

Abstract
Although adenosine analogues such as 5′-N-ethylcarboxamidoadenosine (NECA) relax the rat thoracic aorta in a partially endothelium-dependent manner via adenosine A2A receptors, others such as N6-R-phenylisopropyladenosine (R-PIA) act via an endothelium-independent, antagonist-insensitive mechanism. The role of cyclic nucleotides in these relaxations was investigated in isolated aortic rings using inhibitors of adenylate and guanylate cyclases as well as subtype-selective phosphodiesterase inhibitors.
The adenylate cyclase inhibitor 9-(tetrahydro-2-furanyl)-9H-purin-6-amine (SQ 22536; 100 μM) significantly inhibited responses to NECA, but not responses to R-PIA. The type IV (cyclic AMP-selective) phosphodiesterase inhibitor 4-[(3-butoxy-4-methoxyphenyl)methyl]-2-imidazolidinone (RO 20-1724; 30 μM) significantly enhanced responses to NECA and to a lesser extent those to R-PIA.
The guanylate cyclase inhibitor 1H-[1,2,4]oxadiazolo[4,3a]quinoxalin-1-one (ODQ; 100 μM) significantly inhibited responses to NECA and acetylcholine but not responses to R-PIA. The selective phosphodiesterase V (cyclic GMP-selective) inhibitors, zaprinast (10 μM) and 4-{[3′,4′-(methylenedioxy)benzyl]amino}-6-methoxyquinazoline (MMQ; 1 μM), had no significant effect on responses to either NECA or R-PIA, but enhanced responses to acetylcholine.
These results are consistent with the effects of NECA being via activation of endothelial receptors to release NO which stimulates guanylate cyclase, as well as smooth muscle receptors coupled to stimulation of adenylate cyclase. The lack of effect of zaprinast and MMQ on responses to NECA are likely to be due to simultaneous activation of both adenylate and guanylate cyclases in the smooth muscle, as cyclic AMP reduces the sensitivity of phosphodiesterase V to inhibitors. These results also suggest that the effects of R-PIA are via neither of these mechanisms.
Keywords: Rat aorta, adenosine, relaxations, cyclic nucleotides, nitric oxide, A2A receptors
Introduction
Adenosine is an endogenous vasodilator released during conditions of metabolic stress or hypoxia, and is thought to play an important role in matching blood supply to demand. Four G protein-coupled receptors for adenosine, A1, A2A, A2B and A3, have been cloned and characterized pharmacologically, and of these it is the A2A and A2B receptors which generally mediate vasodilation (see Collis & Hourani, 1993; Ralevic & Burnstock, 1998). However, in a number of blood vessels there is evidence for a vasodilator response to some adenosine analogues which is not mediated by any of the known receptors and is insensitive to adenosine antagonists (for review see Prentice, 2001). Such antagonist-resistant responses have been reported in the guinea-pig, rat, hamster and frog aorta (Collis & Brown, 1983; Lewis et al., 1994; Prentice & Hourani, 1996; 2000; Knight & Burnstock, 1996) and rat mesenteric artery (Prentice et al., 1997), as well as in some non-vascular tissues such as the guinea-pig trachea (Brackett & Daly, 1991) and taenia caeci (Prentice et al., 1995).
In the rat thoracic aorta for example the overall potency order for adenosine agonists of 5′-N-ethylcarboxamidoadenosine (NECA)>2-(p-(2-carboxyethyl)phenylethylamino)-5′-N-ethylcarboxamidoadenosine (CGS 21680)>N6-R-phenylisopropyladenosine (R-PIA)>N6-cyclopentyladenosine (CPA)⩾adenosine is consistent with the presence of an A2A receptor. However, responses to NECA are antagonized by the A1 and A2 antagonist 8-sulphophenyltheophylline (8-SPT) more powerfully than responses to CPA, and responses to R-PIA or to adenosine itself were unaffected (Lewis et al., 1994; Prentice & Hourani, 1996). Similar agonist-dependent inhibition was also seen with the selective A2A receptor antagonist 4-(2-[7-amino-2-(2-furyl)[1,2,4]-triazolo[2,3a][1,3,5]-triazin-5-ylamino]ethyl)phenol (ZM 241385), confirming that while the responses to NECA were via A2A receptors those to R-PIA were not (Prentice & Hourani, 1996). In addition, use of the NO synthase inhibitor NG-nitro-L-arginine methyl ester (L-NAME) or mechanical removal of the endothelium inhibited but did not abolish the responses to NECA, suggesting the presence of an A2A receptor on the endothelium acting to release NO as well as further A2A receptors on the smooth muscle, while responses to R-PIA were unaffected, suggesting that the mechanism or receptor through which R-PIA acts is located solely on the smooth muscle (Prentice & Hourani, 1996).
