

Abstract
Clinical studies have shown enhancement of cyclosporine toxicity when co-administered with the immunosuppressant sirolimus. We evaluated the biochemical mechanisms underlying the sirolimus/cyclosporine interaction on rat brain metabolism using magnetic resonance spectroscopy (MRS) and compared the effects of sirolimus with those of the structurally related RAD.
Two-week-old rats (25 g) were allocated to the following treatment groups (all n=6): I. control, II. cyclosporine (10 mg kg−1 d−1), III. sirolimus (3 mg kg−1 d−1), IV. RAD (3 mg kg−1 d−1), V. cyclosporine+sirolimus and VI. cyclosporine+RAD. Drugs were administered by oral gavage for 6 days. Twelve hours after the last dose, metabolic changes were assessed in brain tissue extracts using multinuclear MRS.
Cyclosporine significantly inhibited mitochondrial glucose metabolism (glutamate: 78±6% of control; GABA: 67±12%; NAD+: 76±3%; P<0.05), but increased lactate production. Sirolimus and RAD inhibited cytosolic glucose metabolism via lactate production (sirolimus: 81±3% of control, RAD: 69±2%; P<0.02). Sirolimus enhanced cyclosporine-induced inhibition of mitochondrial glucose metabolism (glutamate: 60±4%; GABA: 59±8%; NAD+: 45±5%; P<0.02 versus cyclosporine alone). Lactate production was significantly reduced. In contrast, RAD antagonized the effects of cyclosporine (glutamate, GABA, and NAD+, not significantly different from controls).
The results can partially be explained by pharmacokinetic interactions: co-administration increased the distribution of cyclosporine and sirolimus into brain tissue, while co-administration with RAD decreased cyclosporine brain tissue concentrations. In addition RAD, but not sirolimus, distributed into brain mitochondria.
The combination of cyclosporine/RAD compares favourably to cyclosporine/sirolimus in regards to their effects on brain high-energy metabolism and tissue distribution in the rat.
Keywords: Cyclosporine toxicity, sirolimus, everolimus, SDZ-RAD, pharmacodynamic drug interactions, tissue distribution, brain metabolism, mitochondrial glucose metabolism, oxidative phosphorylation, magnetic resonance spectroscopy (MRS)
Introduction
The calcineurin inhibitor cyclosporine (INN: ciclosporin, Figure 1) is used as an immunosuppressant after transplantation and for the treatment of immune diseases (Kahan, 1989; Faulds et al., 1993). The clinical use of cyclosporine is limited by its toxicity in combination with its narrow therapeutic index (Kahan, 1989). Neurotoxicity is one of the most serious side effects and incidences between 25 and 55% have been reported in transplant patients (Hauben, 1996; Gijtenbeek et al., 1999). Since first reported by Calne et al. (1979), a wide range of cyclosporine-associated neurological symptoms have been described, ranging from mild symptoms such as tremor, agitation, insomnia, anxiety, amnesia, headache, and paraesthesia to severe neurotoxicity resulting in seizures, cortical blindness, aphasia, ataxia, and stroke-like episodes (Hauben, 1996; Gijtenbeek et al., 1999). The incidence of cyclosporine neurotoxicity is increased in patients with high cyclosporine blood levels, but neurotoxicity also occurs during long-term treatment with cyclosporine blood concentrations within the therapeutic target range (Hauben, 1996; Gijtenbeek et al., 1999).
Figure 1.
Structures of cyclosporine (A), sirolimus (B) and RAD (C). Numbering of the macrolide immunosuppressants sirolimus and RAD follows the IUPAC guidelines (IUPAC, 1993). (C) shows the region of the RAD molecule different from sirolimus.
In in vitro studies, we showed that cyclosporine primarily inhibits mitochondrial energy production in the brain (Serkova et al., 1996; 1999). Reduction of high-energy phosphate concentrations by cyclosporine was also found in kidney (Henke et al., 1992; Massicot et al., 1997), liver (Salducci et al., 1996), and intestine (Madsen et al., 1995; Gabe et al., 1998).
Recently, the macrolide immunosuppressants sirolimus (rapamycin, Figure 1) and its 40-O-(2-hydroxyethyl) derivative RAD (INN: everolimus, Figure 1) have emerged as promising combination partners for cyclosporine. In the lymphocyte, sirolimus and RAD bind to FK-binding proteins and the immunophilin/sirolimus complex and probably also the immunophilin/RAD complex bind to mTOR, the mammalian target of rapamycin (Gummert et al., 1999). Inhibition of mTOR arrests the interleukin-2 driven T-cell proliferation at the G1- S-interface (Sehgal, 1998). Since the calcineurin inhibitor cyclosporine and sirolimus/RAD inhibit subsequent steps in the T-cell-mediated immune response, combination of cyclosporine and sirolimus or RAD results in synergistic immunosuppressive activity (Kahan, 1997; Stepkowski et al., 1996; Schuurman et al., 1997; Hausen et al., 2000).
Cyclosporine and sirolimus/RAD have different toxicity spectra, with the only exception being hyperlipidemia (Brattström et al., 1998). Although sirolimus by itself is not nephrotoxic, animal (Andoh et al., 1996) and clinical studies (Kahan & The Rapamune US Study Group, 2000) have shown that sirolimus enhances cyclosporine nephrotoxicity. We showed in isolated, perfused rat brain slices that sirolimus and cyclosporine synergistically reduce high-energy phosphate concentrations (Serkova et al., 1999). In the same in vitro model, RAD showed the opposite effect and antagonized the cyclosporine-mediated inhibition of mitochondrial oxidative phosphorylation (Serkova et al., 2000a). However, these in vitro studies exclusively focused on high-energy phosphate metabolism. It is still unclear how sirolimus enhances the inhibition of oxidative phosphorylation by cyclosporine and why the structurally related RAD and sirolimus have opposite effects on high-energy metabolism in the presence of cyclosporine. in vitro (Serkova et al., 1999; 2000a) potential pharmacokinetic and distribution interactions are ignored (Serkova et al., 2000b). In addition, in these in vitro studies brain tissue was exposed to the study drugs for a maximum period of 2 h, a time period most likely too short to detect potential effects of the study drugs on protein expression.
Here, we treated rats with cyclosporine, sirolimus, and RAD as well as sirolimus or RAD in combination with cyclosporine for 6 days. The effects of the study drugs on brain metabolism were evaluated using multinuclear magnetic resonance spectroscopy (MRS). To assess pharmacokinetic interactions, concentrations of the study drugs in blood, brain tissue and brain mitochondria were quantified using h.p.l.c./ mass spectrometry. It was our goal to evaluate and compare the effects of sirolimus and RAD on brain cell metabolism in the presence of cyclosporine.
