

Abstract
In porcine coronary arteries, smooth muscle hyperpolarizations produced by the nitric oxide donor, NOR-1, and the prostacyclin analogue, iloprost, were compared with those induced by substance P and bradykinin and attributed to the endothelium-derived hyperpolarizing factor (EDHF).
In the presence of 300 μM L-nitroarginine and 10 μM indomethacin, iloprost-induced hyperpolarizations were partially inhibited by 10 μM glibenclamide whereas those to NOR-1, substance P and bradykinin were unaffected.
Hyperpolarizations produced by maximally-effective concentrations of NOR-1 and NS1619 were identical (to −65 mV). They were significantly less than those generated by either substance P or bradykinin (to approximately −80 mV) and were abolished by iberiotoxin 100 nM, a concentration which had essentially no effect on responses to substance P or bradykinin.
Incubation of segments of intact arteries for 16–22 h in bicarbonate-buffered Krebs solution had little effect on EDHF responses to substance P or bradykinin. In contrast, after incubation for this period of time in HEPES-buffered Tyrode solution or Krebs containing 10 mM HEPES the EDHF response to substance P was abolished and that to bradykinin was markedly reduced. The residual bradykinin-induced hyperpolarization following incubation in Tyrode solution was inhibited by iberiotoxin and by 10 μM 17-octadecynoic acid.
We conclude that substance P activates only the EDHF pathway in the presence of nitric oxide synthase and cyclo-oxygenase inhibitors. Incubation in HEPES-buffered Tyrode solution abolishes the EDHF responses to substance P and bradykinin to reveal an additional hyperpolarizing mechanism, associated with the opening of K+ channels, activated only by bradykinin.
Keywords: EDHF, iberiotoxin, porcine coronary artery, gap junctions, HEPES, bradykinin, prostacyclin, substance P, nitric oxide donor, hyperpolarization
Introduction
In small arteries and arterioles it is now well established that agonists which interact with vascular endothelial cell receptors can hyperpolarize and relax the underlying smooth muscle. It has long been assumed that the endothelium is stimulated to release an endothelium-derived hyperpolarizing factor (EDHF) which diffuses across the myo-endothelial space to exert its effects by opening smooth muscle K+ channels (see reviews by Edwards & Weston, 1998; Félétou & Vanhoutte, 1999). However, the identity of this ‘factor' remains unknown and in view of the dissimilar pharmacology of the EDHF responses in different species and blood vessels, the possibility that several endogenous substances are involved cannot be discounted.
In most vascular preparations, a characteristic feature of the EDHF response is its full inhibition by charybdotoxin+apamin but only slight inhibition by iberiotoxin+apamin (Zygmunt & Högestätt, 1996; Petersson et al., 1997; Chataigneau et al., 1998; Eckman et al., 1998; Yamanaka et al., 1998; Edwards et al., 2000). However, there are reports that iberiotoxin alone can abolish the EDHF response in bovine and porcine coronary arteries (Gebremedhin et al., 1998; Popp et al., 1998; Fisslthaler et al., 2000). These observations contrast with our own findings that iberiotoxin alone does not inhibit EDHF in the porcine coronary artery (Edwards et al., 2000).
Other endothelium-derived factors such as nitric oxide and prostacyclin can be liberated from the endothelium by agonists which induce an EDHF-mediated response (Parkington et al., 1993; Murphy & Brayden, 1995; Zygmunt et al., 1998; Quignard et al., 2000). The aim of the present study was to determine whether or not such factors are responsible for the reported iberiotoxin-sensitive hyperpolarization of the coronary artery (Gebremedhin et al., 1998; Popp et al., 1998; Fisslthaler et al., 2000). Since substance P and bradykinin may have different effects on porcine coronary artery endothelial K+ channels (Frieden et al., 1999), both agonists were employed in this study and their effects were compared with those of iloprost (a prostacyclin analogue) and NOR-1 (a nitric oxide donor). Some of these findings have been briefly reported (Weston et al., 2001).
Methods
Experiments were performed on porcine left descending coronary arteries which were obtained from the local abattoir and transported to the laboratory in ice-cold Krebs solution. The delay in removal of the heart from the animal and dissection of the coronary artery was approximately 30 min. The coronary arteries were dissected free, cleaned of adherent fat and connective tissue, and maintained at room temperature (24°C) in the physiological salt solution appropriate to that experiment.
Micro-electrode experiments
Intact vessels were opened longitudinally and pinned to the Sylgard base of a heated bath (volume 10 ml) and, unless otherwise stated, were superfused (10 ml min−1), at 37°C, either with Krebs (gassed with 95% O2/5% CO2) or with HEPES-buffered Tyrode solution (gassed with O2), each containing 300 μM NG-nitro-L-arginine (L-NA) and 10 μM indomethacin. Smooth muscle or endothelial cells were impaled using micro-electrodes filled with 3 M KCl (resistance 40–80 MΩ). Successful impalements were signalled by a sudden change in membrane potential which remained stable for at least 2 min before the experiment was commenced. In some experiments the endothelial layer was removed by gentle rubbing and the lack of endothelium in each blood vessel segment was confirmed by the absence of a response to substance P or bradykinin. Bradykinin, levcromakalim, NOR-1 and substance P were each added as bolus injections directly into the bath in quantities calculated to give (transiently) the final concentrations indicated. Iberiotoxin was added to the reservoir of Krebs solution superfusing the bath. To examine the effects of cytochrome P450 inhibition on responses to bradykinin, endothelium-intact segments of artery were first incubated for 20–22 h at room temperature (24°C) in oxygenated HEPES-buffered Tyrode solution. They were then bathed for a further 75 min in HEPES-buffered Tyrode solution containing the suicide inhibitor 17-ODYA (10 μM). The effects of bradykinin were subsequently determined in the absence of 17-ODYA.
Recordings were made using a conventional high impedance amplifier (Intra 767; WPI Instruments) and 50 Hz interference at the amplifier output was selectively removed using an active processing circuit (Humbug; Digitimer). The membrane potential was monitored and analysed using a MacLab system in conjunction with Chart/4 software (AD Instruments).
Drugs and solutions
Krebs solution comprised (mM): NaCl 118, KCl 3.4, CaCl2 2.5, KH2PO4 1.2, MgSO4 1.2, NaHCO3 25, glucose 11.1 and was bubbled with 95% O2 and 5% CO2. For some experiments, HEPES was added to the Krebs solution to a final concentration of 10 mM. The pH of the HEPES-containing Krebs solution was adjusted to 7.4 with NaOH and the solution was gassed, as before, with 95% O2 and 5% CO2. The composition of the HEPES-buffered Tyrode solution was (mM): NaCl 140, KCl 4.7, CaCl2 1.3, MgCl2 1.0, HEPES 10, glucose 11.1. The pH of the solution was adjusted to 7.4 using NaOH and the solution was oxygenated during the incubation period.