The A2A receptor is coupled via GS to stimulation of adenylate cyclase (although coupling to other effectors has also been suggested in some cases; see Ralevic & Burnstock, 1998), and it is a reasonable assumption that it causes vasodilation by increasing cyclic AMP levels. However, an endothelium-dependent mechanism suggests that cyclic GMP, formed as a result of NO release, may also be involved, and indeed, an increase in cyclic GMP but not of cyclic AMP in response to adenosine has been reported in the rat aorta (Moritoki et al., 1990). To investigate further the mechanism(s) by which NECA and R-PIA cause relaxation of the rat aorta and in particular the role of cyclic nucleotides in these responses, we used 9-(tetrahydro-2-furanyl)-9H-purin-6-amine (SQ 22536) and 1H-[1,2,4]oxadiazolo[4,3a]quinoxalin-1-one (ODQ) as inhibitors of adenylate and guanylate cyclase respectively, 4-[(3-butoxy-4-methyoxyphenyl)methyl]-2-imidazolidinone (RO 20-1724) as an inhibitor of the cyclic AMP-selective phosphodiesterase (PDE) IV and zaprinast and 4-{[3′,4′-(methylenedioxy)benzyl]amino}-6-methoxyquinazoline (MMQ) as inhibitors of the cyclic GMP-selective PDE V.
Methods
Male Wistar albino rats (Bantin and Kingman, Hull, U.K.) weighing approximately 200 – 250 g, were killed by cervical dislocation. The abdominal cavity was opened up and the thoracic aorta excised. Tissues were placed directly into Krebs – Henseleit solution (mM): NaCl, 118; KCl, 4.7; NaHCO3, 25; D-glucose, 11; MgSO4.7H2O, 0.45; K3PO4, 1.2; CaCl2.2H2O, 2.5. Excess fat and connective tissue were trimmed from the arteries which were then cut into rings approximately 3 mm long. The rings were carefully mounted between two stainless steel wires and suspended in 3.5 ml organ baths containing Krebs – Henseleit solution maintained at 37°C and continually gassed with 95% O2/CO2. Preparations were allowed to equilibrate for approximately 30 min under an initial resting tension of 1 g. Tissue viability was tested using 0.1 μM phenylephrine, a concentration which should elicit approximately 85% maximum contraction. All tissues were tested for the presence of functional endothelium using 1 μM acetylcholine to oppose the vasoconstriction induced by 0.1 μM phenylephrine. Tissues in which acetylcholine induced less than a 25% decrease in contraction were rejected. Tissues were washed several times with Krebs solution, incubated for a period of 60 min in the presence or absence of an inhibitor or the appropriate vehicle, then contracted again with phenylephrine and cumulative relaxant concentration response (E/[A]) curves constructed. Only one E/[A] curve was constructed in each preparation and tissues for the control and inhibitor-treated curves were taken from the same animal. Responses were recorded isometrically using Grass FT03 force displacement transducers and displayed on a Grass polygraph (model 79). Relaxant responses were expressed as a percentage decrease in the contraction to phenylephrine. Where possible midpoint location ([A50]), upper asymptote (α) and midpoint slope parameter estimates (nH) were obtained by logistic curve fitting (see Prentice et al., 1995) and for display purposes the average logistic fitting parameters were used to generate a line upon which the average data points were superimposed. The effect of drug treatment on the curve fitting parameters (slope, p[A50] and α) was assessed by one way analysis of variance, and P values of less than 0.05 were considered to be statistically significant. Data are presented as mean±s.e.mean of results obtained using tissues from at least three animals.
MMQ was obtained from Calbiochem, (Nottingham, U.K.), and all other drugs were obtained from Sigma-Aldrich Company, (Poole, U.K.). Zaprinast was made up in 100% dimethylsulphoxide (DMSO) at a stock concentration of 10−3 M, MMQ was made up in 50% DMSO/50% distilled water at a stock concentration of 10−3 M, ODQ was dissolved in 20% DMSO/80% distilled water at a stock concentration of 10−2 M, R-PIA in 0.06 M HCl at a stock concentration of 10−2 M and RO 20-1724 initially in a minimal volume of ethanol (10 μl) then made up in distilled water to give a stock concentration of 10−2 M (<1% ethanol). All other drugs were made up in distilled water at a stock concentration of 10−2 M, and further dilutions of all drugs were made in distilled water.