Methods
Materials
Cyclosporine and RAD were kindly provided by Novartis Pharma AG (Basel, Switzerland). Sirolimus was purchased from Sigma (St. Louis, MO, U.S.A.). Stock solutions (10 g l−1) of each drug were prepared in absolute ethanol (Aldrich Chemicals, Milwaukee, WI, U.S.A.). Perchloric acid (PCA, 60%) as well as potassium hydroxide (KOH) for the extraction of brain tissue homogenates were from Aldrich Chemicals (Milwaukee, WI, U.S.A.). Deuterated chemicals (D2O, NaOD, and DCl) for MRS were purchased from Cambridge Isotope Laboratories, Inc. (Andover, MA, U.S.A.) and NMR tubes of 5 mm diameter from Wilmad (Buena, NJ, U.S.A.). Cell metabolites in brain extracts were measured using a 500 MHz Bruker NMR spectrometer (Bruker Instruments Inc., Fremont, CA, U.S.A.). The system was controlled and data were processed using WINNMR software (Bruker Instruments Inc. Fremont, CA, U.S.A.). Methanol and zinc sulphate were purchased from Fisher Scientific (Fair Lawn, NJ, U.S.A.). Percoll and other chemicals for mitochondria isolation were from Aldrich Chemicals (Milwaukee, WI, U.S.A.). Concentrations of the study drug were measured using an h.p.l.c./h.p.l.c.-mass spectrometry (MS) system consisting of the following components (all series 1100 h.p.l.c. components, Hewlett-Packard, Palo Alto, CA, U.S.A.): h.p.l.c. I: G1311A quaternary pump, G1322A degasser and G1329A autosampler equipped with a G1330A thermostat; h.p.l.c. II: G1312A binary pump, G1322A degasser, G1316A column thermostat and G1946A mass selective detector. The two h.p.l.c. systems were connected via a 7240 Rheodyne 6-port switching valve mounted on a step motor (Rheodyne, Cotati, CA, U.S.A.). The system was controlled and data were processed using ChemStation Software Revision A.06.01 (Hewlett-Packard, Palo Alto, CA, U.S.A.). A 10·2 mm extraction column (Keystone Scientific, Bellefonte, PA, U.S.A.) filled with Hypersil ODS-1 of 10 μm particle size (Shandon, Chadwick, U.K.) and a 50·4.6 mm analytical column filled with C8 material of 3.5 μm particle size (Zorbax XDB C8, Hewlett-Packard, Palo Alto, CA, U.S.A.) were used. The internal standard cyclosporin D was a kind gift from Novartis (Basel, Switzerland) and 28,40-O-diacetyl rapamycin was synthesized and purified as described by Streit et al. (1996).
Animal studies
All animal protocols were reviewed and approved by the University of California, San Francisco, Committee on Animal Research.
The animals received humane care in compliance with the ‘Principles of Laboratory Animal Care' formulated by the National Society for Medical Research and the ‘Guide for the Care and Use of Laboratory Animals', published by the National Institutes of Health (NIH publication No. 80-123, revised 1985).
Seventy-two Wistar rats (two weeks old, 28±4 g body weight) were purchased from Charles River, Inc. (Wilmington, MA, U.S.A.) and were allocated to six treatment groups: (I) vehicle (control, n=12); (II) 10 mg kg−1 cyclosporine (group Cs, n=12); (III) 3 mg kg−1 sirolimus (group SRL, n=12); (IV) 3 mg kg−1 RAD (group RAD, n=12); (V) 10 mg kg−1 cyclosporine+3 mg kg−1 sirolimus (group Cs+SRL, n=12), (VI) 10 mg kg−1 cyclosporine+3 mg kg−1 RAD (group Cs+RAD, n=12). Stock solutions of the study drugs were dissolved in whole milk (1 : 10, final concentration 1 g l−1). The study drugs were administered by oral gavage once daily. The final ethanol concentration was 10%. Animals in the control group were treated with 13 ml 10% ethanol/milk vehicle kg−1 body weight. This corresponded to the highest alcohol doses administered during the study (groups Cs+SRL and Cs+RAD). The doses were chosen on basis of a previous dose finding study (results not shown) with the goal to find oral doses of the study drugs resulting in brain tissue concentrations in the range of those that have previously been shown to have significant effects on the cell metabolism of isolated brain slices (Serkova et al., 1999; 2000a).
During our study, the animals were closely monitored, including daily evaluations of appearance, grooming, appetite, behaviour, activity and body weight. At the end of the 6-day study period and 12 h after the last study drug dose, the animals were decapitated and whole blood and brain tissues were collected. Blood samples were drawn into tubes with EDTA as anticoagulant. Tissues were immediately frozen in liquid nitrogen and stored at −80°C until PCA extraction (n=6 per group) or h.p.l.c./h.p.l.c.-MS analysis (n=6 per group). Samples were analysed within 20 days.
Measurement of brain cell metabolites using MRS
Frozen brain tissues were weighed; 2 g (wet weight) of each tissue sample was homogenized in a mortar grinder in the presence of liquid nitrogen and extracted with 6 ml ice-cold PCA (12%). The extracts were centrifuged, the aqueous phase was removed and neutralized using KOH. The samples were centrifuged once again and the aqueous phase was lyophilized overnight. The lyophilized brain PCA extracts were reconstituted in 400 ml deuterium oxide (D2O) and adjusted to pH 7 using DCl and NaOD. The experiments with 1H-, 13C-, and 31P-MRS were carried out as described previously (Serkova et al., 1996). Trimethylsilyl propionic-2,2,3,3,-d4 acid (TSP) was used as an external standard for quantification in 1H-MRS spectra. 1H chemical shifts were referenced to TSP at 0 p.p.m. The use of an internal standard for 1H-MRS allowed quantification based on areas under the 1H-signals of single metabolites. The C3 peak of lactate at 21 p.p.m. was used as the chemical shift reference in 13C-MRS. For 31P-MRS, either phosphocreatine (PCr) at −2.33 p.p.m. or, if present, α-ATP at −9.90 p.p.m. were used as shift references. High-energy phosphate metabolism in the brain is a closed system, which means that a reduced concentration of one metabolite automatically leads to an increase of one or more other metabolites. Therefore, we calculated the relative concentrations of high-energy phosphates as the ratio of the 31P-MRS peak area of a metabolite to the total area of all high-energy phosphate metabolites quantified.
Isolation of brain mitochondria
After treatment with the study drugs or their combinations for 6 days, the concentrations of cyclosporine, sirolimus, and RAD in rat brain mitochondria were determined using h.p.l.c./h.p.l.c.-MS analysis (vide infra). Brain mitochondria from rats treated with the vehicle or the study drugs were isolated by a modified procedure as described previously (Kristal & Dubinsky, 1997). Briefly: after decapitation, brains were rapidly dissected and placed in 10 ml ice-cold isolation buffer containing 350 mM sucrose, 2 mM HEPES and 1 mM EGTA (pH 7.4). After homogenization using a manual homogenizer, the brain suspension was centrifuged at 900 ×g to remove cellular debris and nuclei for 10 min. The supernatant was decanted into a centrifuge tube and re-centrifuged under the same conditions. The supernatant was transferred into another centrifuge tube and was centrifuged at 10,000 ×g for 10 min resulting in an olive-green mitochondrial precipitate. The mitochondrial fraction was homogenized in 1 part Percoll/7 parts buffer. The homogenate was layered over preformed discontinuous Percoll gradients consisting of a bottom layer of 40% Percoll in buffer and a top layer of 26% Percoll in buffer. The gradients were centrifuged at 31,000×g for 45 min. The mitochondrial fraction, which was concentrated at the interface of the two layers, was collected, re-constituted in buffer, and centrifuged again. The supernatant was removed, and the pellet was reconstituted in 0.5 ml of 1 M KH2PO4 buffer pH 7.4. Mitochondria were extracted and analysed using h.p.l.c./h.p.l.c.-MS as described below.