Drugs
Synthetic iberiotoxin (Latoxan, France), Nω-nitro-L-arginine (L-NA) and HEPES (N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulphonic acid)) were dissolved directly in the bath solution to the appropriate final concentration. All other drugs were prepared as concentrated stock solutions and subsequently diluted with bath solution. Bradykinin was dissolved in methanol. Glibenclamide, iloprost (Schering AG), indomethacin, levcromakalim (SmithKline Beecham) and 17-ODYA (17-octadecynoic acid; Tocris Cookson) were dissolved in ethanol. Concentrated stocks of 1-EBIO (1-ethyl-2-benzimidazolinone; Aldrich), NOR-1 ((±)-(E)-methyl-2-[(E)-hydroxyimino]-5-nitro-6-methoxy-3-hexeneamide; Calbiochem) and NS1619 (1-(2′-hydroxy-5′ trifluoromethylphenyl)-5-trifluoromethyl-2(3H)benzimidazolone; Research Biochemicals International) were prepared in dimethylsulphoxide. Substance P (RBI) was dissolved in de-ionized water. Unless otherwise stated, all compounds were obtained from Sigma.
Statistics
All results are expressed as mean±s.e.mean. Student's t-test (for paired or unpaired observations as appropriate) was used to assess the probability that differences between mean values had arisen by chance; P<0.05 was considered to be significant.
Results
Comparison of NOR-1-, prostacyclin- and EDHF-induced hyperpolarizations
Without phenylephrine
When applied to segments of the intact coronary artery at 10 min intervals, the nitric oxide donor NOR-1 elicited reproducible hyperpolarizations of the smooth muscle (Figure 1a). In the absence of L-NA and indomethacin, the resting membrane potential of the smooth muscle was more depolarized in the absence of the endothelium (−48.6±0.5 mV, n=4) than in intact sections of arteries from the same animals (−56.2±0.9 mV, P<0.001). The lowest concentration of NOR-1 used (3 μM) produced a hyperpolarization, raising the membrane potential to a similar level in the absence (−60.6±0.9 mV, n=4) or presence (−60.4±0.8 mV, n=4) of the endothelium, although the absolute change in potential was greater in de-endothelialized (12.0±0.8 mV) than in intact preparations (4.1±0.2 mV). There was no difference between the magnitude of the hyperpolarization induced by 10 μM (14.8±0.5 mV, n=4) and 30 μM NOR-1 (15.1±0.6 mV, n=4) in endothelium-denuded vessels. Similarly, in intact vessels the magnitude of the hyperpolarization induced by 30 μM NOR-1 (8.3±0.6 mV, n=4) was not significantly different from that produced by 10 μM NOR-1 (7.7±0.5 mV, n=4). In intact vessels, the peak membrane potential following exposure to substance P (30 nM; −74.2±0.6 mV, n=4) and bradykinin (30 nM; −72.8±0.8 mV, n=4) was significantly greater than that produced by application of NOR-1 (30 μM; −64.2±0.6 mV, n=4) (Figure 1).
Figure 1.
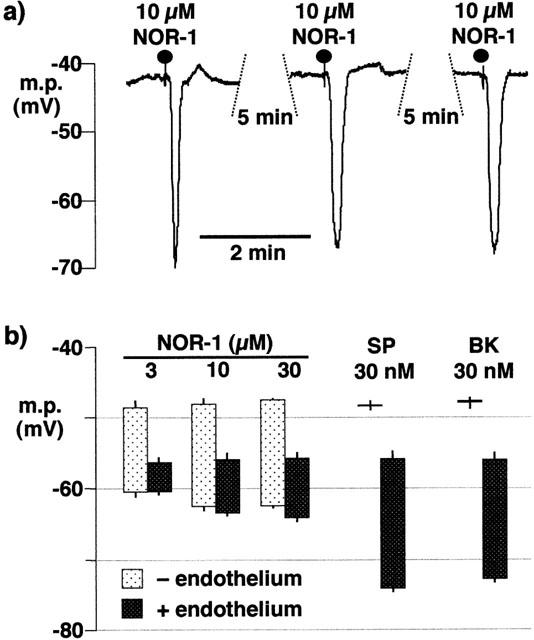
Effects of NOR-1, substance P and bradykinin on porcine coronary artery smooth muscle membrane potential. (a) In the presence of the endothelium, NOR-1 elicited reproducible hyperpolarizations. (b) Comparison of hyperpolarizing effects of NOR-1, substance P and bradykinin in the presence (+) or absence (−) of the endothelium. Each column represents the membrane potential (m.p.),+or−s.e.mean (n=4), before and after exposure to bolus doses of substance P (SP; 300 pmol), NOR-1 (30–300 nmol) or bradykinin (BK; 300 pmol) calculated to give, transiently, the final bath concentrations indicated.
In intact vessels, and in the presence of indomethacin alone (resting membrane potential −54.9±0.4 mV, n=4), substance P (100 nM), NOR-1 (10 μM) and bradykinin (30 nM) each produced a hyperpolarization (Figure 2). Additional exposure to 300 μM L-NA, depolarized the smooth muscle (to −47.2±0.4 mV) but did not modify the potential to which either substance P or NOR-1 was able to hyperpolarize. In contrast, although the magnitude of the hyperpolarization induced by bradykinin was not reduced by the presence of L-NA, the potential which was attained after application of 30 nM bradykinin (−70.8±1.7 mV, n=4) was significantly reduced in comparison to that in the absence of L-NA (−77.7±0.6 mV, n=4). The magnitude of the response to substance P was greater than that induced by either NOR-1 or bradykinin (Figure 2). In the presence of 100 nM iberiotoxin, the increase in membrane potential induced by 10 μM NOR-1 was essentially abolished whereas the responses to 100 nM substance P and 30 nM bradykinin were not significantly affected (Figure 2).
Figure 2.
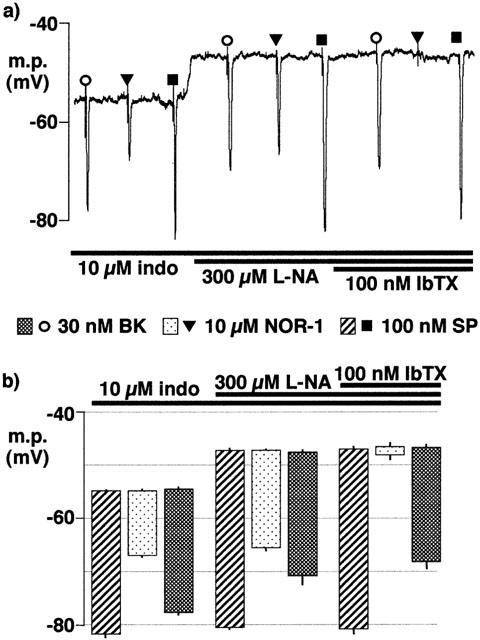
Effects of nitro-L-arginine (L-NA) and iberiotoxin on smooth muscle hyperpolarization induced by substance P, NOR-1 and bradykinin in segments of intact porcine coronary arteries. 10 μM indomethacin (indo) was present throughout. (a) Typical trace showing hyperpolarizations induced by bolus application of substance P (SP), NOR-1 and bradykinin (BK) in the absence and presence of iberiotoxin (IbTX). (b) Graphical representation of data from four separate experiments of the types shown in (a) in which each column represents the membrane potential (m.p.),+or−s.e.mean, before and after exposure to bolus doses of substance P (SP; 1 nmol), NOR-1 (100 nmol) or bradykinin (BK; 300 pmol) calculated to give, transiently, the final bath concentrations indicated. Responses are shown before (control) or after exposure to 100 μM nitro-L-arginine and, in its continued presence, to 100 nM iberiotoxin.