Results
At the concentrations used none of the inhibitors or the vehicles significantly affected either the basal tone of the blood vessel rings or the contractions to phenylephrine (0.1 μM) (results not shown).
The adenylate cyclase inhibitor SQ 22536 (100 μM) caused a significant rightward shift of the concentration-response curve to NECA (p[A]50=6.58±0.17 in the absence and 5.89±0.23 in the presence of SQ 22536, P<0.05) (Figure 1a), but did not inhibit the responses to R-PIA. If anything, a small leftward shift in the concentration-response curve to R-PIA was observed, but this was not statistically significant (p[A]50=5.01±0.16 in the absence and 5.39±0.32 in the presence of SQ 22536, P>0.05) (Figure 1b). Responses to adenosine were also unaffected by SQ 22536 (10 μM) (p[A]50=4.86±0.12 in the absence and 4.55±0.15 in the presence of SQ 22536, P>0.05) (Figure 1c).
Figure 1.
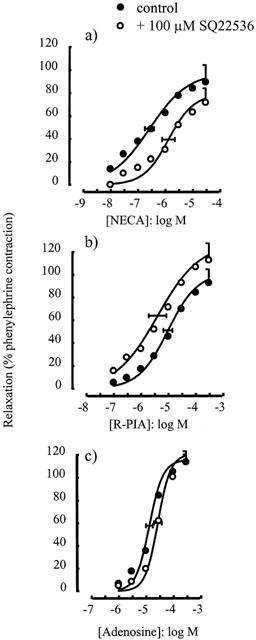
Relaxations of the rat isolated thoracic aorta induced by (a) NECA, (b) R-PIA and (c) adenosine alone or in the presence of SQ 22563 (100 μM). Data points are mean responses (per cent relaxation of contraction induced by 0.1 μM phenylephrine) and the curves through the data were generated by use of logistic fitting parameters. Average p[A]50 and α values are marked, together with their associated s.e.mean (n=4 – 7). For abbreviations see text.
The type IV phosphodiesterase inhibitor RO 20-1724 (30 μM) caused a significant leftward shift of the concentration-response curve to NECA (p[A]50=6.36±0.29 in the absence and 7.09±0.17 in the presence of RO 20-1724, P<0.05) (Figure 2a). RO 20-1724 also caused a small but significant leftward shift of the concentration-response curve to R-PIA (p[A]50=5.48±0.06 in the absence and 5.98±0.08 in the presence of RO 20-1724, P<0.05) (Figure 2b).
Figure 2.
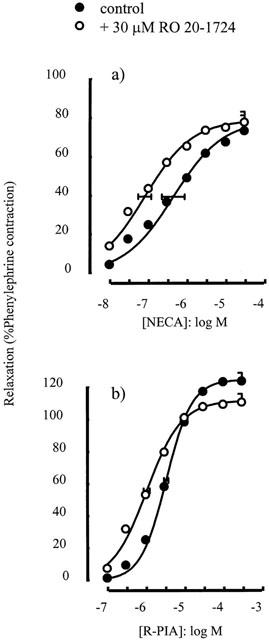
Relaxations of the rat isolated thoracic aorta induced by (a) NECA and (b) R-PIA alone or in the presence of RO 20-1724 (30 μM). Data points are mean responses (per cent relaxation of contraction induced by 0.1 μM phenylephrine) and the curves through the data were generated by use of logistic fitting parameters. Average p[A]50 and α values are marked, together with their associated s.e.mean (n=5 – 9). For abbreviations see text.
The guanylate cyclase inhibitor ODQ (100 μM) caused a significant rightward shift in the concentration-response curve to NECA (p[A]50=6.17±0.32 in the absence and 5.25±0.12 in the presence of ODQ, P<0.05) (Figure 3a) and greatly inhibited the response to acetylcholine (p[A]50=7.03±0.31 in the absence and 5.20±0.21 in the presence of ODQ, P<0.05) (Figure 3d) but did not affect the concentration-response curve to R-PIA (p[A]50=5.53±0.16 in the absence and 5.17±0.14 in the presence of ODQ, P>0.05) (Figure 3b) or to adenosine (p[A]50=5.24±0.26 in the absence and 4.87±0.13 in the presence of ODQ, P>0.05) (Figure 3c).
Figure 3.