Measurement of tissue distribution of immunosuppressants by h.p.l.c./h.p.l.c.-MS
We used an h.p.l.c.-MS assay with automated online sample extraction (h.p.l.c./h.p.l.c.-MS) for quantification of the study drugs in blood, tissue and mitochondria, which was a modification of the assay we described previously (Christians et al., 2000). Brain tissues (1.5 g wet weight) were thawed and weighed. Brain samples were homogenized with 2 ml KH2PO4 buffer pH 7.4 (1 M) using an electric homogenizer. For protein precipitation, 200 μl methanol/1 M ZnSO4 (80/20 v v−1) was added to each 100 μl sample (homogenized brain, blood or isolated mitochondria). Cyclosporin D and 28,40-O-diacetyl rapamycin were added as internal standards for cyclosporine and sirolimus/RAD, respectively, resulting in final concentrations of 100 μg l−1. After centrifugation, 100 μl of the supernatant was injected onto the extraction column. Samples were washed with a mobile phase of 40% methanol and 60% 0.1% formic acid supplemented with 1 μmol l−1 sodium formate. The flow was 5 ml min−1 and the temperature for the extraction column was set to 65°C. After 0.75 min, the switching valve was activated and the analytes were eluted in the backflush mode from the extraction column onto analytical column (flow 0.5 ml min−1). The mobile phase consisted of 90% methanol and 10% 0.1% formic acid supplemented with 1 μmol l−1 sodium formate. The mass spectrometer was run in the selected ion mode and positive ions [M+Na]+ were recorded. For all matrices, the analytical recovery was >90%. In blood, brain and isolated mitochondria, the assay was linear from 1 μg l−1 (lower limit of quantification) to 500 μg l−1 for cyclosporine and 0.25 μg l−1 to 100 μg l−1 for the macrolides (r2>0.99).
Data analysis
All results are reported as means±standard deviation (s.d.). The results of the control and immunosuppressant-treated groups were compared using unpaired Student's t-test or analysis of variance in combination with Duncan grouping as post hoc test (SPSS, version 9.0, SPSS Inc., Chicago, IL, U.S.A.).
Results
Clinical monitoring
On study day 6, the average weight (±s.d.) of vehicle treated animals in the control group was 166±12% of the initial body weight (Figure 2). In contrast, cyclosporine-treated animals (group Cs) showed no weight gain (91±10% of the initial body weight). Animals treated with sirolimus or RAD (groups SRL and RAD) were in better condition than the cyclosporine-treated animals and gained weight, however, slower than the control group (122±8 and 122±9%, respectively). Co-administration of cyclosporine and sirolimus had the most negative effect on the parameters recorded including body weight (85±8% of the initial body weight). In comparison to cyclosporine alone, animals treated with a combination of cyclosporine and RAD lost slightly but significantly less weight than those treated with cyclosporine alone (97±13%). On day 6, body weights differed significantly (P<0.001) among groups as evaluated by analysis of variance in combination with Duncan grouping: control >RAD=SRL>Cs+RAD>Cs>Cs+SRL.
Figure 2.
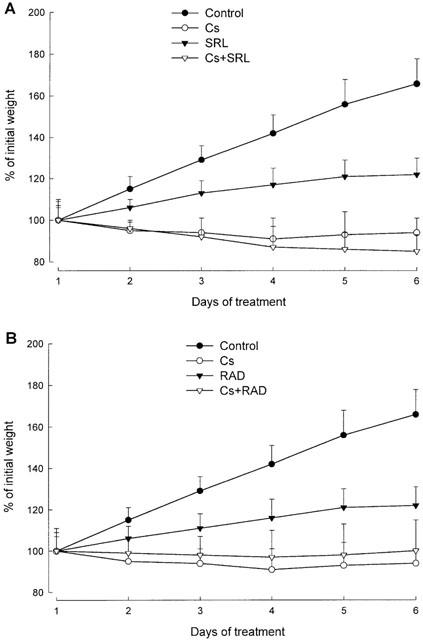
Effect of the study drugs and their combinations on weight gain during the observation period. Data (mean±standard deviation, n=12) shows the per cent change at the beginning of each study day in comparison with the weight at day 1 prior to dosing (=100%). The study drugs were administered by oral gavage for 6 days. The rats (2-weeks old, mean weight 28±4 g) received the following doses: control, 13 ml vehicle (10% ethanol in milk); Cs, 10 mg kg−1 d−1 cyclosporine; SRL, 3 mg kg−1 d−1 sirolimus; RAD, 3 mg kg−1 d−1 RAD; Cs+SRL, 10 mg kg−1 d−1 cyclosporine+3 mg kg−1 d−1 sirolimus; Cs+RAD, 10 mg kg−1 d−1 cyclosporine+3 mg kg−1 d−1 RAD. (A) Comparison of groups Cs, SRL and Cs+SRL; (B) comparison of groups Cs, RAD and Cs+RAD. The results of the control group and Cs are shown in both (A) and (B) to facilitate comparison.
Effect of the study drugs on brain cell metabolism
1H-MRS
In control rats, the following cell metabolites could be quantified: the neuronal marker N-acetyl aspartate (NAA), the excitatory amino acid neurotransmitters glutamate and aspartate, the inhibitory neurotransmitter γ-amino butyric acid (GABA), the cellular osmolytes taurine and myo-inositol as well as lactate (Table 1). In comparison to the controls, cyclosporine (group Cs) decreased the brain tissue concentrations of NAA (59% of control, P<0.002, Table 1) and those of the neurotransmitters glutamate (78%; P<0.02), and GABA (67%, P<0.05). Lactate concentrations increased to 140% (P<0.002). The effects of sirolimus (group SRL) and RAD (group RAD) were similar. In contrast to cyclosporine, the macrolide immunosuppressants decreased lactate concentrations (sirolimus: 81%, P<0.02; RAD: 69%, P<0.002). Sirolimus and RAD also slightly decreased the brain concentration of NAA and increased the concentrations of glutamine (Table 1).
Table 1.
The effects of the study drugs and their combinations on concentrations of neurotransmitters and cell metabolites in rat brain extracts analysed by 1H-mrs
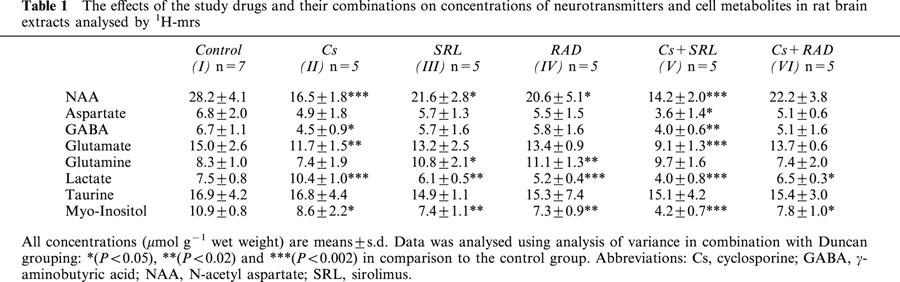
Co-administration of cyclosporine and sirolimus (group Cs+SRL) had the most significant effect. Among the treatment groups, brain tissue extracts from animals in the Cs+SRL group contained the lowest concentrations of NAA (50% of control, P<0.002), of glutamate (60%, P<0.002), of GABA (59%, P<0.02), and of lactate (53%, P<0.002). In comparison to the controls, co-administration of cyclosporine and RAD, however, did not significantly affect most of the parameters measured with the exception of a slight decrease in lactate and myo-inositol concentrations (Table 1).