In the presence of L-NA and indomethacin, and in endothelium-intact segments of porcine coronary artery, iloprost (100 nM) hyperpolarized the smooth muscle to a greater extent than NOR-1 (10 μM). The effect of iloprost was only partially sensitive to inhibition by 10 μM glibenclamide (an inhibitor of the ATP-sensitive K-channel, KATP), but was abolished by subsequent exposure to 100 nM iberiotoxin in the continued presence of the sulphonylurea (Figure 3). Under these conditions the response to 10 μM levcromakalim (an opener of KATP) was fully inhibited by 10 μM glibenclamide whereas the response to bradykinin (30 nM) was not modified by the sulphonylurea or by subsequent exposure to iberiotoxin (Figure 3).
Figure 3.
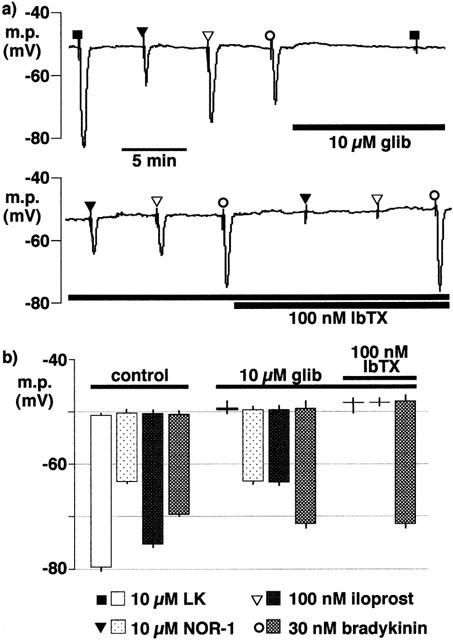
Effects of glibenclamide (glib) and iberiotoxin (IbTX) on smooth muscle hyperpolarization induced by levcromakalim (LK), NOR-1, iloprost and bradykinin in segments of intact porcine coronary arteries in the presence of 300 μM nitro-L-arginine+10 μM indomethacin. (a) Continuous trace showing typical responses. (b) Graphical representation of data from four separate experiments of the types shown in (a) in which each column represents the membrane potential (m.p.),+or−s.e.mean, before and after exposure bolus doses of levcromakalim (100 nmol), NOR-1 (100 nmol), iloprost (1 nmol) or bradykinin (300 pmol) calculated to give, transiently, the final bath concentrations indicated.
With phenylephrine
In the presence of L-NA (300 μM) and indomethacin (10 μM), phenylephrine (1 μM), a spasmogen typically employed in tension studies, depolarized the membrane potential to −44.4±0.5 mV (n=4). Under these conditions, the hyperpolarizations induced by substance P (100 nM), NOR-1 (3–30 μM) and bradykinin (10–100 nM) were compared with those induced by a maximally-effective concentration of the BKCa-channel opener, NS1619 (33 μM; Figure 4). The peak membrane potential in the presence of 100 nM bradykinin (−76.4±1.0 mV, n=4) was similar to that in the presence of 100 nM substance P (−78.4±0.6 mV, n=4) but significantly more negative than in the presence of NS1619 (−64.3±0.3 mV, n=4) or of a maximally-effective concentration of NOR-1 (30 μM, −63.6±0.3 mV, n=4). Iberiotoxin abolished the effect of NS1619 and of the nitric oxide donor but had no effect on the response to the highest dose of bradykinin employed (Figure 4).
Figure 4.
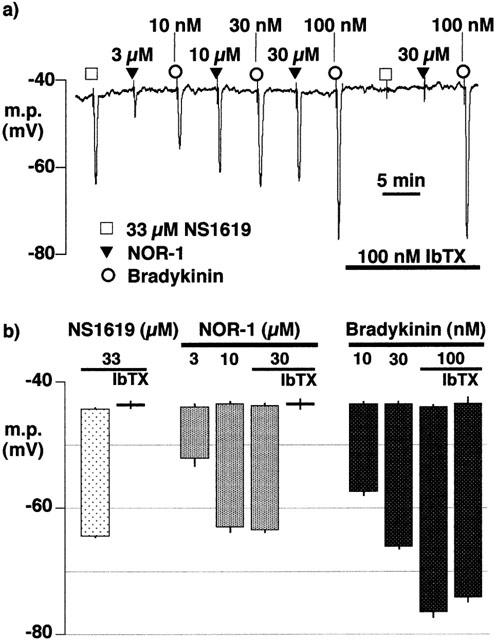
Effects of iberiotoxin on smooth muscle hyperpolarization induced by NS1619, NOR-1 and bradykinin in segments of intact porcine coronary arteries surface in the presence of 1 μM phenylephrine, 300 μM nitro-L-arginine and 10 μM indomethacin. (a) Typical trace in which hyperpolarizations induced by bolus application of increasing concentrations of NOR-1 and bradykinin were compared with those induced by the BKCa channel opener, NS1619 in the absence and presence of iberiotoxin (IbTX). (b) Graphical representation of data from four separate experiments of the types shown in (a) in which each column represents the membrane potential (m.p.),+or−s.e.mean, before and after exposure to bolus doses of NS1619 (330 nmol), NOR-1 (30–300 nmol) or bradykinin (BK; 0.1–1 nmol) calculated to give, transiently, the final bath concentrations indicated. The effects of 100 nM iberiotoxin on the highest concentrations of NS1619 and NOR-1 are also shown.
Comparison of smooth muscle EDHF responses in HEPES- or bicarbonate-buffered physiological salt solutions
Krebs solution
After incubation at room temperature for between 16 and 22 h in the bicarbonate-buffered Krebs solution, both substance P (100 nM) and bradykinin (30 nM and 100 nM) elicited smooth muscle hyperpolarizations. The magnitude of the hyperpolarizations to 100 nM substance P (−27.1±1.8 mV, n=4) and to 30 nM bradykinin (−14.0±2.7, n=4) were similar to those obtained in freshly-isolated preparations (−27.0±0.6 mV, n=4, and −16.8±0.5 mV, n=4, respectively). The responses to bradykinin (30 nM and 100 nM) were unaffected by 100 nM iberiotoxin, a concentration which abolished the hyperpolarization elicited by the opener of BKCa, NS1619 (33 μM) (Figure 5). Levcromakalim was used as an opener of an iberiotoxin-insensitive K+ channel to rule out non-selective modifications of K+ channels by prolonged maintenance of the tissue in vitro.
