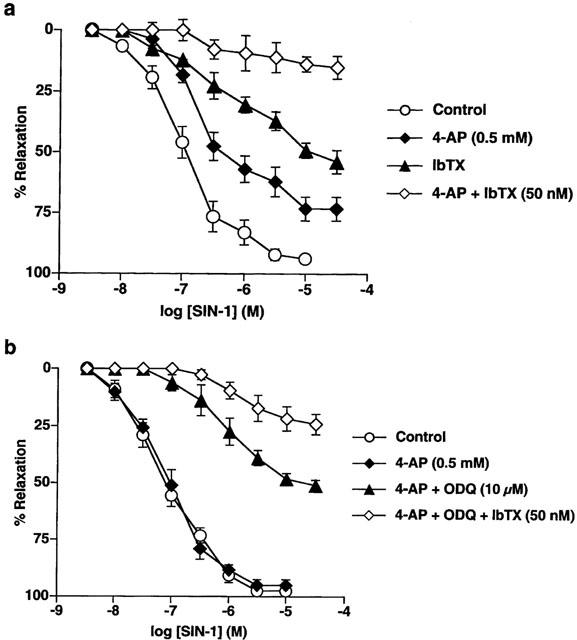
Mean concentration-response curves for SIN-1-evoked relaxation in (a) endothelium intact arteries where relaxation was abolished by either 4-AP (0.5 mM) or IbTX (50 nM) alone, an action which was additive. (b) In denuded arteries, 4-AP was without effect unless it was combined with ODQ or ODQ and IbTX. All points are the means of six observations with the s.e.mean shown by the vertical lines.
In endothelium-denuded arteries, IbTX, either in the presence or absence of ODQ, did not significantly inhibit relaxation to SIN-1 (n=4 in each case). In contrast, although application of 4-AP alone did not alter relaxations to SIN-1 in endothelium-denuded tissues (n=6), in the presence of ODQ, exposure to 4-AP did significantly inhibit relaxation to the NO donor, reducing the maximal response by around 45% to 51±2% (n=6; P<0.01). Furthermore, in the continued presence of ODQ, addition of both 4-AP and IbTX caused a further attenuation of relaxation reducing the maximal relaxation to SIN-1 by around 75% to 24±4.5% (n=6; P<0.01). Mean concentration-response curves showing the effect of IbTX and 4-AP, alone and in combination, on relaxation to SIN-1 in denuded arteries are shown in Figure 5b.
Discussion
The major novel observations in the present study are that smooth muscle hyperpolarization contributes to relaxation with both authentic NO and SIN-1 in the rat isolated small mesenteric artery and that both BKCa and KDR channels may play a role in this response. The hyperpolarization appears to be independent of cyclic GMP, and presumably involves a direct activation of the potassium channels. These data also indicate that the contribution of this pathway depends on the source of NO. In the case of SIN-1, the mechanism driving relaxation can be modified by the endothelium.
A previous study in the rat mesenteric artery, showed that authentic NO (0.1–1 μmoles) could stimulate a glibenclamide-sensitive hyperpolarization of the smooth muscle cell resting membrane potential. However, when the arteries were depolarized and contracted with noradrenaline, no change in membrane potential was observed (Garland & Mcpherson, 1992). Our data indicate that although low doses of authentic NO evoke relaxation of rat isolated mesenteric arteries through the formation of cyclic GMP with no change in membrane potential, higher doses of NO (10–150 μM) can cause significant smooth muscle hyperpolarization in phenylephrine-stimulated arteries. This change in membrane potential can make a significant contribution to relaxation to high doses of NO. The change was blocked with ChTX but not with ODQ, indicating a direct cyclic GMP-independent activation of ChTX-sensitive potassium channels.