13C-MRS
Although the natural abundance of 13C-isotopes is only 1.1%, intensities of 13C-MRS signals in our brain extracts were sufficient for reliable quantification of 13C-groups of glucose and the following intermediates of the glucose pathways: aspartate, lactate, GABA, glutamate, glutamine, and oxaloacetate. In addition, the 13C-signals of creatine, myo-inositol, NAA and taurine were detected. Representative 13C-MRS spectra of rat brain PCA extracts from rats of the control, Cs, SRL and RAD groups are shown in Figure 3.
Figure 3.
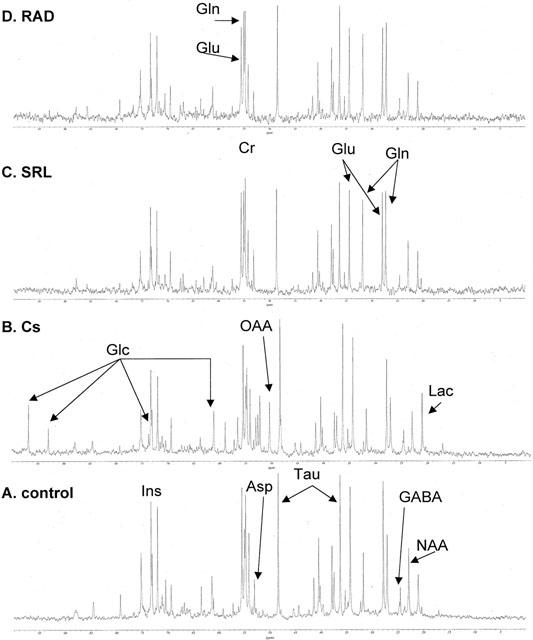
Representative 13C-MRS spectra of brain PCA extracts from rats in the control (A) and mono-treatment groups Cs (B), SRL (C), and RAD (D). Rats were treated with the following doses by oral gavage for 6 days: (A) control, 13 ml vehicle (10% ethanol in milk, total n=7), (B) Cs, 10 mg kg−1 d−1 cyclosporine (n=5), (C) SRL, 3 mg kg−1 d−1 sirolimus (n=5) and (D) RAD, 3 mg kg−1 d−1 RAD (n=5). Abbreviations: Asp, aspartate; Cr, creatine; Cs, cyclosporine; GABA, γ-aminobutyric acid; Glc, glucose; Gln, glutamine; Glu, glutamate; Ins, myo-inositol; Lac, lactate; NAA, N-acetyl aspartate; OAA, oxaloacetate; SRL, sirolimus; Tau, taurine.
In comparison to the controls, brain extracts of cyclosporine treated animals showed reduced signal intensities of Krebs cycle-dependent metabolites such as glutamate, glutamine, GABA, and NAA (Figure 3). This reduction was accompanied by glucose (at 92.9 and 96.8 p.p.m.) and oxaloacetate (at 50 p.p.m.) signals above the detection limit, which were not present in the spectra of the controls. Additionally, the 13C-signal of lactate, an important anaerobic glucose metabolite, was slightly increased. Glucose and oxaloacetate signals were undetectable in the 31C-spectra of brain extracts from rats in the SRL and RAD groups. Lactate signals in these spectra were consistently smaller than in spectra of the control group. Characteristic changes after treatment with sirolimus or RAD were the significantly increased intensities of glutamine signals in comparison to the controls. Due to the lack of an internal standard, absolute concentrations of metabolites based on the 13C-MRS spectra could not be determined. We calculated the ratios of 13C-signal intensities of Krebs cycle metabolites (aerobic glucose metabolism)/glycolysis intermediates (anaerobic glucose metabolism):
Krebs cycle/glycolysis (aerobic/anaerobic glucose metabolism) ratio=
The ratios were 9.5±1.1 for the control group (n=7, mean±s.d.), 5.6±0.9 for the Cs group (n=5), 14.1±1.6 for the SRL group (n=5) and 14.7±2.9 for the RAD group (n=5).
Co-administration of cyclosporine and sirolimus resulted in a reduction of all intermediates of glucose metabolism, from both Krebs cycle as well as from anaerobic glycolysis (Figure 4B) resulting in an aerobic/anaerobic glucose metabolite ratio of 13.0±1.3. In comparison, glucose metabolism was less affected when cyclosporine and RAD were co-administered (ratio: 9.8±1.4, Figure 4C). The ratios were significantly different with P<0.002 (Duncan grouping, SRL=RAD=Cs+SRL>control=Cs+RAD).
Figure 4.
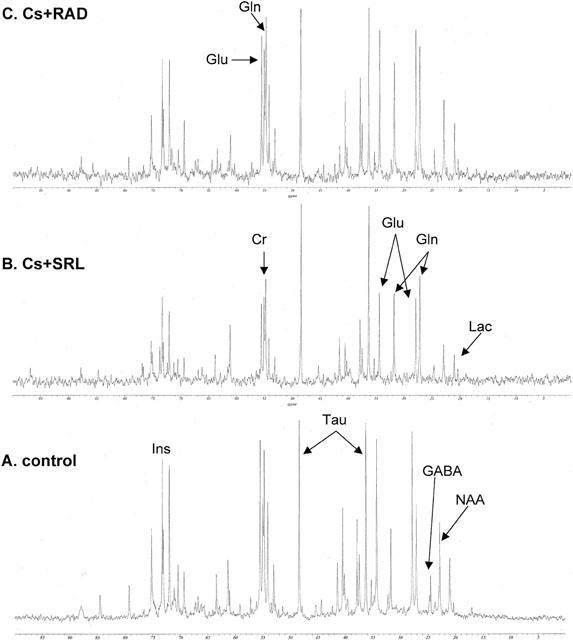
Representative 13C-MRS spectra of brain PCA extracts from rats in the control group (A) and after co-administration of cyclosporine and sirolimus (group Cs+SRL, B) and cyclosporine and RAD (group Cs+RAD, C). Rats were treated with the following doses by oral gavage for 6 days: (A) control, 13 ml vehicle (10% ethanol in milk, total n=7), (B) Cs+SRL, 10 mg kg−1 d−1, cyclosporine+3 mg kg−1 d−1 sirolimus (n=5) and (C) Cs+RAD, 10 mg kg−1 d−1 cyclosporine+3 mg kg−1 d−1 RAD. Abbreviations: Cr, creatine; Cs, cyclosporine; GABA, γ-aminobutyric acid; Gln, glutamine; Glu, glutamate; Ins, myo-inositol; Lac, lactate; NAA, N-acetyl aspartate; SRL, sirolimus; Tau, taurine.