Figure 5.
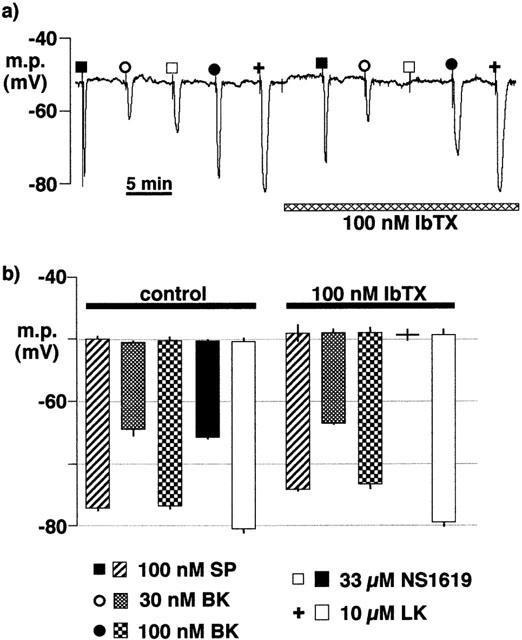
Effects of overnight incubation of intact porcine coronary arteries in Krebs solution (22–26 h, room temperature) on smooth muscle hyperpolarizations induced by substance P (SP), bradykinin (BK), NS1619 and levcromakalim (LK) in the absence and presence of iberiotoxin (IbTX). During the recording, the artery segments were bathed in Krebs solution containing 300 μM nitro-L-arginine +10 μM indomethacin. (a) Typical trace. (b) Graphical representation of data from experiments of the types shown in (a) in which each column represents the membrane potential (m.p.),+or−s.e.mean, before and after exposure to bolus doses of substance P (1 nmol), bradykinin (0.3–1 nmol), NS1619 (330 nmol), or levcromakalim (100 nmol) calculated to give, transiently, the final bath concentrations indicated; (Note: n=2 for substance P+iberiotoxin, thus error bar indicates individual results; n=4 for all other results). Responses are shown before (control) or after exposure to 100 nM iberiotoxin.
HEPES-buffered Tyrode solution
In contrast to the EDHF-induced responses in preparations incubated in Krebs solution, the response to substance P was abolished and those to the two concentrations of bradykinin were reduced in vessel segments which had been maintained overnight in HEPES–Tyrode solution. Furthermore, under these experimental conditions, the responses to bradykinin (30 and 100 nM), like that to NS1619, were abolished by iberiotoxin (Figure 6). In contrast, the hyperpolarization induced by levcromakalim was not modified by incubation in the HEPES-buffered solution and remained insensitive to iberiotoxin (Figure 6).
Figure 6.
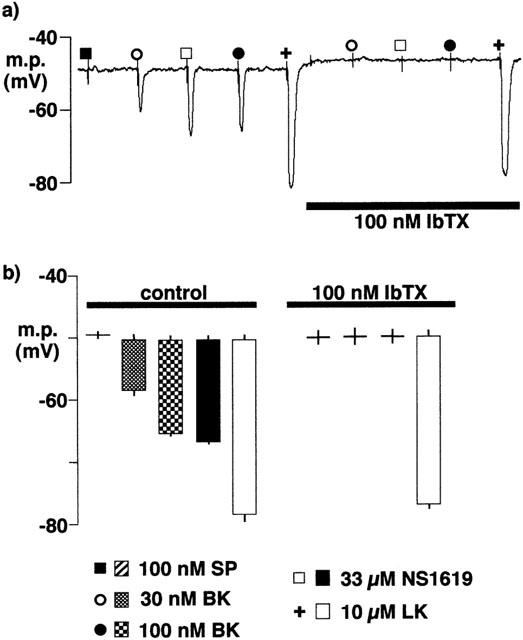
Effects of overnight incubation of intact porcine coronary arteries in Tyrode solution (22–26 h, room temperature) on smooth muscle hyperpolarizations induced by substance P (SP), bradykinin (BK), NS1619 and levcromakalim (LK). During the recording, the artery segments were bathed in Tyrode solution containing 300 μM nitro-L-arginine +10 μM indomethacin. (a) Typical trace. (b) Graphical representation of data from experiments of the types shown in (a) in which each column represents the membrane potential (m.p.),+or−s.e.mean (n=4), before and after exposure to bolus doses of substance P (1 nmol), bradykinin (0.3–1 nmol), NS1619 (330 nmol) or levcromakalim (100 nmol) calculated to give, transiently, the final bath concentrations indicated. Responses are shown before (control) or after exposure to 100 nM iberiotoxin.
Following overnight incubation in HEPES-buffered Tyrode solution, some intact artery segments were incubated for a further 75 min (at room temperature) in the presence of 10 μM 17-ODYA. In these preparations, smooth muscle EDHF responses were subsequently determined in the absence of 17-ODYA. Substance P was, as before, without effect and the hyperpolarization to bradykinin (30 and 100 nM) did not differ in magnitude from that produced by the vehicle, methanol, alone (Figure 7).
Figure 7.
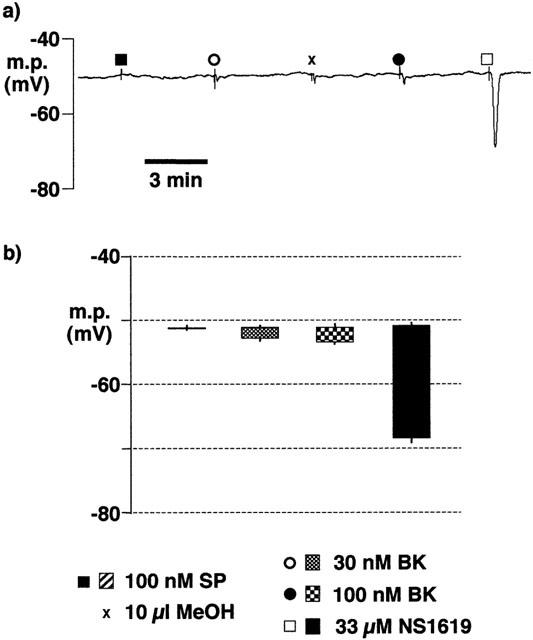
Effect of 17-ODYA (10 μM for 75 min at room temperature) on smooth muscle hyperpolarizations induced by substance P (SP), bradykinin (BK) and NS1619 following overnight incubation of intact porcine coronary arteries in HEPES-buffered Tyrode solution (22–26 h, room temperature). During the recording, the artery segments were bathed in Tyrode solution containing 300 μM nitro-L-arginine +10 μM indomethacin. (a) Typical trace showing responses to agonists and to the bradykinin vehicle, methanol (MeOH). (b) Graphical representation of data from experiments of the types shown in (a) in which each column represents the membrane potential (m.p.),+or−s.e.mean (n=4), before and after exposure to bolus doses of substance P (1 nmol), bradykinin (0.3–1 nmol and NS1619 (330 nmol) calculated to give, transiently, the final bath concentrations indicated.