A similar relationship to concentration of authentic NO and membrane hyperpolarization has been reported in other vessels such as rabbit femoral and guinea-pig uterine arteries (Tare et al., 1990; Plane et al., 1995). In contrast, in the rabbit carotid artery relaxation to all concentrations of NO was associated with smooth muscle hyperpolarization (Plane et al., 1998). At the other extreme, NO failed to evoke a significant change in the smooth muscle membrane potential of either stimulated or unstimulated segments of the rabbit basilar artery, even with concentrations approaching 150 μM (Plane & Garland, 1993). These observations may reflect a wide variation in the sensitivity of target potassium channels to NO, or in the mechanism available for contraction in these different vessels. In smaller arteries, such as the mesenteric, agonist-induced contraction is known to depend largely upon membrane depolarization and the entry of extracellular calcium through voltage-dependent calcium channels (Nilsson, 1998). Thus, relaxation may reflect a greater influence of hyperpolarization on calcium entry through voltage-sensitive channels, compared with other arteries where voltage-independent mechanisms predominate. Also, ChTX-sensitive channels are voltage-dependent, open probability increasing with depolarization. Therefore, in vessels such as the mesenteric artery where contraction is closely linked to smooth muscle membrane potential, agonist-induced depolarization would be expected to increase the open probability of ChTX-sensitive channels and presumably therefore enhance their sensitivity to modulation by NO.
In contrast to the dose-dependent effects of authentic NO on smooth muscle membrane potential, relaxation in the rat mesenteric artery to all concentrations of the NO donor SIN-1 were accompanied by smooth muscle hyperpolarization. Changes in membrane potential to SIN-1 were not modified by the soluble guanylyl cyclase inhibitor, ODQ, but were inhibited with ChTX. This indicates that, as with authentic NO, hyperpolarization to SIN-1 may involve a direct activation of ChTX-sensitive potassium channels.
ChTX can inhibit large-conductance calcium-activated potassium channels (BKCa) and thus these findings extend and lend a functional significance to our recent observations on single smooth muscle cells from the same artery, where authentic NO and SIN-1 both appeared to activate ChTX-sensitive BKCa channels independently of cyclic GMP formation (Mistry & Garland, 1998). How this activation occurs is not clear, but NO, or a reactive intermediate, has been suggested to increase BKCa channel activity by nitrosylating sulphydryl groups on the channels or a closely associated protein (Bolotina et al., 1994; Ahern et al., 1999; Lang et al., 2000). So a variation in the ability of NO to activate potassium channels in different arteries may reflect the expression of a range of channel sub-units or of accessory proteins.
In addition to BKCa, ChTX can also inhibit both intermediate conductance calcium-activated potassium channels (IKCa; Brugnara et al., 1995) and voltage-sensitive, delayed rectifier potassium channels (KDR; Kaczorowski et al., 1996). Thus, the fact that relaxation to SIN-1 was less sensitive to IbTX, a selective inhibitor of BKCa, than to ChTX, may indicate a role for other potassium channels in the response to NO. To date, IKCa has only been reported in proliferating vascular smooth muscle cells and a role for these channels in contractile cells has yet to be demonstrated (Neylon et al., 1999). However, IKCa may well be present on endothelial cells (Edwards et al., 1998). This localization may help to explain the endothelium-dependent component to the action of SIN-1.
Previous studies have shown that 4-AP, can reduce NO-induced dilatation in pulmonary and umbilical arteries, and that cyclic GMP-independent activation of KDR channels may contribute to NO-evoked responses in these vessels (Zhao et al., 1997; Lovren & Triggle, 2000). In the present study, application of 4-AP alone, at a concentration selective for inhibition of KDR, attenuated relaxations to SIN-1 in endothelium-intact strips but was without effect in denuded tissues. However, in both endothelium-intact and denuded arteries, the combination of 4-AP and IbTX caused a similar level of inhibition of SIN-1-evoked relaxations to that observed with ChTX. These data indicate that the inhibitory effects of ChTX on relaxation to NO in mesenteric arteries may be due to an action on both BKCa and KDR channels. Furthermore, in endothelium-denuded tissues, the inhibitory effects of 4-AP and IbTX were observed in the presence of ODQ indicating that the stimulatory effect of NO on these channels may occur via a direct, cyclic GMP-independent mechanism.
