

Abstract
The acyl-CoA:cholesterol acyltransferase (ACAT) enzyme is thought to be responsible for foam cell formation and the subsequent progression of atherosclerosis. The apolipoprotein E and low density lipoprotein receptor double knockout (apoE/LDLr-DKO) mouse is an animal model that develops severe hyperlipidaemia and atherosclerosis.
Here we have examined the effect of oral administration of an ACAT inhibitor, F-1394, on atherosclerosis in apoE/LDLr-DKO mice fed a regular chow diet.
In en face analysis, a dose of 10, 30, or 100 mg kg−1 day−1 F-1394 for 10 weeks reduced the extent of lesions visible in the aorta by 24, 28 and 38%, respectively, as detected by staining with oil red O, without affecting serum cholesterol level in these mice. At the highest dose 100 mg kg−1 day−1 of F-1394, the reduction was statistically significant.
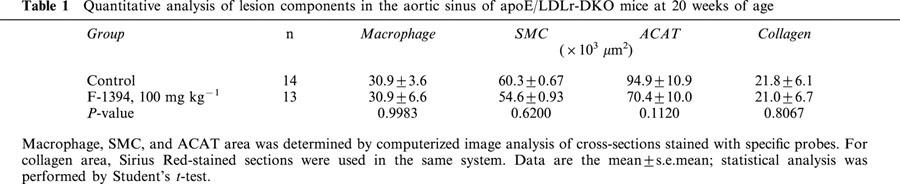
For quantitative analysis of the cellular and non-cellular components comprising the lesions at the aortic sinus, the effects of an oral dose of 100 mg kg−1 day−1 F-1394 for 15 weeks were studied. There was a significant reduction (31.9%) in the oil-red O-stained area in cross-sections of the aortic sinus. In addition, the neointimal area, as well as levels of ACAT-1 protein tended to be decreased (15.2 and 25.8%, respectively, not significant). However, the areas containing macrophages, smooth muscle cells, and collagen were not affected by F-1394.
In vitro, F-1394 attenuated foam cell formation in mouse peritoneal macrophages.
These results indicate that ACAT may be primarily responsible for lipid accumulation in atherosclerotic lesions, and that its inhibition diminishes the lipid deposition via a direct effect on macrophages in the arterial wall.
Keywords: ACAT, ACAT-inhibitor, F-1394, atherosclerosis, apolipoprotein E, low density lipoprotein receptor, knockout mouse, macrophages, foam cells, smooth muscle cells
Introduction
Coronary heart disease (CHD) is a major cause of morbidity and mortality in much of the world (Breslow, 1997). In the past 10 years, a great deal of information has accumulated on the molecular mechanisms underlying hypercholesterolaemia-initiated atherosclerosis. In these conditions, low density lipoprotein (LDL) is thought to be modified in the subendothelial space, and this modified lipoprotein activates endothelial cells, which then attract circulating monocytes (Cybulsky & Gimbrone, 1991; Kume et al., 1992; Witztum & Steinberg, 1991). The monocytes enter the vessel wall, differentiate into macrophages, and then endocytose the modified lipoproteins via a scavenger receptor pathway. This unrestricted uptake, which is not regulated by the intracellular cholesterol level, eventually leads to the formation of lipid-filled foam cells–the initial step in premature atherosclerosis (Brown & Goldstein, 1983; Ross, 1999). In this process, cholesterol esterification via the acyl-CoA:cholesterol acyltransferase (ACAT)-1 enzyme in macrophages is thought to be a major step in foam cell formation (Brown & Goldstein, 1983; Miyazaki et al., 1998). Thus, inhibition of ACAT would be expected to improve or limit the development of atherosclerosis (Matsuda, 1994; Sliskovic & White, 1991). Many types of ACAT inhibitor have therefore been tested for their direct effect on atherosclerosis development in different atherosclerotic models using various protocols (Bocan et al., 1991; 1993; Murakami et al., 1995; Nicolosi et al., 1998). However, these models do not seem accurately to reflect human hyperlipidaemia.
Recently, genetically altered hypercholesterolaemic animal models such as apolipoprotein E (apoE) -deficient mice have been produced, which develop a full range of atherosclerotic lesions (Plump et al., 1992; Zhang et al., 1992), from fatty streaks to raised fibrous plaques which are similar to those seen in humans (Nakashima et al., 1994). Thus, these mice are considered to represent a suitable model to study atherogenesis, as well as the effectiveness of drugs on disease (Breslow, 1996). To date, however, there have been no studies investigating the influence of ACAT-1 disruption by pharmacological inhibitors on the development of atherosclerosis in these mouse models.
More recently, apoE and LDL-receptor (LDLr) double-knockout (apoE/LDLr-DKO) mice have been created (Ishibashi et al., 1994), representing a new mouse model that develops severe hyperlipidaemia and atherosclerosis (Bonthu et al., 1997). It has been reported that, even on a regular chow diet, the progression of atherosclerosis is usually more marked in apoE/LDLr-DKO mice than in mice deficient for apoE alone (Witting et al., 1999). Thus, the apoE/LDLr-DKO mouse is a suitable model in which to study the anti-atherosclerotic effect of compounds without having to feed the animals an atherogenic diet.