Krebs solution plus 10 mM HEPES
Incubation of segments of endothelium-intact arteries in Krebs solution containing 10 mM HEPES also abolished the response to substance P (Figure 8). In these vessel segments the magnitude of the hyperpolarizations induced by 30 and 100 nM bradykinin (−8.8±0.9 mV and −16.1±0.4 mV, respectively, n=4) were not significantly different from those elicited in preparations which had been incubated in HEPES-buffered Tyrode solution (−8.3±1.6 mV and −14.8±0.8 mV, respectively, n=4).
Figure 8.
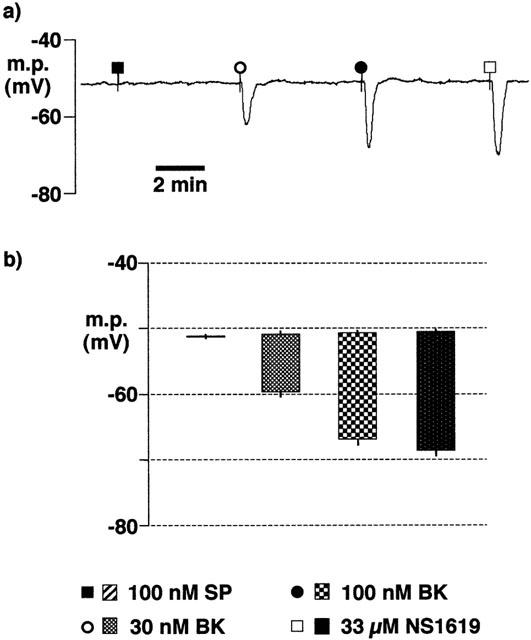
Effects of overnight incubation of intact porcine coronary arteries in Krebs solution containing 10 mM HEPES (22–26 h, room temperature) on smooth muscle hyperpolarizations induced by substance P (SP), bradykinin (BK) and NS1619. During the experiment, tissues were bathed in bicarbonate-buffered Krebs solution containing 300 μM nitro-L-arginine+10 μM indomethacin+10 mM HEPES. (a) Typical trace. (b) Graphical representation of data from experiments of the types shown in (a) in which each column represents the membrane potential (m.p.),+or−s.e.mean (n=4), before and after exposure to bolus doses of substance P (1 nmol), bradykinin (0.3–1 nmol and 1-NS1619 (330 nmol) calculated to give, transiently, the final bath concentrations indicated.
Effect of HEPES-buffered Tyrode solution on endothelial cell hyperpolarization
After incubation at room temperature for between 16–22 h in HEPES-buffered Tyrode solution, the endothelial cell hyperpolarizations to 100 nM substance P (−26.8±0.6 mV, n=4) or to 600 μM 1-EBIO (−22.6±0.8 mV, n=4) (Figure 9) were not significantly different from those obtained in freshly-dissected arteries which had been maintained in Krebs solution (−27.8±0.8 mV, n=4 and −24.1±1.0 mV, n=4, respectively).
Figure 9.
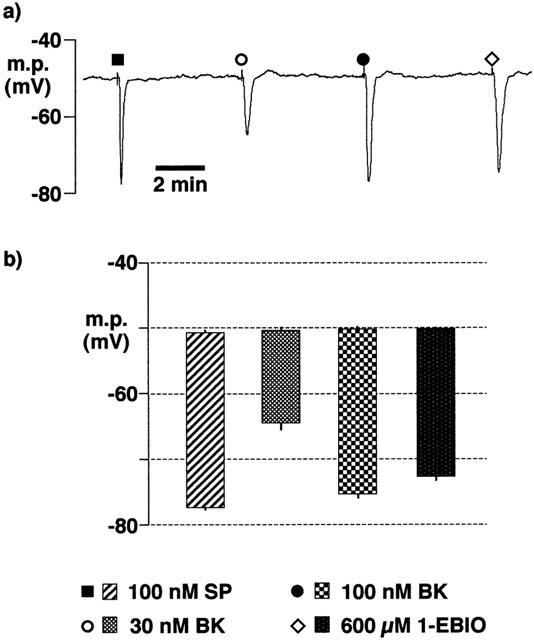
Effects of overnight incubation of intact porcine coronary arteries in HEPES-buffered Tyrode solution (22–26 h, room temperature) on endothelial cell hyperpolarizations induced by substance P (SP), bradykinin (BK) and 1-EBIO. During the experiment, tissues were bathed in Tyrode solution containing 300 μM nitro-L-arginine +10 μM indomethacin. (a) Typical trace showing agonist-induced hyperpolarizations. (b) Graphical representation of data from experiments of the types shown in (a) in which each column represents the membrane potential (m.p.),+or−s.e.mean (n=4), before and after exposure to bolus doses of substance P (1 nmol), bradykinin (0.3–1 nmol and 1-EBIO (6 μmol) calculated to give, transiently, the final bath concentrations indicated.
Discussion
Background
In the porcine coronary artery, smooth muscle hyperpolarizations induced by bradykinin and sensitive to iberiotoxin have been described. Since these experiments were conducted in the presence of cyclo-oxygenase and NO-synthase inhibitors, it was concluded that they were mediated by EDHF which exerted its actions by opening the large conductance, calcium-sensitive potassium channel, BKCa (Popp et al., 1996; 1998; Fisslthaler et al., 2000). However, these findings contrast with the failure of iberiotoxin to inhibit EDHF in many other blood vessels (see Edwards & Weston, 1998 or Félétou & Vanhoutte, 1999) and in another investigation using porcine coronary artery (Edwards et al., 2000). The present study was thus designed to determine the reason(s) for these divergent results.
Possible involvement of nitric oxide
Nitric oxide can open BKCa (Robertson et al., 1993; Bolotina et al., 1994; Peng et al., 1996; Shin et al., 1997; Lang et al., 2000; Quignard et al., 2000) and some nitric oxide release from rabbit carotid arteries still occurs in the presence of nitric oxide synthase inhibitors (Cohen et al., 1997). The possibility that the iberiotoxin-sensitive hyperpolarizations described by Popp et al. (1998) and Fisslthaler et al. (2000) were in fact mediated by nitric oxide rather than by EDHF was thus considered.
In the present study, the nitric oxide donor NOR-1 hyperpolarized the smooth muscle of pig coronary artery in the absence of the endothelium, a finding consistent with results obtained using nitric oxide donors in other blood vessels (Murphy & Brayden, 1995; Parkington et al., 1995; Corriu et al., 1996; Cohen et al., 1997). Responses to NOR-1 were reproducible and there was no evidence of tachyphylaxis under the experimental conditions employed.