The molecular identity of the channels that contribute to KDR currents in mesenteric artery smooth muscle cells is unclear. The pore-forming sub-unit Kv1.5 is widely expressed in vascular tissue, including mesenteric arteries (Xu et al., 1999), and is thought to contribute to KDR currents in vascular smooth muscle cells isolated from a number of vessels (Overturf et al., 1994; Clément-Chomienne et al., 1999). Although sensitive to low concentrations of 4-AP (IC50 around 200 μM; Overturf et al., 1994; Clément-Chomienne et al., 1999), Kv1.5 homomultimeric channels, are resistant to inhibition by ChTX and the presence of one Kv1.5 sub-unit can confer resistance to the toxin on heteromultimeric channels (Russell et al., 1994). However, there is now evidence for the expression of other ChTX-sensitive pore-forming sub-units in vascular smooth muscle cells and, although the ChTX-sensitivity of KDR currents in rat mesenteric artery smooth muscle cells has yet to be investigated, expression of both Kv1.2 and Kv1.3 channel proteins in smooth muscle cells from this artery has recently been demonstrated (Xu et al., 1999).
In the rabbit carotid artery, the ability of authentic NO and SIN-1 to cause hyperpolarization and relaxation was similar in both endothelium-intact and denuded arteries (Plane et al., 1998). This is consistent with a number of other studies, predominantly involving larger arteries, in which no differences were reported between the relaxant actions of authentic NO in intact and denuded preparations (Huang et al., 1988; Tare et al., 1990). In contrast, the present data from the rat mesenteric artery reveal that a functional endothelium in some way decreased the ability of authentic NO to evoke hyperpolarization and relaxation. This characteristic was not shared with SIN-1, which induced responses in intact and denuded vessels with equal potency. However, the importance of the change in membrane potential to the relaxation induced with SIN-1 could be modulated by the endothelium. In endothelium-intact segments, the hyperpolarization underpinned relaxation, whereas in endothelium-denuded arteries full relaxation could still be achieved without a change in membrane-potential. In the rabbit mesenteric artery, endothelium-dependent modulation has also been described with NO-induced responses. Smooth muscle hyperpolarization to either SIN-1 or sodium nitroprusside was only observed in the absence of the endothelium or if the generation of endothelium-derived NO was inhibited (Murphy & Brayden, 1995). The authors suggested that this might reflect a desensitization of the hyperpolarizing response by the endogenous release of NO.
Our data suggest this is not the case in rat mesenteric arteries. In endothelium-intact vessels, relaxation to SIN-1 can be almost totally accounted for by cyclic GMP-independent, ChTX-sensitive hyperpolarization. Thus, it appears that an intact endothelial cell layer in some way reduces the ability of SIN-1 to stimulate relaxation via the membrane potential-independent, cyclic GMP-mediated pathway. Previous studies have shown that the basal release of endothelium-derived NO depresses the sensitivity of soluble guanylyl cyclase to the nitrovasodilators, sodium nitroprusside and SIN-1 (Busse et al., 1989; Moncada et al., 1991; Brandes et al., 2000). This effect can be reversed by acute inhibition of NO synthase (Brandes et al., 2000). However, our previous studies have demonstrated that SIN-1's ability to increase cyclic GMP is not significantly different in the presence or absence of basal NO release (Plane et al., 1996). This suggests that the endothelium-dependent modulation of the relaxation pathway occurs downstream of the formation of cyclic GMP, possibly at the level of the phosphodiesterases.
In conclusion, in rat small mesenteric arteries both authentic NO and SIN-1 can evoke smooth muscle hyperpolarization and relaxation, but with different characteristics. Authentic NO did evoke ChTX-sensitive smooth muscle hyperpolarization, but only in high concentrations. This was in contrast to the glibenclamide-sensitive hyperpolarization of the resting membrane potential in this artery evoked with low concentrations of NO (Garland & Mcpherson, 1992). However, SIN-1 evoked smooth muscle hyperpolarization in the same concentration-range as relaxation, and in the presence of an intact endothelium the change in membrane potential was the primary drive to relaxation. These differences in the response characteristics between authentic NO and NO generated from SIN-1 may reflect the action of additional breakdown products from SIN-1 (Feelisch & Stamler, 1996). As such, they emphasise the importance of using authentic NO in any investigation of the mechanisms responsible for smooth muscle relaxation.