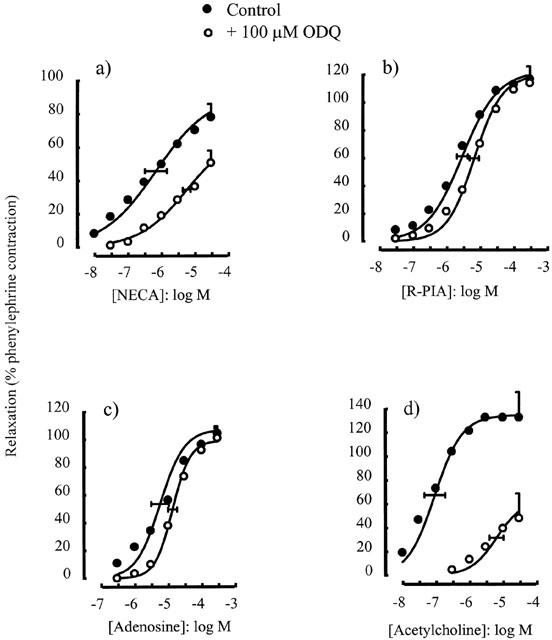
Relaxations of the rat isolated thoracic aorta induced by (a) NECA, (b) R-PIA, (c) adenosine and (d) acetylcholine alone or in the presence of ODQ (100 μM). Data points are mean responses (per cent relaxation of contraction induced by 0.1 μM phenylephrine) and the curves through the data were generated by use of logistic fitting parameters. Average p[A]50 and α values are marked, together with their associated s.e.mean (n=4 – 7). For abbreviations see text.
The selective inhibitor of phosphodiesterase V, zaprinast (10 μM), had no significant effect on the concentration-response curve for either NECA (p[A]50=6.34±0.23 in the absence and 6.45±0.29 in the presence of zaprinast, P>0.05) (Figure 4a) or R-PIA (p[A]50=5.57±0.05 in the absence and 5.27±0.13 in the presence of zaprinast, P>0.05) (Figure 4b), but enhanced responses to acetylcholine (α=75.0±8.3% in the absence and 122.9±15.1% in the presence of zaprinast, P<0.05) (Figure 4c) and sodium nitroprusside (p[A]50=8.80±0.06 in the absence and 9.72±0.06 in the presence of zaprinast, P<0.05) (Figure 4d). Similar results were observed with MMQ (1 μM), which had no significant effect on responses to either NECA (p[A]50=6.57±0.27 in the absence and 7.17±0.21 in the presence of MMQ, P>0.05) (Figure 5a) or to R-PIA (p[A]50=5.07±0.04 in the absence and 5.16±0.14 in the presence of MMQ, P>0.05) (Figure 5b) but enhanced responses to acetylcholine (p[A]50=5.94±0.10 in the absence and 7.35±0.05 in the presence of MMQ, P<0.05 and α=62.7±5.4% in the absence and 86.7±5.5% in the presence of MMQ, P<0.05) (Figure 5c).
Figure 4.
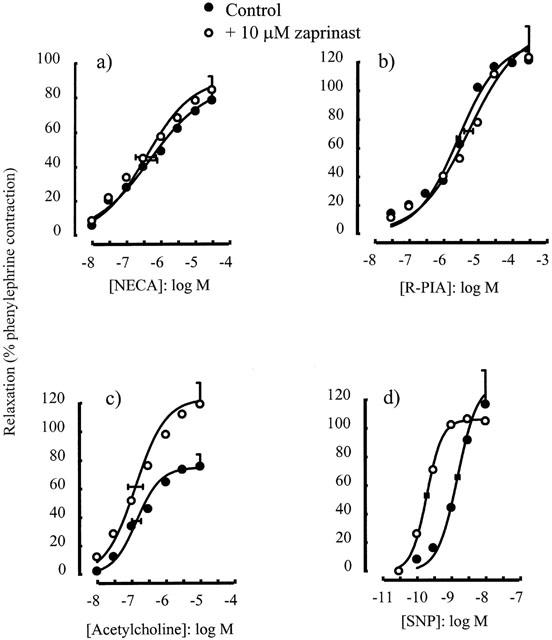
Relaxations of the rat isolated thoracic aorta induced by (a) NECA, (b) R-PIA, (c) acetylcholine and (d) SNP alone or in the presence of zaprinast (10 μM). Data points are mean responses (per cent relaxation of contraction induced by 0.1 μM phenylephrine) and the curves through the data were generated by use of logistic fitting parameters. Average p[A]50 and α values are marked, together with their associated s.e.mean (n=4 – 7). For abbreviations see text.
Figure 5.