Here we have tested the direct effect of (1S,2S)-2-[3-(2,2-dimethylpropyl)-2-nonylureido]cyclohexane-1-yl 3-[(4R)-N-(2,2,5,5-tetramethyl-1,3-dioxane-4-carbonyl)amino]propionate (F-1394), which potently inhibits both ACAT-1 and ACAT-2 (Kusunoki et al., 1995b), on the development of atherosclerosis in apoE/LDLr-DKO mice fed a regular chow diet.
Methods
Animals and diet
ApoE/LDLr-DKO mice were created by mating apoE-deficient mice and LDLr-knockout mice, as described previously (Ishibashi et al., 1994). The genetic background of these mice was derived from the 129/Ola and C57BL/6J strains. ApoE/LDLr-DKO mice were maintained in a temperature- and humidity-regulated animal facility (22±2°C, 55±15%) with controlled lighting (12-h light/dark cycle). They had free access to both tap water and a commercial regular chow diet (CRF-1; Oriental Yeast) throughout the study. All experiments were performed on both male and female mice, and approved by the Animal Ethics Committee of Fujirebio Incorporated.
Study protocol
To determine how the ACAT enzyme affects atherosclerosis formation, an orally active ACAT inhibitor, compound F-1394 (Fujirebio Inc., Tokyo, Japan), was used to inhibit ACAT in vivo (Aragane et al., 1998; Kusunoki et al., 1995a). For oral administration, F-1394 was suspended in 0.5% sodium carboxymethylcellulose solution.
For en face analysis, the apoE/LDLr-DKO mice were divided into four groups at 5 weeks of age, and each group was given F-1394 at a dose of 0, 10, 30, or 100 mg kg−1 day−1 for 10 weeks. For detailed histological analysis of the aortic sinus, the DKO mice were divided into two groups at 5 weeks of age, and each group was given F-1394 at a dose of 0 or 100 mg kg−1 day−1 for 10–15 weeks. At the end of each experiment, blood was collected from the animals after overnight fasting.
Tissue preparation and histochemistry
After the mice were sacrificed under ether anaesthesia, the heart and the whole aorta were perfused with phosphate-buffered formaldehyde (10%, pH 7.4) containing 5% sucrose, before being removed and fixed in the same solution.
The whole aorta was used for en face lipid staining (Tangirala et al., 1995). After the adventitia had been removed, the aorta was cut open longitudinally and stained with oil red O to visualize the extent of the lipid deposition.
For the analysis of the aortic sinus, the heart was embedded in OTC compound (Milles Inc.), and serial cross-sections 8 μm thick were cut by cryostat from any lesions seen in the aortic sinus. The serial sections were stained with oil red O and with Mayer's hematoxylin, Elastica van Gieson, or Sirius red (Junqueira et al., 1979; Kratky et al., 1996) to visualize lipid, fibrous extracellular matrixes, or collagen (type I, II, and III), respectively. Macrophages, smooth muscle cells (SMC), and ACAT-1 protein in a given section were visualized by anti-mouse macrophage monoclonal antibody (BM8; BMA Biochemicals), anti-human SMC monoclonal antibody (1A4; Dako), and anti-mouse ACAT-1 monoclonal antibody (KF-620; Fujirebio Inc.), respectively. These immunostained sections were then counterstained with methyl green.
Evaluation of atherosclerotic lesions
Images from a cross-section or from the aorta were collected in one of two ways: either using a microscope (Nikon, Japan) fitted with a 3-CCD Color Camera (KY-F55MP; Victor) or using a video-microscope (PV-10; Olympus). Quantification of the images was carried out using Mac Scope software (Mitani Corporation) after carefully outlining a specific area of the image.
Serum lipid measurement
Serum total cholesterol (TC) and triglyceride (TG) levels were measured by enzymatic/colorimetric methods (Cholesterol E-HA Test Wako; Triglyceride E-HA Test Wako; Wako Pure Chemical Industries, Osaka, Japan). The high density lipoprotein cholesterol (HDL-C) level was also determined enzymatically after selective precipitation of all lipoproteins except HDL (HDL cholesterol Precipitation Reagent Set; Wako Pure Chemical Industries).
Peritoneal macrophage assay
Human plasma was obtained from healthy volunteers, and LDL (d=1.019–1.063 g ml−1) and HDL3 (d=1.125–1.210 g ml−1) were isolated by ultracentrifugation as described previously (Havel et al., 1955). Acetylated LDL (acLDL) was prepared according to the method of Basu et al. (1976). Peritoneal macrophages were collected from male ddY mice (Sankyo Labo Service), and cells that adhered to the culture dish were used in this study. Briefly, macrophages (2×106 cells) were plated and preincubated for 2 h in Dulbecco's modified Eagle's medium (DMEM) containing 10% foetal bovine serum (FBS, Gibco) and antibiotics. Then, the following 24-h incubations were carried out in DMEM containing 0.1% bovine serum albumin (BSA): (i) to induce foam cells, 25 μg ml−1 acLDL was added to the media; and (ii) to determine the effect of ACAT inhibition on foam cell formation, 25 μg ml−1 acLDL and 5 μg ml−1 HDL3 with or without F-1394 dissolved in dimethylsulfoxide (final concentration, 0.1%) was added to the media. After incubation, intracellular lipids were extracted with n-hexane/2-isopropanol (3 : 2, v v−1), and TC and free cholesterol (FC) were determined enzymatically. Esterified cholesterol (EC) was calculated by subtraction of FC from TC.