31P-MRS
The results of the 31P-MRS analyses are summarized in Table 2. Treatment with cyclosporine reduced the relative concentrations of nucleoside triphosphates (NTP) to 34%, those of phosphocreatine to 30% and those of nicotinamide adenine dinucleotide (NAD+) to 76% of the controls. In parallel, relative nucleoside diphosphate (NDP) concentrations were 2.4 fold and nucleoside monophosphate (NMP) concentrations 20.8 fold higher than in the control group. Sirolimus and RAD inhibited high-energy phosphate metabolism to a significantly smaller extent than cyclosporine (Table 2). Compared to the controls, sirolimus decreased NTP to 61% of the controls and increased relative NDP concentrations to 161% and NMP concentrations 5 fold. RAD decreased NTP concentration to 77% of controls, increased NDP concentrations to 171% but had no statistically significant effect on NMP concentrations. Both sirolimus and RAD did not significantly affect phosphocreatine or NAD+ concentrations.
Table 2.
The effects of cyclosporine, sirolimus and RAD, as well as co-administration of cyclosporine/sirolimus and cyclosporine/RAD on brain phosphate metabolism

Again, co-administration of sirolimus and cyclosporine resulted in the most significant changes. At the end of the study period, the average relative NTP concentration in brains of animals in group Cs+SRL was 22%, that of phosphocreatine 22% and that of NAD+ 45% of the controls. Consequently, relative NDP and NMP concentrations were significantly higher than in the controls. Again, co-administration of cyclosporine and RAD compared favorably with the cyclosporine/sirolimus combination (group Cs+SRL) and also with cyclosporine mono-treatment (group Cs). In the Cs+RAD group relative brain concentrations of NTP were reduced to 55% of the controls. However, there was no statistically significant difference between relative phosphocreatine and NAD+ concentrations between group Cs+RAD and the controls (Table 2). Comparison of the six study groups using analysis of variance indicated significant differences in the relative concentrations of the key parameters NTP, phosphocreatine and NAD+ (all P<0.0002). Duncan grouping gave the following results: NTP, control>RAD>SRL>Cs+RAD>Cs>Cs+SRL; phosphocreatine: control=SRL=RAD=Cs+RAD>Cs>Cs+SRL; NAD+: control=SRL=RAD=Cs+RAD>Cs>Cs+SRL.
Blood and brain tissue concentrations of immunosuppressants
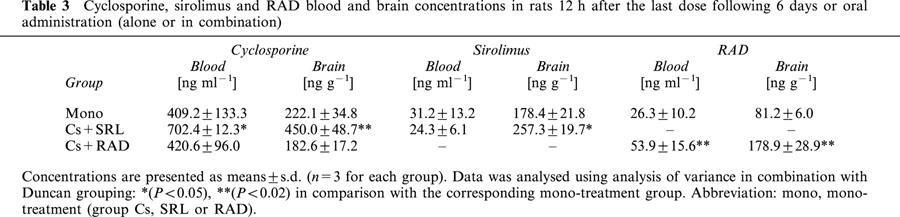
The concentrations of the study drugs in blood and brain tissue 12 h after the last dose (C12h) are shown in Table 3. Comparison of the brain-to-blood partition coefficients showed that co-administration of cyclosporine and sirolimus increased brain penetration for both cyclosporine (0.68±0.03 in group Cs+SRL versus 0.56±0.08 in group Cs, mean±s.d., P<0.05) and sirolimus (10.9±2.0 in group Cs+SRL versus 6.0±1.6 in group SRL, P<0.01). In contrast, co-administration of cyclosporine and RAD decreased the cyclosporine brain-to-blood partition coefficient (0.44±0.06 in group Cs+RAD versus 0.56±0.08 in group Cs, P<0.05). The brain-to-blood partition coefficient for RAD was not affected by the presence of cyclosporine (3.4±0.5 in group Cs+RAD versus 3.4±1.2 in group RAD, statistically not significant).
Table 3.
Cyclosporine, sirolimus and RAD blood and brain concentrations in rats 12 h after the last dose following 6 days or oral administration (alone or in combination)
Distribution into brain mitochondria
At the end of the study period, RAD and cyclosporine, but not sirolimus, were found in rat brain mitochondria (limit of detection of the h.p.l.c/h.p.l.c.-MS assay: 0.25 μg l−1). In group Cs, the cyclosporine concentrations in mitochondria were 28%, and in the RAD group, RAD concentrations in the mitochondria were 82% of those in the brain tissue (Tables 3 and 4). Addition of RAD reduced cyclosporine concentrations in brain mitochondria by 65% (P<0.005 group Cs+RAD versus group Cs), while cyclosporine increased RAD penetration into mitochondria 2.5 fold (P<0.0001 group Cs+RAD versus group RAD).
Table 4.
Cyclosporine, sirolimus and RAD concentrations in isolated brain mitochondria of rats after oral treatment with the study drugs or their combinations for 6 days

Even though sirolimus itself could obviously not pass mitochondrial membranes (Table 4), it increased cyclosporine distribution into brain mitochondria 3.1 fold (P<0.001 group Cs+SRL versus group Cs).
Discussion
Our results show that, at the doses used, sirolimus enhances the negative effects of cyclosporine on rat brain metabolism in vivo. In contrast, RAD has either no effect or improves brain glucose and high-energy phosphate metabolism in the presence of cyclosporine.
We carried out our study in 14-day-old rather than adult rats for several reasons. The brains of young rats are more resistant against hypoxia compared to those of adult rats during sample preparation and extraction. Our previous in vitro studies used rat brain slices of young rats for the same reason. The use of young rats in our in vivo study facilitated comparison with our in vitro results (Serkova et al., 1999; 2000a) without being confounded by potential age-dependent differences. In adult cynomolgus monkeys, steady-state concentrations of cyclosporine in brain were found to be 4.7 fold lower than those in blood (Serkova et al., 2000b). The distribution of cyclosporine into the brain is mostly limited by the ATP-binding cassette transporter p-glycoprotein as part of the blood-brain barrier (Schinkel et al., 1996). In 14-day-old rats, the blood-brain barrier is not yet fully developed. This allowed us to reach cyclosporine concentrations in the range of those known to significantly change brain metabolism in vitro in isolated brain slices (Serkova et al., 1999; 2000a) without being limited by peripheral organ toxicity.
As mentioned above, it can be expected that the metabolic changes after short-term exposure (Serkova et al., 1999; 2000a) differ from those after long-term exposure since changes of cell metabolism, such as mechanisms that involve modulation of protein expression, may take significantly longer than 2 h to reach full effects. However, our results show that the rat brain cells were unable to compensate for the effects of the study drugs during long-term exposure and that the changes of high energy phosphate metabolism observed in vivo here were similar to our previous in vitro studies (Serkova et al., 1999; 2000a).