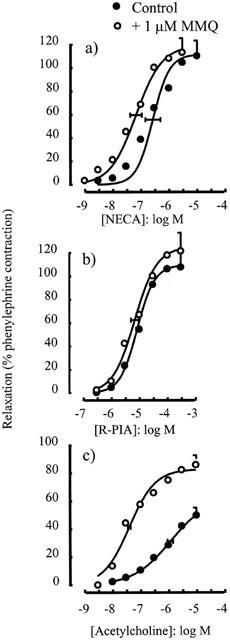
Relaxations of the rat isolated thoracic aorta induced by (a) NECA, (b) R-PIA and (c) acetylcholine alone or in the presence of MMQ (1 μM). Data points are mean responses (per cent relaxation of contraction induced by 0.1 μM phenylephrine) and the curves through the data were generated by use of logistic fitting parameters. Average p[A]50 and α values are marked, together with their associated s.e.mean (n=3 – 8). For abbreviations see text.
Discussion
The results presented here support a model whereby NECA acts through increases in both cyclic AMP and cyclic GMP, whereas the effects of R-PIA are largely independent of cyclic nucleotides. The inhibition by the adenylate cyclase inhibitor SQ 22536 of the responses to NECA shows that, as expected, the relaxations mediated via the A2A receptor are dependent on stimulation of adenylate cyclase, while the lack of effect of SQ 22536 suggests that stimulation of adenylate cyclase is not essential for relaxation induced by R-PIA, which we have previously shown to act in this tissue by an unknown mechanism independently of activation of adenosine A1, A2A, A2B or A3 receptors (Prentice & Hourani, 1996). That the responses to adenosine were also not blocked by SQ 22536 supports our previous finding that the vasodilations induced by this endogenous agonist in this tissue are also largely via an unknown mechanism which does not involve activation of the known cloned adenosine receptors (Prentice & Hourani, 1996). This result is also consistent with a previous study which showed no increase in cyclic AMP levels in rat thoracic aorta in response to adenosine (10 μM) (Moritoki et al., 1990). The enhancement by RO 20-1724 of the responses to NECA confirms the involvement of cyclic AMP in these responses, as this compound is an inhibitor of type IV phosphodiesterase, which selectively degrades cyclic AMP (see Beavo, 1995). This compound also slightly enhanced the responses to R-PIA, which indicates that although the major effect of R-PIA is not via the A2A receptor there may be a small A2A component to its action which is capable of being enhanced by RO 20-1724.
The inhibition by the guanylate cyclase inhibitor ODQ of relaxations induced by NECA confirms the suggestion (Prentice & Hourani, 1996) that this agonist acts at least partly via endothelial release of NO, which is known to activate guanylate cyclase within the vascular smooth muscle to cause relaxation. The responses to NECA were not inhibited to the same extent as those for acetylcholine, which acts entirely via the release of NO, confirming that NECA does not act exclusively on the endothelium but also has direct effects on the smooth muscle. The responses to R-PIA were unaffected by ODQ, confirming that this analogue does not act via the endothelium but has a different mechanism of action to NECA. Responses to adenosine were also unaffected, confirming our previous conclusions that this endogenous agonist acts via a non-endothelium-dependent mechanism (Prentice & Hourani, 1996), although Moritoki et al. (1990) reported an increase in cyclic GMP in the rat aorta in response to adenosine. However, this effect was reduced with age (Moritoki et al., 1990), and as our rats were adult (>8 weeks), this could explain the discrepancy.
The enhancement by zaprinast of the relaxations caused by the endothelium-dependent vasodilator acetylcholine or by the NO donor sodium nitroprusside confirmed the ability of this compound to enhance NO-mediated responses by inhibiting breakdown of cyclic GMP. The lack of effect of zaprinast on responses to R-PIA confirmed that NO release and the stimulation of guanylate cyclase are not involved in the relaxations caused by this analogue. However, the lack of effect of zaprinast on relaxation caused by NECA was unexpected, as our previous results (Prentice & Hourani, 1996) show that at least part of the response to NECA is endothelium-dependent and due to the release of NO, and the results obtained with ODQ showed that this is via stimulation of guanylate cyclase. A more selective inhibitor of phosphodiesterase V, MMQ, also failed to enhance responses to NECA while enhancing responses to acetylcholine. The most likely explanation for this is that NECA causes the concomitant elevation of both cyclic AMP and cyclic GMP in the vascular smooth muscle, the cyclic AMP elevation being a direct consequence of the activation of smooth muscle A2A receptors while the elevation of cyclic GMP is a result of NO release following activation of endothelial A2A receptors. There are known to be many and complex interactions between the two cyclic nucleotide signalling pathways (see e.g. Pelligrino & Wang, 1998), and in particular cyclic AMP has been reported to reduce the sensitivity of phosphodiesterase V to zaprinast via stimulation of protein kinase A which phosphorylates the enzyme (Burns & Pyne, 1992; Burns et al., 1992). Thus when both cyclic AMP and cyclic GMP are raised by an agonist, although inhibition of either adenylate cyclase or of guanylate cyclase will inhibit the relaxation and inhibition of the cyclic AMP-selective phosphodiesterase IV will enhance it, zaprinast will be unable to enhance the effect of the agonist because it is not able to inhibit phosphodiesterase V due to phosphorylation of this enzyme by protein kinase A. Although the effect of protein kinase A on the sensitivity of phosphodiesterase V to other inhibitors, such as MMQ, has not to our knowledge been reported, the results obtained here showing a lack of effect of MMQ on NECA-induced relaxations suggests that it is affected in the same way as zaprinast.