Statistical analysis
The results are expressed as mean±standard error (s.e.mean). Statistical differences among the groups were assessed using Williams' multiple range test, and differences between groups were assessed using Student's t-test or Aspin–Welch's test (SAS software package). Values of P<0.05 were considered to be significant.
Results
En face analysis of the aorta
In the en face lipid staining analysis, oil red O stained 8.01±1.41% of the luminal surface of the whole aorta in the control group. In contrast, administration of F-1394 diminished lipid deposition in the aorta in a dose-dependent manner; at the highest dose of F-1394, lipid deposition was significantly reduced by 38.1% (Figure 1). The mean serum TC level in the control group was 732±44 mg dl−1, which was not affected by the administration of F-1394.
Figure 1.
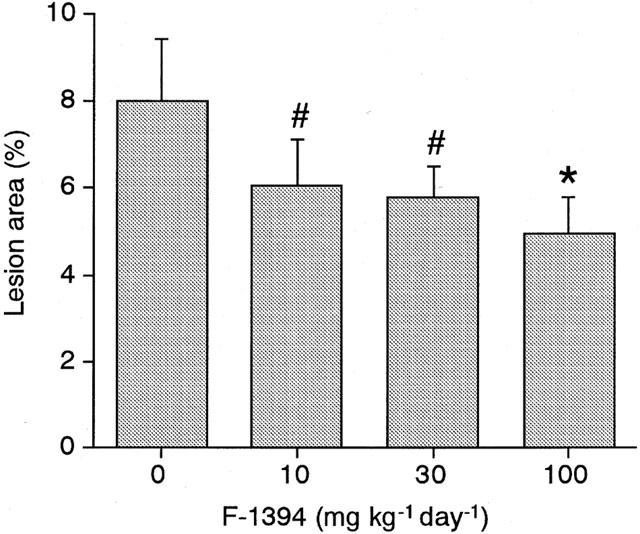
Quantitative analysis of en face lipid-stained area in whole aorta from apoE/LDLr DKO-mice at 15 weeks of age. Each column is the mean±s.e.mean of results from eight mice. Statistical analysis was performed by Williams' multiple range testing. #P<0.10; *P<0.05 vs control.
Atherosclerosis at the aortic sinus
Quantitative analysis of the atherosclerotic lesions found in the aortic sinus in the control group determined the neointimal area to be approximately 320×103 μm2 and 490×103 μm2 at 15 and 20 weeks of age, respectively (Figure 2A). In the F-1394-treated mice, the neointimal area was slightly reduced (by 8.4 and 15.2% at 15 and 20 weeks of age, respectively), but this difference did not reach statistical significance. F-1394 treatment reduced the size of the area stained with oil red O by 30.7% (P=0.217) and 31.9% (P=0.027) in mice at 15 and 20 weeks of age, respectively (Figure 2B); this difference did reach statistical significance for the 20-week-old group. ACAT protein in the lesions was located in an area of 94.9±10.9×103 μm2 in sections from the control group. F-1394 treatment reduced ACAT protein in the lesions by 26%, but this difference did not reach significance (P=0.1120) (Table 1). The other lesion components that we examined, namely macrophages, SMC, and collagen, were not altered by F-1394 treatment (Table 1).
Figure 2.
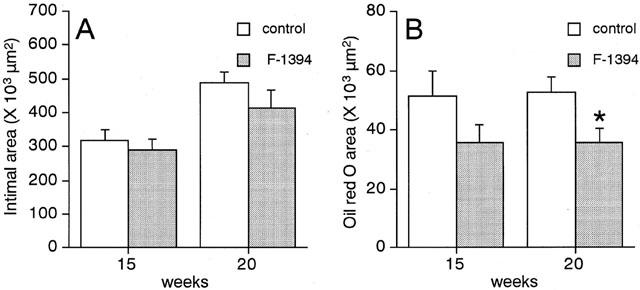
Quantitative analysis of the extent of neointima (A) and oil red O-positive area (B) in the aortic sinus from apoE/LDLr-DKO mice. Each column is the mean±s.e.mean of results from 13–14 mice. Statistical analysis was performed by Student's t-test. *P<0.05 vs corresponding control.
Table 1.
Quantitative analysis of lesion components in the aortic sinus of apoE/LDLr-DKO mice at 20 weeks of age
Typical cross-sections of the aortic sinus from control and F-1394-treated mice are shown in Figure 3. Lipid staining was confined to the subendothelial space in sections from the F-1394-treated group, whereas lipid deposition was clearly present throughout the entire neointima in the control group. Macrophages, neutral lipids and ACAT protein were colocalized in these sections. In contrast, collagen was not colocalized with these components in lesions from either group. SMC were just visible in the lesions from either group.