Smooth muscle hyperpolarizations induced by NOR-1 were then compared with those induced by bradykinin and substance P in segments of intact coronary artery in the presence of indomethacin and L-NA. Three concentrations of NOR-1 and of bradykinin were used to determine the maximum hyperpolarization which could be achieved by these agents. Only a single concentration (100 nM) of substance P was employed since this hyperpolarized the smooth muscle cells to close to the theoretical potassium equilibrium potential (EK). The hyperpolarization generated by the maximally-effective concentration of NOR-1 raised the membrane potential to approximately −65 mV, a value essentially identical to that produced by a maximally-effective concentration of the BKCa opener, NS1619.
Hyperpolarizations induced by NOR-1 were significantly smaller than those produced by either bradykinin or substance P, both of which raised the membrane potential to close to EK. On exposure to iberiotoxin, the effects of NOR-1 and those of NS1619 were abolished, indicating that these were mediated by the opening of BKCa. In contrast, those to bradykinin and substance P were little affected.
From these studies it therefore seems unlikely that the production of nitric oxide by bradykinin due to the incomplete inhibition of NO-synthase by L-NA can account for the reported iberiotoxin sensitivity of EDHF-induced hyperpolarizations (Popp et al., 1996; Fisslthaler et al., 2000).
Possible involvement of prostacyclin
Prostacyclin is also an endothelium-derived hyperpolarizing factor, although its actions are reported to be primarily glibenclamide-sensitive (Parkington et al., 1995). However, to exclude the possibility that this agent was somehow involved in the hyperpolarizations described by Popp et al. (1996; 1998) and by Fisslthaler et al. (2000), experiments were performed using iloprost, a stable prostacyclin analogue. Iloprost hyperpolarized the smooth muscle cells of endothelium-intact pig coronary artery, an action which was partially inhibited by glibenclamide and which was abolished when the preparation was exposed to both glibenclamide and iberiotoxin. This contrasts with the findings in the guinea-pig coronary artery in which the effect of iloprost was abolished by glibenclamide alone (Parkington et al., 1995; Corriu et al., 1996). However, in the rat tail artery, prostacyclin and iloprost activate not only KATP, but also BKCa (Schubert et al., 1996; 1997). Thus, iloprost may also release an additional hyperpolarizing factor from the endothelium (possibly nitric oxide) which is responsible for the iberiotoxin-sensitivity of one component of the response observed in the present study.
Collectively, these results suggest that prostacyclin release could theoretically have contributed to the observed iberiotoxin-sensitivity of EDHF responses (Popp et al., 1998; Fisslthaler et al., 2000). However, the presence of diclofenac in their experiments together with the glibenclamide-sensitive component of iloprost-induced hyperpolarizations observed in the present study render it unlikely that prostacyclin involvement offers an explanation for the findings of Popp et al. (1998) and Fisslthaler et al. (2000).
Basic experimental conditions–involvement of HEPES
One difference between the porcine coronary artery studies in which the ‘EDHF' response was found to be iberiotoxin-insensitive (Edwards et al., 2000) and those in which it was abolished by iberiotoxin (Fisslthaler et al., 2000) lies in the experimental protocol. In the former study experiments were performed on arteries dissected and maintained in a bicarbonate-buffered Krebs solution, whereas in the latter a HEPES-buffered Tyrode solution was employed. Thus, the effects of incubation of coronary arteries either in a HEPES-buffered physiological salt solution or in a bicarbonate-buffered Krebs solution on ‘EDHF' responses were examined.
After 16–22 h incubation in a bicarbonate-buffered (Krebs) solution, both bradykinin and substance P induced iberiotoxin-resistant hyperpolarizations of the smooth muscle and these were similar in magnitude to those produced in freshly-isolated preparations. However, no substance P effect could be observed after similar exposure to the HEPES-buffered (Tyrode) solution. Furthermore, under these conditions, bradykinin-induced hyperpolarizations were relatively small (raising the membrane potential to approximately −65 mV) and these were fully inhibited by exposure to iberiotoxin.
The absence of HCO3− from the physiological solution was recently proposed to underlie the iberiotoxin-sensitivity of endothelium-dependent hyperpolarizations in porcine coronary arteries which were bathed in HEPES-buffered Tyrode solution (Chataigneau et al., 2001). These workers speculated that differences in intracellular pH maintenance and consequent changes in ion channel activity or regulation were responsible. However, HEPES and other taurine-based buffers inhibit gap junctions by a pH-independent action which may involve direct binding of the buffer amino-sulphonate moiety to the gap junction protein (Bevans & Harris, 1999).
Gap junctions almost certainly play a role in the EDHF-mediated response in several vessels including the porcine coronary artery (Kühberger et al., 1994; Chaytor et al., 1998; Dora et al., 1999; Edwards et al., 1999, 2000; Yamamoto et al., 1999). The data obtained in the present study, in which there was loss of ‘EDHF-like' responses in the presence of bicarbonate plus HEPES, clearly suggest that the underlying cause is due to the presence of HEPES, rather than to the absence of bicarbonate. The smooth muscle EDHF response to substance P was lost following exposure to HEPES, whereas the endothelial cell hyperpolarization to this agonist was retained. Collectively, these data are consistent with an inhibitory action of HEPES at myo-endothelial gap junctions in the porcine coronary artery.
Exposure to HEPES revealed a secondary action of bradykinin–endothelium-dependent hyperpolarization of smooth muscle cells via activation of BKCa. Several studies have provided evidence that bradykinin's action may be mediated, at least in part, by a cytochrome P450 metabolite, probably the epoxyeicosatrienoic acid, 11,12-EET (Hayabuchi et al., 1998; Frieden et al., 1999; Edwards et al., 2000). This eicosanoid is known to hyperpolarize vascular smooth muscle by opening BKCa (Edwards et al., 2000). Furthermore, 17-ODYA, a suicide substrate inhibitor of cytochrome P450 epoxygenase (Zou et al., 1994), abolishes the endothelium-dependent opening of smooth muscle BKCa channels and partially inhibits the relaxation to bradykinin whereas it has no effect on the relaxant response to substance P (Hayabuchi et al., 1998; Frieden et al., 1999). In the present study, 17-ODYA abolished the residual endothelium-dependent smooth muscle hyperpolarization to bradykinin following incubation in HEPES-buffered Tyrode solution. This finding strongly favours the identity of the additional hyperpolarizing factor released by bradykinin but not by substance P (and which has a pharmacology distinct from that of ‘EDHF') as an epoxyeicosatrienoic acid, probably 11,12-EET as proposed by Fisslthaler et al. (2000).