Overall the results of this study are consistent with a model in which NECA acts via A2A receptors to cause both direct stimulation of adenylate cyclase in smooth muscle and also stimulation of NO release from endothelial cells which causes an increase in cyclic GMP in the smooth muscle (see Figure 6). The mechanism by which A2A receptor activation in endothelial cells causes NO release is not clear, but it could be via an increase in cyclic AMP levels. The A2A receptor is generally coupled via Gs to stimulation of adenylate cyclase, and adenosine receptor stimulation has been shown to result in increased levels of cyclic AMP in cultured endothelial cells (see e.g. Legrand et al., 1990; Côte et al., 1993; Iwamoto et al., 1994; Anwar et al., 1999). Colforsin, a forskolin analogue which acts as a direct stimulant of adenylate cyclase, has been reported to release NO from cultured rat aortic endothelial cells (Mori et al., 1999). Other agonists acting via Gs-coupled receptors (the β receptor and the receptor for α-CGRP) have also been shown to cause endothelium-dependent relaxation of the rat thoracic aorta (Gray & Marshall, 1992a, 1992b). Until recently the ability of cyclic AMP directly to activate endothelial NO synthase was not widely accepted (see e.g. Pelligrino & Wang, 1998), although it was suggested in pig endothelial cells that cyclic AMP via protein kinase A may activate large-conductance K+ channels and that the consequent hyperpolarization enhances agonist-induced entry of Ca2+ (Graier et al., 1993). However, Butt et al. (2000) showed very recently that cyclic AMP dependent protein kinase could directly phosphorylate endothelial NO synthase and activate it in a calcium-independent manner. It is not possible to say from our results whether or not the endothelium-dependent effects of NECA and the release of NO are dependent on stimulation of endothelial adenylate cyclase, but it seems a possible explanation. On the other hand, the rather small effect of SQ 22536 on the relaxations induced by NECA could suggest that an adenylate cyclase-independent mechanism is involved.
Figure 6.
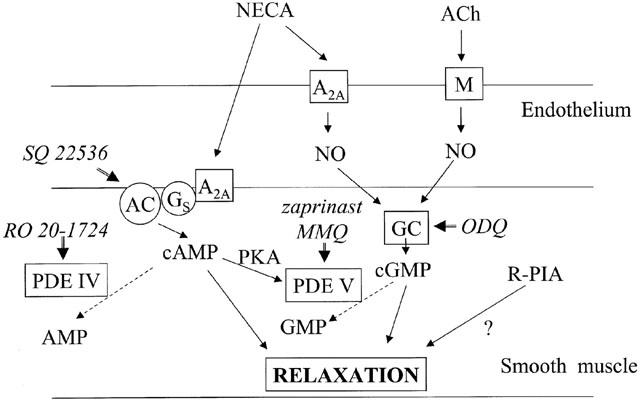
Proposed mechanisms for relaxation of the rat thoracic aorta by NECA, compared to the mechanisms for acetylcholine and R-PIA. For abbreviations see text.
Apart from a small enhancement by RO 20-1724 which is likely to be due to a small effect of R-PIA on the A2A receptor, none of the inhibitors used had any effect on responses to R-PIA confirming our previous conclusions (Prentice & Hourani, 1996) that the major action of this adenosine analogue is different from that of NECA. The results here confirm that R-PIA does not act via release of NO with the consequent stimulation of guanylate cyclase, or by stimulation of adenylate cyclase, and the mechanism by which it acts is still therefore unknown. It should be noted that the effects of adenosine were also not blocked by inhibition of adenylate or guanylate cyclases, again confirming our previous conclusions in this and in other tissues (Prentice & Hourani, 1996; 2000; Prentice et al., 1997) that the effects of this endogenous agonist are also largely not mediated via the known adenosine receptors. This unknown mechanism may therefore be of physiological relevance, as it may be involved in the beneficial effects of adenosine acting as a homeostatic mediator to match energy supply and demand.