Figure 3.
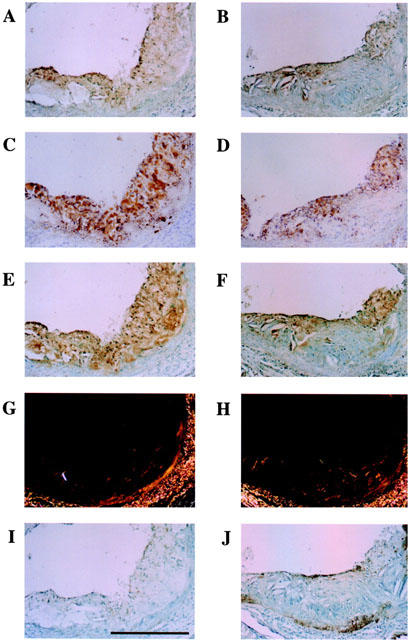
Photomicrographs showing serial cross-sections of aortic sinus from apoE/LDLr-DKO mice at 20 weeks of age. Left side, control group (A, C, E, G and I); right side, F-1394-treated group (B, D, F, H and J). A and B, macrophage (BM8); C and D, neutral lipid (oil red O stain); E and F, ACAT (KF-620); G and H, collagen (Sirius red); I and J, SMC (1A4). Scale is 100 μm for all photographs. Magnification,×25.
In control mice at 20 weeks of age, serum TC, non HDL-C, HDL-C, and TG levels were 538±30, 525±29, 13±2 and 69±7 mg dl−1, respectively. The repeated administration of F-1394 did not affect these values. In mice at 15 weeks of age, the levels of most of these serum lipids were much greater than at 20 weeks, but there were no differences in levels between control and F-1394-treated mice (Table 2).
Table 2.
Body weight and serum lipid levels in apoE/LDLr-DKO mice
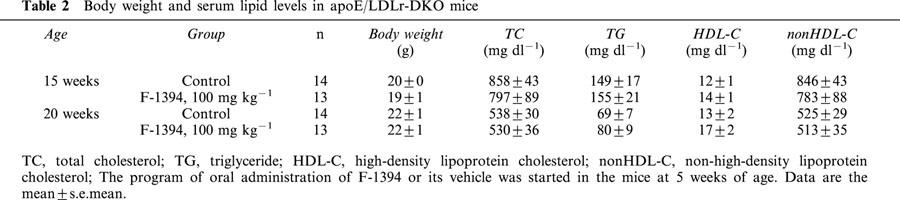
At the end of the F-1394 dosing period (at 20 weeks of age), the mean body weight of both control and F-1394-treated mice was 22 g (Table 2). Moreover, the F-1394-treated mice displayed no abnormalities, such as alopecia or dermal hypertrophy during the dosing period.
Foam cell formation in mouse peritoneal macrophages
Figure 4 shows the effect of F-1394 on the cholesterol content of foam cells. In normal macrophages, the intracellular concentrations of TC, FC, and EC were 61.5±2.6, 61.5±1.8, and 0.0±0.6 nmol mg protein−1, respectively. In acLDL-induced foam cells, the concentrations of TC and FC were 2.02 fold and 1.54 fold higher, respectively, and the EC content in particular was increased markedly (to 29.7±1.0 nmol mg protein−1). HDL significantly decreased the concentrations of TC and EC (by 18.0 and 37.4%, respectively), but not the FC concentration found in acLDL-induced foam cells. Moreover, further reductions in TC and EC concentrations were observed in the HDL/F-1394-treated foam cells; the reduction observed was dependent on the concentration of F-1394. At the highest concentration of F-1394 used (1.0 μmol l−1), the concentrations of TC and EC were 61 and 12.3%, respectively, of the concentrations seen in the foam cells treated only with HDL. The FC concentration was not affected by F-1394 at 0.3 μmol l−1 or less, but was significantly reduced (by 12.3%) following treatment with F-1394 at 1.0 μmol l−1.
Figure 4.
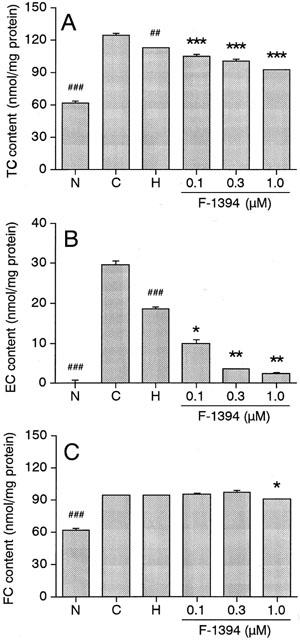
Effect of F-1394 on accumulation of TC (A), EC (B), and FC (C) in mouse peritoneal macrophages. Left columns: N, normal macrophages; C, control macrophages (foam cells); H, HDL-treated control macrophages; right columns; F-1394- and HDL-treated control macrophages. Each column is the mean±s.e.mean of results from triplicate assays. Statistical analysis was performed by Student's t-test or Aspin–Welch's test. ##P<0.01; ###P<0.001 vs control macrophages (C). *P<0.05, **P<0.01, ***P<0.001 vs HDL-treated control macrophages (H).