Conclusions
The results of the present study show that the classical EDHF pathway in porcine coronary artery does not involve a cytochrome P450-derived metabolite as previously claimed (Fisslthaler et al., 2000; Fleming et al., 2001). Instead, the data indicate that bradykinin stimulates not only the EDHF pathway but also one which involves cytochrome P450. In contrast, substance P only activates the classical EDHF response through a HEPES-sensitive mechanism which may involve myo-endothelial gap junctions.
Acknowledgments
The generous gift of iloprost by Schering AG, Berlin is gratefully acknowledged. We are very grateful to Dalehead Abattoir, Ashton-under-Lyne for the supply of fresh pig hearts. This study was supported by grants from the British Heart Foundation (G. Edwards, M.J. Gardener, G.R. Richards, A.H. Weston) and the Medical Research Council (C.D. Glen).
Abbreviations
- BKCa
large conductance calcium-sensitive K+ channel
- 1-EBIO
1-ethyl-2-benzimidazolinone
- EDHF
endothelium-derived hyperpolarizing factor
- HEPES
N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulphonic acid)
- KATP
ATP-sensitive K+ channel
- L-NA
Nω-nitro-L-arginine
- NOR-1
(±)-(E)-methyl-2-[(E)-hydroxyimino]-5-nitro-6-methoxy-3-hexeneamide
- NS1619
1-(2′-hydroxy-5′ trifluoromethylphenyl)-5-trifluoromethyl-2(3H)benzimidazolone
- 17-ODYA
17-octadecynoic acid
References
- BEVANS C.G., HARRIS A.L. Regulation of connexin channels by pH. Direct action of the protonated form of taurine and other aminosulfonates. J. Biol. Chem. 1999;274:3711–3719. doi: 10.1074/jbc.274.6.3711. [DOI] [PubMed] [Google Scholar]
- BOLOTINA V.M., NAJIBI S., PALACINO J.J., PAGANO P.J., COHEN R.A. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle cells. Nature. 1994;368:850–853. doi: 10.1038/368850a0. [DOI] [PubMed] [Google Scholar]
- CHATAIGNEAU T., FELETOU M., DUHAULT J., VANHOUTTE P.M. Epoxyeicosatrienoic acids, potassium channel blockers and endothelium-dependent hyperpolarization in the guinea-pig carotid artery. Br. J. Pharmacol. 1998;123:574–580. doi: 10.1038/sj.bjp.0701629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHATAIGNEAU T., FLEMING I., BUSSE R.EDHF-mediated responses induced by bradykinin in the porcine coronary artery EDHF 2000 2001Vanhoutte. London: Taylor & Francis (in press)ed. P.M [Google Scholar]
- CHAYTOR A.T., EVANS W.H., GRIFFITH T.M. Central role of heterocellular gap junctional communication in endothelium-dependent relaxations of rabbit arteries. J. Physiol. 1998;508:561–573. doi: 10.1111/j.1469-7793.1998.561bq.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COHEN R.A., PLANE F., NAJIBI S., HUK I., MALINSKI T., GARLAND C.J. Nitric oxide is the mediator of both endothelium-dependent relaxation and hyperpolarization of the rabbit carotid artery. Proc. Natl. Acad. Sci. U.S.A. 1997;94:4193–4198. doi: 10.1073/pnas.94.8.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CORRIU C., FELETOU M., CANET E., VANHOUTTE P.M. Endothelium-derived factors and hyperpolarization of the carotid artery of the guinea-pig. Br. J. Pharmacol. 1996;119:959–964. doi: 10.1111/j.1476-5381.1996.tb15765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DORA K.A., MARTIN P.E.M., CHAYTOR A.T., EVANS W.H., GARLAND C.J., GRIFFITH T.M. Role of heterocellular gap junctional communication in endothelium-dependent smooth muscle hyperpolarization: inhibition by a connexin-mimetic peptide. Biochem. Biophys. Res. Commun. 1999;254:27–31. doi: 10.1006/bbrc.1998.9877. [DOI] [PubMed] [Google Scholar]
- ECKMAN D.M., HOPKINS N., MCBRIDE C., KEEF K.D. Endothelium-dependent relaxation and hyperpolarization in guinea-pig coronary artery: role of epoxyeicosatrienoic acid. Br. J. Pharmacol. 1998;124:181–189. doi: 10.1038/sj.bjp.0701778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDWARDS G., FELETOU M., GARDENER M.J., THOLLON C., VANHOUTTE P.M., WESTON A.H. Role of gap junctions in the responses to EDHF in rat and guinea-pig small arteries. Br. J. Pharmacol. 1999;128:1788–1794. doi: 10.1038/sj.bjp.0703009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDWARDS G., THOLLON C., GARDENER M.J., FELETOU M., VILAINE J., VANHOUTTE P.M., WESTON A.H. Role of gap junctions and EETs in endothelium-dependent hyperpolarization of porcine coronary artery. Br. J. Pharmacol. 2000;129:1145–1154. doi: 10.1038/sj.bjp.0703188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDWARDS G., WESTON A.H.Endothelium-derived hyperpolarizing factor-a critical appraisal Progress in Drug Research 1998Basel BirkhauserVerlag; 107–133.Vol. 50. (ed) E. Jucker. pp [DOI] [PubMed] [Google Scholar]
- FELETOU M., VANHOUTTE P.M. The alternative: EDHF. J. Mol. Cell. Cardiol. 1999;31:15–22. doi: 10.1006/jmcc.1998.0840. [DOI] [PubMed] [Google Scholar]
- FISSLTHALER B., HINSCH N., CHATAIGNEAU T., POPP R., KISS L., BUSSE R., FLEMING I. Nifedipine increases cytochrome P4502C expression and endothelium-derived hyperpolarizing factor-mediated responses in coronary arteries. Hypertension. 2000;36:270–275. doi: 10.1161/01.hyp.36.2.270. [DOI] [PubMed] [Google Scholar]
- FLEMING I., FISSLTHALER B., MICHAELIS U.R., KISS L., POPP R., BUSSE R.The coronary endothelium-derived hyperpolarizing factor (EDHF) stimulates multiple signalling pathways and proliferation in vascular cells Pflügers Arch. 2001(in press) [DOI] [PubMed]
- FRIEDEN M., SOLLINI M., BENY J. Substance P and bradykinin activate different types of KCa currents to hyperpolarize cultured porcine coronary artery endothelial cells. J. Physiol. 1999;519:361–371. doi: 10.1111/j.1469-7793.1999.0361m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GEBREMEDHIN D., HARDER D.R., PRATT P.F., CAMPBELL W.B. Bioassay of an endothelium-derived hyperpolarizing factor from bovine coronary arteries: role of a cytochrome P450 mono-oxygenase. J. Vasc. Res. 1998;35:274–284. doi: 10.1159/000025594. [DOI] [PubMed] [Google Scholar]