Acknowledgments
This work was supported by a Programme Grant from the Wellcome Trust. L.J. Sampson was supported by an MRC studentship. F. Plane is currently a Wellcome Career Development Fellow.
Abbreviations
- 4-AP
4-aminopyridine
- BKCa
large-conductance calcium-activated potassium channels
- cyclic GMP
cyclic guanosine 3′,5′-monophosphate
- ChTX
charybdotoxin
- KDR
delayed rectifier potassium channels
- IbTX
iberiotoxin
- NO
nitric oxide
- ODQ
1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one
- SIN-1
3-morpholino-sydnonimine
References
- AHERN G.P., HSU S.-F., JACKSON M.B. Direct actions of nitric oxide on rat neurohypophysial K+ channels. J. Physiol. 1999;520:165–176. doi: 10.1111/j.1469-7793.1999.00165.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARCHER S.L., HUANG J.M.C., HAMPL V., NELSON D.P., SHULTZ P.J., WEIR E.K. Nitric oxide and cGMP cause vasorelaxation by activation of a charybdotoxin-sensitive potassium channel by cGMP-dependent protein kinase. Proc. Natl. Acad. Sci. U.S.A. 1995;91:7583–7587. doi: 10.1073/pnas.91.16.7583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOLOTINA V.M., NAJIBI S., PALACINO J.J., PAGANO P.J., COHEN R.A. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature. 1994;368:850–853. doi: 10.1038/368850a0. [DOI] [PubMed] [Google Scholar]
- BRANDES R.P., KIM D., SCHMITZ-WINNENTHAL F.H., AMIDI M., GODECKE A., MULSCH A., BUSSE R. Increased nitrovasodilator sensitivity in endothelial nitric oxide synthase knockout mice: role of soluble guanylyl cyclase. Hypertension. 2000;35:231–236. doi: 10.1161/01.hyp.35.1.231. [DOI] [PubMed] [Google Scholar]
- BRUGNARA C., ARMSBY C., DE FRANCESCHI L., CREST M., MARTIN EUCLAIRE M.-F., ALPER S. Ca2+-activated K+ channels of human and rabbit erythrocytes display distinctive patterns of inhibition by venom peptide toxins. J. Membr. Biol. 1995;147:71–82. doi: 10.1007/BF00235398. [DOI] [PubMed] [Google Scholar]
- BUSSE R., POHL U., MULSCH A., BASSENGE E. Modulation of the vasodilator action of SIN-1 by the endothelium. J. Cardiovasc. Pharmacol. 1989;14:S81–S85. doi: 10.1097/00005344-198906152-00015. [DOI] [PubMed] [Google Scholar]
- CLÉMENT-CHOMIENNE O., ISHII K., WALSH M.P., COLE W.C. Identification, cloning and expression of rabbit vascular smooth muscle Kv1.5 and comparison with native delayed rectifier K+ current. J. Physiol. 1999;515:653–667. doi: 10.1111/j.1469-7793.1999.653ab.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COHEN R.A., PLANE F., NAJIBI S., HUK I., MALINSKI T., GARLAND C.J. Nitric oxide is the mediator of both endothelium-dependent relaxation and hyperpolarisation of the rabbit coronary artery. Proc. Natl. Acad. Sci. 1997;94:4193–4198. doi: 10.1073/pnas.94.8.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CORNWELL T.L., PRYZWANSKY K.B., WYATT T.A., LINCOLN T.M. Regulation of the sarcoplasmic reticulum protein phosphorylation by localized cyclic GMP-dependent protein kinase in vascular smooth muscle cells. Mol. Pharmacol. 1991;40:923–931. [PubMed] [Google Scholar]
- EDWARDS G., DORA K.A., GARDENER M.J., GARLAND C.J., WESTON A.H. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- FEELISCH M., STAMLER J.S.Donors of nitrogen oxides Methods In Nitric Oxide Research 1996Chichester: John Wiley & Sons Ltd; 69–114.ed. Feelisch, M. & Stamler, J.S. pp [Google Scholar]