Discussion
In preliminary studies, early small lesions comprising only of foam cells in the aortic sinus were observed in some of the apoE/LDLr-DKO mice at 5 weeks of age, and these lesions stained with oil red O (data not shown). Therefore we started administration of F-1394 to the mice at 5 weeks of age.
As expected, F-1394 significantly reduced the extent of en face lipid staining in the whole aorta in a dose-dependent manner (Figure 1). In addition, F-1394 significantly decreased lipid deposition detected by oil red O in the neointima of the aortic sinus (Figure 2B). In general, ACAT inhibitors are predicted to have an anti-atherosclerotic effect because ACAT-1 catalyzes both cholesterol esterification in macrophages and the subsequent formation of foam cells – a distinctive feature of premature atherosclerosis (Matsuda, 1994). We previously reported that F-1394 strongly inhibited ACAT activity in a murine macrophage cell line (Kusunoki et al., 1995b). In the present study, we found that F-1394 decreased foam cell formation in mouse peritoneal macrophages in a dose-dependent manner in vitro (Figure 4). Furthermore, a previous pharmacokinetic study had shown that the dose of F-1394 administered to mice resulted in a sufficiently high concentration in the circulation to exert these effects in vivo (unpublished data). These findings suggest that the beneficial effect of F-1394 documented in this study is due to diminished ACAT-1 activity in macrophages, decreased foam cell formation, and a subsequent reduction in lipid deposition on the arterial wall. Moreover, this does not seem to be due to a hypolipidaemic effect of F-1394, but to a direct action at the arterial wall, because the drug did not affect the levels of any serum lipids (Table 2).
Previous investigators have proposed that ACAT inhibitors may have a direct action on atherosclerosis in a rabbit model in which atherosclerosis is induced by an atherogenic diet (Bocan et al., 1991; 2000; Tanaka et al., 1994). However, apart from the work of Matsuo et al. (1995), these models do not necessarily reflect the actual hypercholesterolaemic conditions seen in patients with atherosclerosis. Matsuo et al. (1995) have reported an anti-atherosclerotic effect of an ACAT inhibitor, FR-145237, which reduced the lesion index at coronary arteries in rabbits with spontaneous hypercholesterolaemia that was not induced by a hypercholesterolaemic diet. Although this model closely reflects the hypercholesterolaemia seen in humans, these authors did not carry out a detailed analysis of the lesions. Thus, little is known about the changes induced by ACAT inhibitors in the components of lesions in the face of persistent hypercholesterolaemia. Therefore, in the present study, we adopted a ‘human-like' protocol using a spontaneous hypercholesterolaemic mouse model on a regular chow diet to evaluate the direct anti-atherosclerotic effect of the ACAT inhibitor F-1394, and we also performed a detailed histochemical analysis of the lesions, which are similar to those seen in humans (Nakashima et al., 1994).
Although we found no statistically significant differences, the neointimal area and amount of ACAT protein were reduced by 15.2 and 26%, respectively, in the F-1394-treated group. However, the levels of macrophages, SMC, and collagen fibres (types I, II and III) were not affected. There is a number of possibilities to explain the reduced lesion size in F-1394-treated mice. ACAT inhibition might cause atrophy of the macrophages/foam cells, or might affect the extracellular matrix, which would not have been detected by the dye used. Alternatively, ACAT inhibition might prevent migration into the lesions of the other cellular components, which are attracted by factors released from foam cells. In addition, the reduced expression of ACAT-1 protein in the lesions might be attributable to attenuation of foam cell formation by F-1394 treatment in such lesions, but the underlying mechanisms remain unclear. It has been reported that ACAT-1 mRNA and protein are induced during differentiation of monocytes into macrophages, but expression of these products is not induced by foam cell formation in macrophage in vitro (Miyazaki et al., 1998; Wang et al., 1996). It could be that BM8, the anti-macrophage antibody used here, stains not only macrophages abundantly expressing the ACAT-1 protein at the lesions, but also monocytes which would not express this protein. Additionally, attenuation of foam cell formation by ACAT inhibition may cause a delay in the differentiation of macrophages from monocytes and, consequently the number of monocytes is increased at the lesions. Therefore, it might be that ACAT protein is in fact reduced, but the number of BM8-positive cells, both monocytes and macrophages, is not changed by F-1394-treatment. Furthermore, in our preliminary experiments, ACAT protein expression was induced in human vascular smooth muscle cells (VSMC) by cholesterol loading but not in rat VSMC (unpublished data). Hence, regulation of ACAT expression may be complex and differ under various conditions, depending on species, tissue examined, etc.