In conclusion, these results are consistent with the effects of NECA being via activation of endothelial receptors to release NO which stimulates guanylate cyclase, as well as smooth muscle receptors coupled to stimulation of adenylate cyclase, whilst the effects of R-PIA are via neither of these mechanisms.
Acknowledgments
We thank the Wellcome Trust for financial support (Ref 057088) and acknowledge the contribution made by Kevin Foord and Golnaz Tabatabaee to this work.
Abbreviations
- CGS 21680
2-(p-(2-carboxyethyl)phenylethylamino)-5′-N-ethylcarboxamidoadenosine
- CPA
N6-cyclopentyladenosine
- DMSO
dimethylsulphoxide
- L-NAME
NG-nitro-L-arginine methyl ester
- MMQ
4-{[3′,4′-(methylenedioxy)benzyl]amino}-6-methoxyquinazoline
- NECA
5′-N-ethylcarboxamidoadenosine
- ODQ
1H-[1,2,4]oxadiazolo[4,3a]quinoxalin-1-one
- PDE
phosphodiesterase
- PKA
protein kinase A
- RO 20-1724
4-[(3-butoxy-4-methyoxyphenyl)methyl]-2-imidazolidinone
- R-PIA
N6-R-phenylisopropyladenosine
- 8-SPT
8-sulphophenyltheophylline
- SQ 22536
9-(tetrahydro-2-furanyl)-9H-purin-6-amine
- ZM 241385
4-(2-[7-amino-2-(2-furyl)[1,2,4]-triazolo[2,3a][1,3,5]-triazin-5-ylamino]ethyl)phenol
References
- ANWAR Z., ALBERT J.L., GUBBY S.E., BOYLE J.P., ROBERTS J.A., WEBB T.A., BOARDER M.R. Regulation of cyclic AMP by extracellular ATP in cultured brain capillary endothelial cells. Br. J. Pharmacol. 1999;128:465–471. doi: 10.1038/sj.bjp.0702792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BEAVO J.A. Cyclic nucleotide phosphodiesterases: functional implications of multiple isoforms. Physiol. Rev. 1995;75:725–748. doi: 10.1152/physrev.1995.75.4.725. [DOI] [PubMed] [Google Scholar]
- BRACKETT L.E., DALY J.W. Relaxant effects of adenosine analogs on guinea-pig trachea in vitro: xanthine-sensitive and xanthine-insensitive mechanisms. J. Pharm. Exp. Ther. 1991;257:205–213. [PubMed] [Google Scholar]
- BURNS F., PYNE N.J. Interaction of the catalytic subunit of protein kinase A with the lung type V cyclic GMP phosphodiesterase: modulation of non-catalytic binding sites. Biochem. Biophys. Res. Comm. 1992;189:1389–1396. doi: 10.1016/0006-291x(92)90228-d. [DOI] [PubMed] [Google Scholar]
- BURNS F., RODGER I.W., PYNE N.J. The catalytic subunit of protein kinase A triggers activation of the type V cyclic GMP-specific phosphodiesterase from guinea-pig lung. Biochem. J. 1992;283:487–491. doi: 10.1042/bj2830487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUTT E., BERNHARDT M., SMOLENSKI A., KOTSONIS P., FROHLICH L.G., SICKMANN A., MEYER H.E., LOHMANN S.M., SCHMIDT H.H.H.W. Endothelial nitric oxide synthase (type III) is activated and becomes calcium-independent upon phosphorylation by cyclic nucleotide-dependent protein kinases. J. Biol. Chem. 2000;275:5179–5187. doi: 10.1074/jbc.275.7.5179. [DOI] [PubMed] [Google Scholar]
- COLLIS M.G., BROWN C.M. Adenosine relaxes the aorta by interacting with an A2 receptor and an intracellular site. Eur. J. Pharmacol. 1983;96:61–69. doi: 10.1016/0014-2999(83)90529-0. [DOI] [PubMed] [Google Scholar]
- COLLIS M.G., HOURANI S.M.O. Adenosine receptor subtypes. Trends Pharmacol. Sci. 1993;14:360–366. doi: 10.1016/0165-6147(93)90094-z. [DOI] [PubMed] [Google Scholar]
- CÔTE S., VAN SANDE J., BOEYNAEMS J.-M. Enhancement of endothelial cAMP accumulation by adenine nucleotides: role of methylxanthine-sensitive sites. Am. J. Physiol. 1993;264:H1498–H1503. doi: 10.1152/ajpheart.1993.264.5.H1498. [DOI] [PubMed] [Google Scholar]