- GARLAND C.J., MCPHERSON G.A. Evidence that nitric oxide does not mediate the hyperpolarisation and relaxation to acetylcholine in the rat small mesenteric artery. Br. J. Pharmacol. 1992;105:429–435. doi: 10.1111/j.1476-5381.1992.tb14270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GARTHWAITE J., SOUTHAM E., BOULTON C.L., NIELSEN E.B., ACHMIDT K., MAYER B. Potent and selective inhibition of nitric oxide-sensitive guanylyl cyclase by 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one. Mol. Pharmacol. 1995;48:184–188. [PubMed] [Google Scholar]
- HUANG A.H., BUSSE R., BASSENGE E. Endothelium-dependent hyperpolarization of smooth muscle cells in rabbit femoral arteries is not mediated by EDRF (nitric oxide) Naunyn-Schmiedeberg's Archives of Pharmacology. 1988;338:438–442. doi: 10.1007/BF00172124. [DOI] [PubMed] [Google Scholar]
- IGNARRO L.J. Signal transduction mechanisms involving nitric oxide. Biochem. Pharmacol. 1991;41:485–490. doi: 10.1016/0006-2952(91)90618-f. [DOI] [PubMed] [Google Scholar]
- KACZOROWSKI G.J., KNAUS H.G., LEONARD R.J., MCMANUS O.B., GARCIA M.L. High-conductance calcium activated potassium channels; structure, pharmacology and function. J. Bioenerg. Biomembr. 1996;28:255–267. doi: 10.1007/BF02110699. [DOI] [PubMed] [Google Scholar]
- KHAN S.A., MATHEWS W.R., MEISHERI K.D. Role of calcium-activated K+ channels in vasodilation induced by nitroglycerine, acetylcholine and nitric oxide. J. Pharmacol. Exp. Thera. 1993;267:1327–1334. [PubMed] [Google Scholar]
- LANG R.J., HARVEY J.R., McPHEE G.J., KLEMM M.F. Nitric oxide and thiol reagent modulation of Ca2+-activated K+ (BKCa) channels in myocytes of the guinea-pig taenia caeci. J. Physiol. 2000;525:363–376. doi: 10.1111/j.1469-7793.2000.00363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOVREN F., TRIGGLE C. Nitric oxide and sodium nitroprusside-induced relaxation of the human umbilical artery. Br. J. Pharmacol. 2000;131:521–536. doi: 10.1038/sj.bjp.0703588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MISTRY D.K., GARLAND C.J. Nitric oxide (NO)-induced activation of large conductance Ca2+-dependent K+ channels (BKCa) in smooth muscle cells isolated from rat mesenteric artery. Br. J. Pharmacol. 1998;124:1131–1140. doi: 10.1038/sj.bjp.0701940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MONCADA S., REES D.D., SCHULZ R., PALMER R.M.J. Development and mechanism of a specific supersensitivity to nitrovasodilators after inhibition of vascular nitric oxide synthesis in vivo. Proc. Natl. Acad. Sci. U.S.A. 1991;88:2166–2170. doi: 10.1073/pnas.88.6.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MURPHY M.E., BRAYDEN J.E. Nitric oxide hyperpolarizes rabbit mesenteric arteries via ATP-sensitive potassium channels. J. Physiol. 1995;486:47–58. doi: 10.1113/jphysiol.1995.sp020789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NEYLON C.B., LANG R.J., FU Y., BOBIK A., REINHART P.H. Molecular cloning and characterization of the intermediate-conductance Ca2+-activated K+ channel in vascular smooth muscle: relationship between KCa channel diversity and smooth muscle cell function. Circ Res. 1999;85:e33–e43. doi: 10.1161/01.res.85.9.e33. [DOI] [PubMed] [Google Scholar]