In this study, the body weights of the mice increased normally and there were no significant differences between the control group and the F-1394-treated group at either 15 or 20 weeks of age (Table 2). Moreover, the mice did not develop skin diseases, such as dermal hypertrophy or alopecia. Recently, it was reported that an atherogenic diet induced severe toxicity in both ACAT-disrupted apoE- and LDLr-knockout mice. It was proposed that a complete lack of ACAT-1 in macrophages in mice with severe hypercholesterolaemia leads to a massive accumulation of FC and subsequent inflammation in tissues such as brain, skin, eye and aortic wall (Accad et al., 2000; Yagyu et al., 2000). In our study, the serum cholesterol level in the apoE/LDLr-DKO mice would have been much lower than in apoE- or LDLr-deficient mice fed an atherogenic diet; this may be why toxic changes were not observed in our double-knockout mice. However, we have found that the ACAT inhibitor F-1394 did not induce toxicity in apoE-deficient mice fed a western-type diet, although it did have an anti-atherosclerotic effect (unpublished data). Thus, we should emphasize that partial inhibition of ACAT-1 enzyme activity by pharmacological compounds does not cause such toxic alterations as the severe dermal xanthomas observed when there is complete disruption of the enzyme activity.
Esterified cholesterol deposition mediated by the ACAT pathway is thought to be one of the major factors involved in lesion stability in rupture-prone plaques in the coronary artery in humans (Davies, 1996; Falk et al., 1995; Libby, 1995). We have shown here that excess deposition of neutral lipids or EC is significantly reduced by ACAT inhibition in vivo (Figures 1, 2B and 3D) and in vitro (Figure 4). Thus, we postulate that F-1394 might prevent the disruption of unstable plaques as a result of a decrease in EC deposition in foam cells at the coronary arterial wall.
In summary, ACAT inhibition by treatment with F-1394 diminished both lipid deposition and lesion size without affecting the serum cholesterol level, indicating that the ACAT enzyme is involved in foam cell formation and lesion development. These results imply that F-1394 exerts its effects directly on the arterial wall. Thus, ACAT inhibition by F-1394 treatment is a potential new therapeutic strategy for acute coronary syndromes.
Abbreviations
- ACAT
acyl-CoA:cholesterol acyltransferase
- apoE
apolipoprotein E
- EC
esterified cholesterol
- FC
free cholesterol
- HDL -C
high density lipoprotein cholesterol
- LDL
low density lipoprotein
- SMC
smooth muscle cell
- TC
total cholesterol
- TG
triglyceride
References
- ACCAD M., SMITH S.J., NEWLAND D.L., SANAN D.A., KING L.E., JR, LINTON M.F., FAZIO S., FARESE R.V., JR Massive xanthomatosis and altered composition of atherosclerotic lesions in hyperlipidemic mice lacking acyl CoA:cholesterol acyltransferase 1. J. Clin. Invest. 2000;105:711–719. doi: 10.1172/JCI9021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARAGANE K., KUSUNOKI J., KITAMINE T., YAMAURA T., OHNISHI H. Effects of F-1394, an acyl-CoA:cholesterol acyltransferase (ACAT) inhibitor, on ACAT activity in HepG2 cells and on hepatic secretion of lipids in Triton WR-1339-induced hyperlipidemic rats: possible role of hepatic ACAT in very low density lipoprotein secretion. Jpn J. Pharmacol. 1998;76:309–312. doi: 10.1254/jjp.76.309. [DOI] [PubMed] [Google Scholar]
- BASU S.K., GOLDSTEIN J.L., ANDERSON G.W., BROWN M.S. Degradation of cationized low density lipoprotein and regulation of cholesterol metabolism in homozygous familial hypercholesterolemia fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 1976;73:3178–3182. doi: 10.1073/pnas.73.9.3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOCAN T.M., KRAUSE B.R., ROSEBURY W.S., MUELLER S.B., LU X., DAGLE C., MAJOR T., LATHIA C., LEE H. The ACAT inhibitor avasimibe reduces macrophages and matrix metalloproteinase expression in atherosclerotic lesions of hypercholesterolemic rabbits. Arterioscler. Thromb. Vasc. Biol. 2000;20:70–79. doi: 10.1161/01.atv.20.1.70. [DOI] [PubMed] [Google Scholar]