- GRAIER W.F., KUKOVETZ W.R., GROSCHNER K. Cyclic AMP enhances agonist-induced Ca2+ entry into endothelial cells by activation of potassium channels and membrane hyperpolarization. Biochem. J. 1993;291:263–267. doi: 10.1042/bj2910263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRAY D.W., MARSHALL I. Novel signal transduction pathway mediating endothelium-dependent β-adrenoceptor vasorelaxation in rat thoracic aorta. Br. J. Pharmacol. 1992a;107:684–690. doi: 10.1111/j.1476-5381.1992.tb14507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRAY D.W., MARSHALL I. Human α-calcitonin gene-related peptide stimulates adenylate cyclase and guanylate cyclase and relaxes rat thoracic aorta by releasing nitric oxide. Br. J. Pharmacol. 1992b;107:691–696. doi: 10.1111/j.1476-5381.1992.tb14508.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IWAMOTO T., UMEMURA S., TOYA Y., UCHIBORI T., KOGI K., TAKAGI N., ISHII M. Identification of adenosine A2 receptor-cAMP system in human aortic endothelial cells. Biochem. Biophys. Res. Comm. 1994;199:905–910. doi: 10.1006/bbrc.1994.1314. [DOI] [PubMed] [Google Scholar]
- KNIGHT G.E., BURNSTOCK G. The effects of purine compounds on the isolated aorta of the frog Rana temporaria. Br. J. Pharmacol. 1996;117:873–878. doi: 10.1111/j.1476-5381.1996.tb15274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEGRAND A.B., NARAYANAN T.K., RYAN U.A., ARONSTAM R.S., CATRAVAS J.D. Effects of adenosine and analogs on adenylate cyclase activity in cultured bovine aortic endothelial cells. Biochem. Pharmacol. 1990;40:1103–1109. doi: 10.1016/0006-2952(90)90499-b. [DOI] [PubMed] [Google Scholar]
- LEWIS C.D., HOURANI S.M.O., LONG C.J., COLLIS M.G. Characterization of adenosine receptors in the rat isolated aorta. Gen. Pharmacol. 1994;25:1381–1387. doi: 10.1016/0306-3623(94)90162-7. [DOI] [PubMed] [Google Scholar]
- MORI K., HAYABUCHI Y., KURODA Y., NAKAYA Y., TSUCHIYA K., MORITOKI H. Age-related endothelium-dependent vascular relaxation in rat aorta in response to colforsin. Pediatrics Int. 1999;41:673–681. doi: 10.1046/j.1442-200x.1999.01138.x. [DOI] [PubMed] [Google Scholar]
- MORITOKI H., MATSUGI T., TAKASE H., UEDA H., TANAOKA A. Evidence for the involvement of cyclic GMP in adenosine-induced, age-dependent vasodilatation. Br. J. Pharmacol. 1990;100:569–575. doi: 10.1111/j.1476-5381.1990.tb15848.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PELLIGRINO D.A., WANG Q. Cyclic nucleotide crosstalk and the regulation of cerebral vasodilation. Prog. Neurobiol. 1998;56:1–18. doi: 10.1016/s0301-0082(98)00009-4. [DOI] [PubMed] [Google Scholar]
- PRENTICE D.J. Vascular effects of adenosine and its analogues. Drug Dev. Res. 2001;52:346–349. [Google Scholar]
- PRENTICE D.J., HOURANI S.M.O. Activation of multiple sites by adenosine analogues in the rat isolated aorta. Br. J. Pharmacol. 1996;118:1509–1517. doi: 10.1111/j.1476-5381.1996.tb15567.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRENTICE D.J., HOURANI S.M.O. Characterisation of adenosine receptors mediating relaxation in hamster isolated aorta. Naunyn-Schmied. Arch. Pharmacol. 2000;362:427–434. doi: 10.1007/s002100000292. [DOI] [PubMed] [Google Scholar]
- PRENTICE D.J., PAYNE S.L., HOURANI S.M.O. Activation of two sites by adenosine receptor agonists to cause relaxation in rat isolated mesenteric artery. Br. J. Pharmacol. 1997;122:1509–1515. doi: 10.1038/sj.bjp.0701524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRENTICE D.J., SHANKLEY N.P., BLACK J.W. Pharmacological analysis of the interaction between purinoceptor agonists and antagonists in the guinea-pig taenia caecum. Br. J. Pharmacol. 1995;115:549–556. doi: 10.1111/j.1476-5381.1995.tb14967.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RALEVIC V., BURNSTOCK G. Receptors for purines and pyrimidines. Pharmacol. Rev. 1998;50:413–492. [PubMed] [Google Scholar]