- NILSSON H. Interactions between membrane potential and intracellular calcium concentration in vascular smooth muscle. Acta Physiol. Scand. 1998;164:559–566. doi: 10.1046/j.1365-201X.1998.00435.x. [DOI] [PubMed] [Google Scholar]
- OVERTURF K.E., RUSSELL S.N., CARL A., VOGALIS F., HART P.J., HUME J.R., SANDERS K.M., HOROWITZ B. Cloning and characterization of a Kv1.5 delayed rectifier K+ channel from vascular and visceral smooth muscles. Am. J. Physiol. 1994;267:C1231–C1238. doi: 10.1152/ajpcell.1994.267.5.C1231. [DOI] [PubMed] [Google Scholar]
- PLANE F., GARLAND C.J. Differential effects of acetylcholine, nitric oxide and levcromakalim on smooth muscle membrane potential and tone in the rabbit basilar artery. Br. J. Pharmacol. 1993;110:651–656. doi: 10.1111/j.1476-5381.1993.tb13861.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PLANE F., HURRELL A., JEREMY J.Y., GARLAND C.J. Evidence that potassium channels make a major contribution to SIN-1-evoked relaxation of rat isolated mesenteric artery. Br. J. Pharmacol. 1996;119:1557–1562. doi: 10.1111/j.1476-5381.1996.tb16072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PLANE F., PEARSON T., GARLAND C.J. Multiple pathways underlying endothelium-dependent relaxation in the rabbit isolated femoral artery. Br. J. Pharmacol. 1995;115:31–38. doi: 10.1111/j.1476-5381.1995.tb16316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PLANE F., WILEY K.E., JEREMY J.Y., COHEN R.A., GARLAND C.J. Evidence that different mechanisms underlie smooth muscle relaxation to nitric oxide and nitric oxide donors in the rabbit isolated carotid artery. Br. J. Pharmacol. 1998;123:1351–1366. doi: 10.1038/sj.bjp.0701746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROBERTSON B.E., SCHUBERT R., HESCHELER J., NELSON M.T. cGMP-dependent protein kinase activates Ca-activated K channels in cerebral artery smooth muscle cells. Am. J. Physiol. 1993;265:C299–C303. doi: 10.1152/ajpcell.1993.265.1.C299. [DOI] [PubMed] [Google Scholar]
- RUSSELL S.N., OVERTURF K.E., HOROWITZ B. Heterotetramer formation and charybdotoxin sensitivity of two K+ channels cloned from smooth muscle. Am. J. Physiol. 1994;267:C1729–C1733. doi: 10.1152/ajpcell.1994.267.6.C1729. [DOI] [PubMed] [Google Scholar]
- TARE M., PARKINGTON H.C., COLEMAN H.A., NEILD T.O., DUSTING G.J. Hyperpolarization and relaxation of arterial smooth muscle caused by nitric oxide derived from the endothelium. Nature. 1990;346:69–71. doi: 10.1038/346069a0. [DOI] [PubMed] [Google Scholar]
- TRAN N.N.P., SPITZBARTH E., ROBERT A., GIUMMELLY P., ATKINSON J., CAPDEVILLE-ATKINSON C. Nitric oxide lowers the calcium sensitivity of tension in the rat tail artery. J. Physiol. 1998;507:163–174. doi: 10.1111/j.1469-7793.1998.163bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XU C., LU Y., TANG G., WANG R. Expression of voltage-dependent K+ channel genes in mesenteric artery smooth muscle cells. Am. J. Physiol. 1999;277:G1055–G1063. doi: 10.1152/ajpgi.1999.277.5.G1055. [DOI] [PubMed] [Google Scholar]
- ZHAO Y.J., WANG J., RUBIN L.J., YUAN X.J. Inhibition of Kv and Kca channels antagonizes NO-induced relaxation in pulmonary artery. Am. J. Physiol. 1997;272:H904–H912. doi: 10.1152/ajpheart.1997.272.2.H904. [DOI] [PubMed] [Google Scholar]