- HAYABUCHI Y., NAKAYA Y., MATSUOKA S., KURODA Y. Endothelium-derived hyperpolarizing factor activates Ca2+-activated K+ channels in porcine coronary artery smooth muscle cells. J. Cardiovasc. Pharmacol. 1998;32:642–649. doi: 10.1097/00005344-199810000-00018. [DOI] [PubMed] [Google Scholar]
- KÜHBERGER E., GROSCHNER K., KUKOVETZ W.R., BRUNNER F. The role of myoendothelial cell contact in non-nitric oxide-, non-prostanoid-mediated endothelium-dependent relaxation of porcine coronary artery. Br. J. Pharmacol. 1994;113:1289–1294. doi: 10.1111/j.1476-5381.1994.tb17138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LANG R.J., HARVEY J.R., MCPHEE G.J., KLEMM M.F. Nitric oxide and thiol reagent modulation of Ca2+-activated K+ (BKCa) channels in myocytes of the guinea-pig taenia caeci. J. Physiol. 2000;525:363–376. doi: 10.1111/j.1469-7793.2000.00363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MURPHY M.E., BRAYDEN J.E. Nitric oxide hyperpolarizes rabbit mesenteric arteries via ATP-sensitive potassium channels. J Physiol. 1995;486:47–58. doi: 10.1113/jphysiol.1995.sp020789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PARKINGTON H.C., TARE M., TONTA M.A., COLEMAN H.A. Stretch revealed three components in the hyperpolarization of guinea-pig coronary artery in response to acetylcholine. J. Physiol. 1993;465:459–476. doi: 10.1113/jphysiol.1993.sp019687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PARKINGTON H.C., TONTA M.A., COLEMAN H.A., TARE M. Role of membrane potential in endothelium-dependent relaxation of guinea-pig coronary arterial smooth muscle. J. Physiol. 1995;484:469–480. doi: 10.1113/jphysiol.1995.sp020679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PENG W., HOIDAL J.R., FARRUKH I.S. Regulation of Ca2+-activated K+ channels in pulmonary vascular smooth muscle cells–role of nitric oxide. J. Appl. Physiol. 1996;81:1264–1272. doi: 10.1152/jappl.1996.81.3.1264. [DOI] [PubMed] [Google Scholar]
- PETERSSON J., ZYGMUNT P.M., HÖGESTÄTT E.D. Characterization of the potassium channels involved in EDHF-mediated relaxation in cerebral arteries. Br. J. Pharmacol. 1997;120:1344–1350. doi: 10.1038/sj.bjp.0701032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POPP R., BAUERSACHS J., HECKER M., FLEMING I., BUSSE R. A transferable, beta-naphthoflavone-inducible, hyperpolarizing factor is synthesized by native and cultured porcine coronary endothelial cells. J. Physiol. 1996;497:699–709. doi: 10.1113/jphysiol.1996.sp021801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POPP R., FLEMING I., BUSSE R. Pulsatile stretch in coronary arteries elicits release of endothelium-derived hyperpolarizing factor: a modulator of arterial compliance. Circ. Res. 1998;82:696–703. doi: 10.1161/01.res.82.6.696. [DOI] [PubMed] [Google Scholar]
- QUIGNARD J., FELETOU M., CORRIU C., CHATAIGNEAU T., EDWARDS G., WESTON A.H., VANHOUTTE P.M. 3-Morpholinosydnonimine (SIN-1) and K+ channels in smooth muscle cells of the rabbit and guinea pig carotid arteries. Eur. J. Pharmacol. 2000;399:9–16. doi: 10.1016/s0014-2999(00)00372-1. [DOI] [PubMed] [Google Scholar]
- ROBERTSON B.E., SCHUBERT R., HESCHELER J., NELSON M.T. Cyclic-GMP-dependent protein kinase activates Ca-activated K channels in cerebral artery smooth muscle cells. Am. J. Physiol. 1993;265:C299–C303. doi: 10.1152/ajpcell.1993.265.1.C299. [DOI] [PubMed] [Google Scholar]
- SCHUBERT R., SEREBRYAKOV V.N., ENGEL H., HOPP H.H. Iloprost activates KCa channels of vascular smooth muscle cells: role of cAMP-dependent protein kinase. Am. J. Physiol. 1996;271:C1203–C1211. doi: 10.1152/ajpcell.1996.271.4.C1203. [DOI] [PubMed] [Google Scholar]
- SCHUBERT R., SEREBRYAKOV V.N., MEWES H., HOPP H.H. Iloprost dilates rat small arteries: role of K(ATP)- and K(Ca)-channel activation by cAMP-dependent protein kinase. Am. J. Physiol. 1997;272:H1147–H1156. doi: 10.1152/ajpheart.1997.272.3.H1147. [DOI] [PubMed] [Google Scholar]
- SHIN J.H., CHUNG S., PARK E.J., UHM D.Y., SUH C.K. Nitric oxide directly activates calcium-activated potassium channels from rat brain reconstituted into planar lipid bilayer. FEBS Lett. 1997;415:299–302. doi: 10.1016/s0014-5793(97)01144-7. [DOI] [PubMed] [Google Scholar]
- WESTON A.H., GARDENER M.J., FÉLÉTOU M., VANHOUTTE P.M., EDWARDS G. Components of the bradykinin-induced endothelium-dependent hyperpolarization of pig coronary artery. Br. J. Pharmacol. 2001;133:68P. doi: 10.1038/sj.bjp.0704157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAMAMOTO Y., IMAEDA K., SUZUKI H. Endothelium-dependent hyperpolarization and intercellular electrical coupling in guinea-pig mesenteric arterioles. J. Physiol. 1999;514:505–513. doi: 10.1111/j.1469-7793.1999.505ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAMANAKA A., ISHIKAWA T., GOTO K. Characterization of endothelium-dependent relaxation independent of NO and prostaglandins in guinea pig coronary artery. J. Pharmacol. Exp. Ther. 1998;285:480–489. [PubMed] [Google Scholar]
- ZOU A.P., MA Y.H., SUI Z.H., ORTIZ DE MONTELLANO P.R., CLARK J.E., MASTERS B.S., ROMAN R.J. Effects of 17-octadecynoic acid, a suicide-substrate inhibitor of cytochrome P450 fatty acid omega-hydroxylase, on renal function in rats. J. Pharmacol. Exp. Ther. 1994;268:474–481. [PubMed] [Google Scholar]
- ZYGMUNT P.M., HÖGESTÄTT E.D. Role of potassium channels in endothelium-dependent relaxation to nitroarginine in the rat hepatic artery. Br. J. Pharmacol. 1996;117:1600–1606. doi: 10.1111/j.1476-5381.1996.tb15327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZYGMUNT P.M., PLANE F., PAULSSON M., GARLAND C.J., HÖGESTÄTT E.D. Interactions between endothelium-derived relaxing factors in the rat hepatic artery: focus on regulation of EDHF. Br. J. Pharmacol. 1998;124:992–1000. doi: 10.1038/sj.bjp.0701893. [DOI] [PMC free article] [PubMed] [Google Scholar]