Our study gave additional insights into the mechanisms underlying the reduction of high-energy phosphate concentrations by cyclosporine. Previously described mechanisms involved in the cyclosporine-mediated reduction of cellular ATP concentrations include increased radical formation (Uemoto et al., 1989; Wang & Salahudeen, 1994; Wolf et al., 1997) and mitochondrial Ca2+-overload (Salducci et al., 1996). The accumulation of oxaloacetate and lactate indicated that cyclosporine significantly inhibited the Krebs cycle at its entrance, possibly by inhibiting pyruvate dehydrogenase. The increased concentrations of oxaloacetate in combination with the reduction of aspartate and glutamate concentrations may suggest that cyclosporine also inhibited the aspartate/malate shuttle, which transfers cytosolic NADH produced during glycolysis into the mitochondria. However, the cause-effect relationships between the negative effects of cyclosporine on mitochondrial glucose metabolism observed in our study and increased mitochondrial calcium concentrations and enhanced oxygen radical production as reported previously by others (Uemoto et al., 1989; Wang & Salahudeen, 1994; Wolf et al., 1997; Salducci et al., 1996) remains to be evaluated in future studies.
Our in vivo study confirmed previous in vitro results indicating that in contrast to cyclosporine, sirolimus and RAD do not directly affect mitochondrial oxidation (Serkova et al., 2000a). Sirolimus and RAD had little effect on the concentrations of Krebs cycle products; NAD+ concentrations, a marker of oxidative phosphorylation (Erecinska & Wilson, 1982), were not significantly different from the controls. However, sirolimus and RAD reduced lactate concentrations indicating interference of these compounds with cytosolic glycolysis. Quantification of lactate by MRS may not be a reliable parameter, since during sample collection, tissues are hypoxic and aerobic glycolysis is stimulated. We tried to minimize hypoxic effects during sample collection by snap freezing tissues at −80°C. The rather small standard deviations of the lactate concentrations in Table 1 indicated that sample collection and preparation exhibited little variability and that comparison of lactate concentrations with those of the controls yielded valid information. An earlier study showed that sirolimus inhibits cytosolic glycolysis in activated T-lymphocytes by reducing the activity of aldolase A (Wang et al., 1996). Since the MRS assays used in our study did not allow for detection of upstream glycolysis products, it was not possible to decide whether or not aldolase inhibition also was the major mechanism underlying the inhibitory effects of sirolimus, and possibly RAD, on glucose metabolism found in our study. Interestingly, we did not find an increase of oxaloacetate when cyclosporine was combined with sirolimus or RAD (Figure 4). This may indicate that most of the oxaloacetate that accumulated during cyclosporine monotreatment was synthesized from glycolysis products, e.g. via pyruvate carboxylase (Scrutton, 1978; Jitrapakdee & Wallace, 1999) The small, but significant reduction of NTP during treatment with sirolimus and RAD may be explained by the reduced availability of Krebs cycle substrates through glycolysis and the reduced production of cytosolic NADH.
Our results show that enhancement of the cyclosporine-mediated inhibition of high-energy phosphate product is based on a pharmacodynamic as well as on a pharmacokinetic interaction. Cyclosporine decreases the Krebs cycle/ glycolysis ratio, by both reducing the concentration of Krebs cycle products and increasing the concentration of glycolysis products. In addition to a possible accumulation of glycolysis products as a result of inhibiting the Krebs cycle, this may be due to activation of cytosolic glycolysis to partially compensate for the inhibition of mitochondrial high-energy phosphate production by cyclosporine. As indicated by the lack of difference of the Krebs cycle/glycolysis ratio after co-administration of cyclosporine and sirolimus versus sirolimus alone, sirolimus may override a potential stimulating effect of cyclosporine on cytosolic ATP production via glycolysis. Inhibition of a major compensatory mechanism can be expected to enhance the effects of cyclosporine on high-energy phosphate metabolism. In addition, co-administration of cyclosporine and sirolimus increased the blood and brain tissue concentrations of both cyclosporine and sirolimus as well as the mitochondrial concentrations of cyclosporine. Similar cyclosporine/sirolimus drug interactions have been reported in the rat (Napoli et al., 1998). Possible mechanisms include interactions at ATP-binding cassette transporters and cytochrome P4503A enzymes in the small intestine and liver as previously discussed in detail (Crowe & Lemaire, 1998, Lampen et al., 1998; Napoli et al., 1998; Crowe et al., 1999; Serkova et al., 1999; 2000a,2000b).
In contrast to sirolimus, RAD did not affect cyclosporine concentrations in blood and brain tissue but reduced cyclosporine concentrations in brain mitochondria by 65%. Our previous study indicated that in isolated rat brain slices, RAD antagonizes the effects of cyclosporine by stimulating oxidative phosphorylation (Serkova et al., 2000a). Our study here shows that in the presence of cyclosporine, RAD also partially restores the Krebs cycle. The opposite effects of sirolimus and RAD on Krebs cycle and high-energy phosphate metabolism in the presence of cyclosporine can be explained by our finding that at the doses used, RAD, but not sirolimus, distributes into brain mitochondria. These findings are consistent with the results of our previous in vitro study (Serkova et al., 2000a). Our previous in vitro study clearly indicated a pharmacodynamic, synergistic interaction between sirolimus and a pharmacodynamic antagonistic interaction between RAD with cyclosporine-mediated reduction of high-energy phosphate concentrations (Serkova et al., 1999; 2000a). Our study results did not allow us to dedecide whether the opposite effects of sirolimus and RAD on the distribution of cyclosporine into mitochondria or the fact that RAD, but not sirolimus, distributes into mitochondria was the major mechanism responsible for the different effects of RAD and sirolimus on brain metabolism in the presence of cyclosporine in vivo.
As reviewed by Beal (1992) and to our knowledge, there is not a single report in the literature showing that inhibition of anaerobic glycolysis alone causes neuropathological symptoms. This is in agreement with the observation that sirolimus alone causes neither neurotoxicity nor nephrotoxicity. However, drugs that are inhibitors of glycolysis potentially enhance toxicity of other drugs that inhibit mitochondrial oxidation by inhibiting the major pathway to compensate for the inhibition of mitochondrial high-energy production. Concomitant with our study, Kahan & The US Rapamune Study Group (2000) reported that in transplant patients the kidney is the major target organ of the enhancement of cyclosporine toxicity by sirolimus. Preliminary results of a study in adult rats show that the interaction mechanisms of cyclosporine, sirolimus and/or RAD and their effects on cell metabolism identified in brain is consistent with findings in the kidney (Serkova et al., unpublished results). This suggests that the interaction mechanisms identified here may also explain the enhancement of long-term cyclosporine nephrotoxicity by sirolimus observed in sirolimus phase III clinical trials (Kahan & The US Rapamune Study Group, 2000).
On the other hand, the cyclosporine/sirolimus or cyclosporine/RAD dose ratio used here was 3.3 : 1, while the dose ratios in clinical study protocols used today is 20-100 : 1. It remains to be evaluated whether or not the favourable toxicological effects of RAD in comparison to sirolimus in combination with cyclosporine found here will translate into a significant advantage in long-term treated patients and at clinically relevant cyclosporine/RAD dose ratios.