- BOCAN T.M., MUELLER S.B., UHLENDORF P.D., BROWN E.Q., MAZUR M.J., BLACK A.E. Inhibition of acyl-CoA cholesterol O-acyltransferase reduces the cholesteryl ester enrichment of atherosclerotic lesions in the Yucatan micropig. Atherosclerosis. 1993;99:175–186. doi: 10.1016/0021-9150(93)90020-u. [DOI] [PubMed] [Google Scholar]
- BOCAN T.M., MUELLER S.B., UHLENDORF P.D., NEWTON R.S., KRAUSE B.R. Comparison of CI-976, an ACAT inhibitor, and selected lipid-lowering agents for antiatherosclerotic activity in iliac-femoral and thoracic aortic lesions. A biochemical, morphological, and morphometric evaluation. Arterioscler. Thromb. 1991;11:1830–1843. doi: 10.1161/01.atv.11.6.1830. [DOI] [PubMed] [Google Scholar]
- BONTHU S., HEISTAD D.D., CHAPPELL D.A., LAMPING K.G., FARACI F.M. Atherosclerosis, vascular remodeling, and impairment of endothelium- dependent relaxation in genetically altered hyperlipidemic mice. Arterioscler. Thromb. Vasc. Biol. 1997;17:2333–2340. doi: 10.1161/01.atv.17.11.2333. [DOI] [PubMed] [Google Scholar]
- BRESLOW J.L. Mouse models of atherosclerosis. Science. 1996;272:685–688. doi: 10.1126/science.272.5262.685. [DOI] [PubMed] [Google Scholar]
- BRESLOW J.L. Cardiovascular disease burden increases, NIH funding decreases. Nat. Med. 1997;3:600–601. doi: 10.1038/nm0697-600. [DOI] [PubMed] [Google Scholar]
- BROWN M.S., GOLDSTEIN J.L. Lipoprotein metabolism in the macrophage: implications for cholesterol deposition in atherosclerosis. Annu. Rev. Biochem. 1983;52:223–261. doi: 10.1146/annurev.bi.52.070183.001255. [DOI] [PubMed] [Google Scholar]
- CYBULSKY M.I., GIMBRONE M.A., JR Endothelial expression of a mononuclear leukocyte adhesion molecule during atherogenesis. Science. 1991;251:788–791. doi: 10.1126/science.1990440. [DOI] [PubMed] [Google Scholar]
- DAVIES M.J. Stability and instability: two faces of coronary atherosclerosis. The Paul Dudley White Lecture 1995. Circulation. 1996;94:2013–2020. doi: 10.1161/01.cir.94.8.2013. [DOI] [PubMed] [Google Scholar]
- FALK E., SHAH P.K., FUSTER V. Coronary plaque disruption. Circulation. 1995;92:657–671. doi: 10.1161/01.cir.92.3.657. [DOI] [PubMed] [Google Scholar]
- HAVEL R.J., EDER H.A., BRAGDON J.H. The distribution and chemical composition of ultracentrifugally separated lipoproteins in human serum. J. Clin. Invest. 1955;34:1345–1353. doi: 10.1172/JCI103182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ISHIBASHI S., HERZ J., MAEDA N., GOLDSTEIN J.L., BROWN M.S. The two-receptor model of lipoprotein clearance: tests of the hypothesis in “knockout” mice lacking the low density lipoprotein receptor, apolipoprotein E, or both proteins. Proc. Natl. Acad. Sci. U.S.A. 1994;91:4431–4435. doi: 10.1073/pnas.91.10.4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JUNQUEIRA L.C., BIGNOLAS G., BRENTANI R.R. Picrosirius staining plus polarization microscopy, a specific method for collagen detection in tissue sections. Histochem. J. 1979;11:447–455. doi: 10.1007/BF01002772. [DOI] [PubMed] [Google Scholar]
- KRATKY R.G., IVEY J., ROACH M.R. Collagen quantitation by video-microdensitometry in rabbit atherosclerosis. Matrix Biol. 1996;15:141–144. doi: 10.1016/s0945-053x(96)90155-9. [DOI] [PubMed] [Google Scholar]
- KUME N., CYBULSKY M.I., GIMBRONE M.A., JR Lysophosphatidylcholine, a component of atherogenic lipoproteins, induces mononuclear leukocyte adhesion molecules in cultured human and rabbit arterial endothelial cells. J. Clin. Invest. 1992;90:1138–1144. doi: 10.1172/JCI115932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KUSUNOKI J., ARAGANE K., KITAMINE T., HIGASHINAKAGAWA S., KASE N., YAMAURA T., OHNISHI H. Hypocholesterolemic action and prevention of cholesterol absorption via the gut by F-1394, a potent acyl-CoA:cholesterol acyltransferase (ACAT) inhibitor, in cholesterol diet-fed rats. Jpn J. Pharmacol. 1995a;69:53–60. doi: 10.1254/jjp.69.53. [DOI] [PubMed] [Google Scholar]
- KUSUNOKI J., ARAGANE K., YAMAURA T., OHNISHI H. Studies on acyl-CoA: cholesterol acyltransferase (ACAT) inhibitory effects and enzyme selectivity of F-1394, a pantotheic acid derivative. Jpn J. Pharmacol. 1995b;67:195–203. doi: 10.1254/jjp.67.195. [DOI] [PubMed] [Google Scholar]
- LIBBY P. Molecular bases of the acute coronary syndromes. Circulation. 1995;91:2844–2850. doi: 10.1161/01.cir.91.11.2844. [DOI] [PubMed] [Google Scholar]