Acknowledgments
The authors thank Dr Thomas L. James and Dr Vladimir Basus (Department of Pharmaceutical Chemistry, School of Pharmacy, University of California at San Francisco) for their most valuable input and support and Ms Karen Baner for her assistance with the animal experiments. Financial support for this work was obtained from the Alexander von Humboldt-Foundation (grant V-3-FLF-1052812), the Deutsche Forschungsgemeinschaft (grants Ch95/6-2 and Se985/1-1) and Novartis Pharma AG (Basel, Switzerland).
Abbreviations
- AMU
atomic mass units
- CNS
central nervous system
- GABA
γ-amino butyric acid
- MRS
magnetic resonance spectroscopy
- MS
mass spectrometry
- NAA
N-acetyl aspartate
- NAD+
nicotinamide adenine dinucleotide
- NDP
nucleoside diphosphate
- NMP
nucleoside monophosphate
- NTP
nucleoside triphosphate
- OAA
oxaloacetate
- TSP
trimethylsilyl propionic-2,2,3,3,-d4 acid
References
- ANDOH T.F., LINDSLEY J., FRANCESCHINNI N., BENNETT W.M. Synergistic effects of cyclosporine and rapamycin in a chronic nephrotoxicity model. Transplantation. 1996;62:311–316. doi: 10.1097/00007890-199608150-00002. [DOI] [PubMed] [Google Scholar]
- BEAL M.F. Does impairment of energy metabolism result in excitotoxic neuronal death in neurodegenerative illnesses. Ann. Neurol. 1992;31:119–130. doi: 10.1002/ana.410310202. [DOI] [PubMed] [Google Scholar]
- BRATTSTRÖM C., WILCZEK H., TYDEN G., BOTTINGER Y., SÄWE J., GROTH C.G. Hyperlipidemia in renal transplant recipients treated with sirolimus (rapamycin) Transplantation. 1998;65:1272–1274. doi: 10.1097/00007890-199805150-00023. [DOI] [PubMed] [Google Scholar]
- CALNE R.Y., ROLLES K., THIRU S., MCMASTER P., CRADDOCK G.N., AZIZ S., WHITE D.J.G., EVANS D.B., DUNN D.C., HENDERSON R.G., LEWIS P. Cyclosporin A initially as the only immunosuppressant in 34 recipients of cadaveric organs: 32 kidneys, 2 pancreas and 2 livers. Lancet. 1979;8151:1033–1036. doi: 10.1016/s0140-6736(79)92440-1. [DOI] [PubMed] [Google Scholar]
- CHRISTIANS U., JACOBSEN W., SERKOVA N., BENET L.Z., VIDAL C., SEWING K.F., MANNS M.P., KIRCHNER G.I. Automated, fast and sensitive quantification of drugs in blood using LC/LC-MS: Immunosuppressants. J. Chromatogr. B. 2000;748:41–53. doi: 10.1016/s0378-4347(00)00380-7. [DOI] [PubMed] [Google Scholar]
- CROWE A., BRÜLISAUER A., DÜRR L., GINTZ P., LEMAIRE M. Absorption and intestinal metabolism of RAD and rapamycin in rats. Drug Metab. Dispos. 1999;27:627–632. [PubMed] [Google Scholar]
- CROWE A., LEMAIRE M. In vitro and in situ absorption of RAD using a human intestinal cell line (Caco-2) and a single pass perfusion model in rats: comparison with rapamycin. Pharm. Res. 1998;15:1666–1672. doi: 10.1023/a:1011940108365. [DOI] [PubMed] [Google Scholar]
- ERECINSKA M., WILSON D.F. Regulation of cellular energy metabolism. J. Membrane Biol. 1982;70:1–14. doi: 10.1007/BF01871584. [DOI] [PubMed] [Google Scholar]
- FAULDS G., GOA K.L., BENFIELD P. Cyclosporine. A review of its pharmacodynamic properties, and therapeutic use in immunoregulatory disorders. Drugs. 1993;45:953–1040. doi: 10.2165/00003495-199345060-00007. [DOI] [PubMed] [Google Scholar]
- GABE S.M., BJARNASON I., TOLOU-GHAMARI Z., TREDGER J.M., JOHSON P.G., BARCLAY G.R., WILLIAMS R., SILK D.B.A. The effect of tacrolimus (FK506) on intestinal barrier function and cellular energy production in humans. Gastroenterology. 1998;115:67–74. doi: 10.1016/s0016-5085(98)70366-x. [DOI] [PubMed] [Google Scholar]
- GIJTENBEEK J.M., VAN DEN BENT M.J., VECHT C.J. Cyclosporine neurotoxicity: a review. J. Neurol. 1999;246:339–346. doi: 10.1007/s004150050360. [DOI] [PubMed] [Google Scholar]
- GUMMERT J.F., IKONEN T., MORRIS R.E. Newer immunosuppressive drugs: a review. J. Am. Soc. Nephrol. 1999;10:1366–1380. doi: 10.1681/ASN.V1061366. [DOI] [PubMed] [Google Scholar]
- HAUBEN M. Cyclosporine neurotoxicity. Pharmacotherapy. 1996;16:576–583. [PubMed] [Google Scholar]
- HAUSEN B., IKONEN T., BRIFFA N., BERRY G.J., CHRISTIANS U., ROBBINS R.C., HOOK L., SERKOVA N., BENET L.Z., SCHULER W., MORRIS R.E. Combined immunosuppression with cyclosporine (Neoral) and SDZ RAD in non-human primate lung transplantation: systematic pharmacokinetic-based trials to improve efficacy and tolerability. Transplantation. 2000;69:76–86. doi: 10.1097/00007890-200001150-00015. [DOI] [PubMed] [Google Scholar]
- HENKE W., NICKEL E., JUNG K. Cyclosporine A inhibits ATP net uptake of rat kidney mitochondria. Biochem. Pharmacol. 1992;43:1021–1024. doi: 10.1016/0006-2952(92)90608-l. [DOI] [PubMed] [Google Scholar]
- IUPAC . Blackwell Scientific Publications, Boston; 1993. IUPAC Nomenclature of Organic Chemistry. A guide to IUPAC nomenclature of organic components (recommendations 1993) [Google Scholar]
- JITRAPAKDEE S., WALLACE J.C. Structure, function and regulation of pyruvate carboxylase. Biochem. J. 1999;340:1–16. doi: 10.1042/bj3400001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAHAN B.D. Cyclosporine. N. Engl. J. Med. 1989;321:1727–1737. doi: 10.1056/NEJM198912213212507. [DOI] [PubMed] [Google Scholar]
- KAHAN B.D. The synergistic effect of cyclosporine and sirolimus. Transplantation. 1997;63:170. doi: 10.1097/00007890-199701150-00034. [DOI] [PubMed] [Google Scholar]
- KAHAN B.D., THE RAPAMUNE US STUDY GROUP Efficacy of sirolimus compared with azathioprine for reduction of acute renal allograft rejection: a randomized multicentre study. Lancet. 2000;356:194–202. doi: 10.1016/s0140-6736(00)02480-6. [DOI] [PubMed] [Google Scholar]
- KRISTAL B.K., DUBINSKY J.M. Mitochondrial permeability transition in the central nervous system: induction by calcium cycling-dependent and -independent pathways. J. Neurochem. 1997;69:524–538. doi: 10.1046/j.1471-4159.1997.69020524.x. [DOI] [PubMed] [Google Scholar]
- LAMPEN A., ZHANG Y., HACKBARTH I., BENET L.Z., SEWING K.F., CHRISTIANS U. Metabolism and transport of the macrolide immunosuppressant sirolimus in the small intestine. J. Pharmacol. Exp. Ther. 1998;285:1104–1112. [PubMed] [Google Scholar]
- MADSEN K.L., YANCHAR N.L., SIGALET D.L., REIGEL T., FEDORAK R.N. FK506 increases permeability in rat intestine by inhibiting mitochondrial function. Gastroenterology. 1995;109:107–114. doi: 10.1016/0016-5085(95)90274-0. [DOI] [PubMed] [Google Scholar]
- MASSICOT F., MARTIN C., DUTERTRE-CATELLA H., ELOUK-ACHARD S., PHAM-HUY C., THEVENIN M., RUCAY P., WARNET J.M., CLAUDE J.R. Modulation of energy status and cytotoxicity induced by FK506 and cyclosporin A in a renal epithelial cell line. Arch. Toxicol. 1997;71:529–531. doi: 10.1007/s002040050423. [DOI] [PubMed] [Google Scholar]
- NAPOLI K.L., WANG M.E., STEPKOWSKI S.M., KAHAN B.D. Relative tissue distribution of cyclosporine and sirolimus after concomitant peroral administration to the rat: evidence for pharmacokinetic interaction. Ther. Drug Monit. 1998;20:123–133. doi: 10.1097/00007691-199804000-00001. [DOI] [PubMed] [Google Scholar]
- SALDUCCI M.D., CHAUVET-MONGES A.M., TILLEMENT J.P., ALBENGRES E., TESTA B., CARRUPT P., CREVAT A. Trimetazidine reverses calcium accumulation and impairment of phosphorylation induced by cyclosporine A in isolated rat liver mitochondria. J. Pharmacol. Exp. Ther. 1996;277:417–422. [PubMed] [Google Scholar]
- SCHINKEL A.H., WAGENAAR R., MOL C.A.A.M., VAN DEEMTER L. P-glycoprotein in the blood brain barrier of mice influences the brain penetration and pharmacological activity of many drugs. J. Clin. Invest. 1996;97:2517–2524. doi: 10.1172/JCI118699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHUURMAN H.-J., COTTENS S., FUCHS S., JOERGENSEN J., MEERLOO T., SEDRANI R., TANNER M., ZENKE G., SCHULER W. SDZ RAD, a new rapamycin derivative: Synergism with cyclosporine. Transplantation. 1997;64:32–35. doi: 10.1097/00007890-199707150-00007. [DOI] [PubMed] [Google Scholar]
- SCRUTTON M.C. Fine control of the conversion of pyruvate (phosphoenolpyruvate) to oxaloacetate in various species. FEBS Lett. 1978;89:1–9. doi: 10.1016/0014-5793(78)80510-9. [DOI] [PubMed] [Google Scholar]
- SEHGAL S.N. Rapamune (RAPA, rapamycin, sirolimus): mechanism of action immunosuppressive effect results from blockade of signal transduction and inhibition of cell cycle progression. Clin. Biochem. 1998;31:335–340. doi: 10.1016/s0009-9120(98)00045-9. [DOI] [PubMed] [Google Scholar]
- SERKOVA N., BRAND A., CHRISTIANS U., LEIBFRITZ D. Evaluation of the effects of immunosuppressants on neuronal and glial cells in vitro by multinuclear magnetic resonance spectroscopy. Biochim. Biophys. Acta. 1996;1314:93–104. doi: 10.1016/s0167-4889(96)00081-x. [DOI] [PubMed] [Google Scholar]
- SERKOVA N., HAUSEN B., BERRY G.J., JACOBSEN W., BENET L.Z., MORRIS R.E., CHRISTIANS U. Tissue distribution and clinical monitoring of the novel macrolide immunosuppressant RAD and its metabolites in monkey lung transplant recipients: interaction with cyclosporine. J. Pharmacol. Exp. Ther. 2000a;294:323–332. [PubMed] [Google Scholar]
- SERKOVA N., LITT L., JAMES T.L., SADEE W., LEIBFRITZ D., BENET L.Z., CHRISTIANS U. Evaluation of individual and combined neurotoxicity of the immunosuppressants cyclosporine and sirolimus by in vitro multinuclear NMR spectroscopy. J. Pharmacol. Exp. Ther. 1999;289:800–806. [PubMed] [Google Scholar]
- SERKOVA N., LITT L., LEIBFRITZ D., HAUSEN B., MORRIS R.E., JAMES T.L., BENET L.Z., CHRISTIANS U. The novel immunosuppressant RAD protects rat brain slices from cyclosporine-induced reduction of high-energy phosphates. Br. J. Pharmacol. 2000b;129:485–492. doi: 10.1038/sj.bjp.0703079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEPKOWSKI S.M., NAPOLI K.L., WANG M.E., QU X., CHOU T.C., KAHAN B.D. Effects of the pharmacokinetic interaction between orally administered sirolimus and cyclosporine on the synergistic prolongation of heart allograft survival in rats. Transplantation. 1996;62:986–994. doi: 10.1097/00007890-199610150-00018. [DOI] [PubMed] [Google Scholar]
- STREIT F., CHRISTIANS U., SCHIEBEL H.M., NAPOLI K.L., ERNST L., LINCK A., KAHAN B.D., SEWING K.F. Sensitive and specific quantification of sirolimus (rapamycin) and its metabolites in blood of kidney graft recipients by HPLC/electrospray-mass spectrometry. Clin. Chem. 1996;42:1417–1425. [PubMed] [Google Scholar]
- UEMOTO S., TANAKA K., ASONUMA K., OKAMURA R., KITAKADO Y., MATSUOKA S., OZAKI N., OZAWA K., HASHIDA T., INUI K., HORI R. Effect of cyclosporine on oxidative phosphorylation and adenylate energy of regenerating rat liver. Res. Exp. Med. 1989;189:313–320. doi: 10.1007/BF01855036. [DOI] [PubMed] [Google Scholar]
- WANG X., LUO H., PERKS A., WU J. Rapamycin inhibits aldolase A expression during human lymphocyte activation. J. Cell. Biochem. 1996;63:239–251. doi: 10.1002/(sici)1097-4644(19961101)63:2<239::aid-jcb11>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- WANG C., SALAHUDEEN A.K. Cyclosporine nephrotoxicity: Attenuation by an antioxidant-inhibitor of lipid peroxidation in vitro and in vivo. Transplantation. 1994;58:940–946. doi: 10.1097/00007890-199410270-00014. [DOI] [PubMed] [Google Scholar]
- WOLF A., TRENDELENBURG C.F., DIEZ-FERNANDEZ C.F., PRIETO S., HOUY E., TROMMER W.E., CORDIER A. Cyclosporine A-induced oxidative stress in rat hepatocytes. J. Pharmacol. Exp. Ther. 1997;280:1328–1334. [PubMed] [Google Scholar]