- MATSUDA K. ACAT inhibitors as antiatherosclerotic agents: compounds and mechanisms. Med. Res. Rev. 1994;14:271–305. doi: 10.1002/med.2610140302. [DOI] [PubMed] [Google Scholar]
- MATSUO M., ITO F., KONTO A., AKETA M., TOMOI M., SHIMOMURA K. Effect of FR145237, a novel ACAT inhibitor, on atherogenesis in cholesterol-fed and WHHL rabbits. Evidence for a direct effect on the arterial wall. Biochim. Biophys. Acta. 1995;1259:254–260. doi: 10.1016/0005-2760(95)00178-6. [DOI] [PubMed] [Google Scholar]
- MIYAZAKI A., SAKASHITA N., LEE O., TAKAHASHI K., HORIUCHI S., HAKAMATA H., MORGANELLI P.M., CHANG C.C., CHANG T.Y. Expression of ACAT-1 protein in human atherosclerotic lesions and cultured human monocytes-macrophages. Arterioscler. Thromb. Vasc. Biol. 1998;18:1568–1574. doi: 10.1161/01.atv.18.10.1568. [DOI] [PubMed] [Google Scholar]
- MURAKAMI S., NARA Y., YAMORI Y. Hypolipidemic and antiatherosclerotic effects of a novel ACAT inhibitor, HL-004, in stroke-prone spontaneously hypertensive rats. Exp. Mol. Pathol. 1995;63:23–32. doi: 10.1006/exmp.1995.1027. [DOI] [PubMed] [Google Scholar]
- NAKASHIMA Y., PLUMP A.S., RAINES E.W., BRESLOW J.L., ROSS R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler. Thromb. 1994;14:133–140. doi: 10.1161/01.atv.14.1.133. [DOI] [PubMed] [Google Scholar]
- NICOLOSI R.J., WILSON T.A., KRAUSE B.R. The ACAT inhibitor, Cl-1011 is effective in the prevention and regression of aortic fatty streak area in hamsters. Atherosclerosis. 1998;137:77–85. doi: 10.1016/s0021-9150(97)00279-7. [DOI] [PubMed] [Google Scholar]
- PLUMP A.S., SMITH J.D., HAYEK T., AALTO-SETALA K., WALSH A., VERSTUYFT J.G., RUBIN E.M., BRESLOW J.L. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell. 1992;71:343–353. doi: 10.1016/0092-8674(92)90362-g. [DOI] [PubMed] [Google Scholar]
- ROSS R. Atherosclerosis–an inflammatory disease. N. Engl. J. Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- SLISKOVIC D.R., WHITE A.D. Therapeutic potential of ACAT inhibitors as lipid lowering and anti-atherosclerotic agents. Trends Pharmacol. Sci. 1991;12:194–199. doi: 10.1016/0165-6147(91)90546-5. [DOI] [PubMed] [Google Scholar]
- TANAKA H., OHTSUKA I., KOGUSHI M., KIMURA T., FUJIMORI T., SAEKI T., HAYASHI K., KOBAYASHI H., YAMADA T., HIYOSHI H., et al. Effect of the acyl-CoA:cholesterol acyltransferase inhibitor, E5324, on experimental atherosclerosis in rabbits. Atherosclerosis. 1994;107:187–201. doi: 10.1016/0021-9150(94)90020-5. [DOI] [PubMed] [Google Scholar]
- TANGIRALA R.K., RUBIN E.M., PALINSKI W. Quantitation of atherosclerosis in murine models: correlation between lesions in the aortic origin and in the entire aorta, and differences in the extent of lesions between sexes in LDL receptor-deficient and apolipoprotein E-deficient mice. J. Lipid Res. 1995;36:2320–2328. [PubMed] [Google Scholar]
- WANG H., GERMAIN S.J., BENFIELD P.P., GILLIES P.J. Gene expression of acyl coenzyme A:cholesterol acyltransferase is upregulated in human monocytes during differentiation and foam cell formation. Arterioscler. Thromb. Vasc. Biol. 1996;16:809–814. doi: 10.1161/01.atv.16.6.809. [DOI] [PubMed] [Google Scholar]
- WITTING P.K., PETTERSSON K., OSTLUND-LINDQVIST A.M., WESTERLUND C., ERIKSSON A.W., STOCKER R. Inhibition by a coantioxidant of aortic lipoprotein lipid peroxidation and atherosclerosis in apolipoprotein E and low density lipoprotein receptor gene double knockout mice. FASEB J. 1999;13:667–675. doi: 10.1096/fasebj.13.6.667. [DOI] [PubMed] [Google Scholar]
- WITZTUM J.L., STEINBERG D. Role of oxidized low density lipoprotein in atherogenesis. J. Clin. Invest. 1991;88:1785–1792. doi: 10.1172/JCI115499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAGYU H., KITAMINE T., OSUGA J., TOZAWA R., CHEN Z., KAJI Y., OKA T., PERREY S., TAMURA Y., OHASHI K., OKAZAKI H., YAHAGI N., SHIONOIRI F., IIZUKA Y., HARADA K., SHIMANO H., YAMASHITA H., GOTODA T., YAMADA N., ISHIBASHI S. Absence of ACAT-1 attenuates atherosclerosis but causes dry eye and cutaneous xanthomatosis in mice with congenital hyperlipidemia. J. Biol. Chem. 2000;275:21324–21330. doi: 10.1074/jbc.M002541200. [DOI] [PubMed] [Google Scholar]
- ZHANG S.H., REDDICK R.L., PIEDRAHITA J.A., MAEDA N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science. 1992;258:468–471. doi: 10.1126/science.1411543. [DOI] [PubMed] [Google Scholar]