

Abstract
Nitric oxide (NO) regulates cyclo-oxygenase (COX) activity in various cell systems and reports conflict in regard to its stimulatory versus inhibitory role. Incubation of human umbilical vein endothelial cells (HUVEC) with SIN-1 (3-morpholinosydnonimine), a donor of NO, resulted in a rapid and dose-dependent increase in the expression of COX-2 as analysed by Western and Northern blotting.
Incubation of HUVEC with SIN-1 and interleukine (IL)-1α resulted in increased induction of COX-2 compared with IL-1α alone and corresponded to an additive effect. The COX-2 induction was dependent on a de novo synthesis since cycloheximide, an inhibitor of protein synthesis, blocked the enzyme expression. The increase in COX-2 expression was not accompanied by a corresponding change in prostaglandin (PG) production. However, the COX activity was partially recovered when immunoprecipitated COX-2 was incubated with arachidonic acid and haematin.
Peroxynitrite, a highly reactive nitrogen molecule derived from the interaction of NO and superoxide anion, significantly increased COX-2 expression. Under these conditions and within the limit of detection of the antibody, selective antibody for nitrotyrosine failed to detect nitrated COX-2 in immunoprecipitated COX-2 when cells where incubated with SIN-1 or SIN-1+IL-1α.
Ro 31-8220, a specific inhibitor of protein kinase (PK) C, blocked the induction of COX-2. Also, SB203580, the selective inhibitor of p38 MAP kinase, strongly blocked the induction of COX-2 by SIN-1 in the presence or absence of IL-1α, whereas the MEK-1 inhibitor, PD 98059, affected it to a lesser extent. These data demonstrate that SIN-1 induces COX-2 in HUVEC in the absence of PG formation and suggest a complex regulation of COX-2 expression and PG formation by NO in endothelial cells.
Keywords: Nitric oxide (NO), SIN-1, COX-2, endothelial cells, prostacyclin, MAP kinases
Introduction
Prostacyclin, (prostaglandin, PG, I2) is a potent vasodilator and anti-aggregatory substance and is rapidly synthesized upon receptor-mediated stimulation of endothelial cells. This involves release of arachidonic acid by phospholipases and transformation into PGG2 and PGH2 via a cyclo-oxygenase (COX) and further into PGI2 via a PGI2 synthase (Smith, 1992). Although one of the main limiting rates in PG synthesis relates to phospholipases, recent studies have demonstrated that modulation of expression of COX is an important regulatory step (Smith & Dewitt, 1996).
Two different isoforms of COX have been demonstrated in mammalian cells: COX-1 which is constitutively expressed in a variety of cells such as platelets, vascular cells, fibroblasts or epithelial cells, and COX-2 which is an inducible enzyme and product of a primary response gene (Kujubu et al., 1991; O'banion et al., 1991). After activation of vascular cells by cytokines, growth factors or mitogens, COX-2 is rapidly expressed (i.e. 1–2 h) resulting in an enhanced capacity of cells to generate PG (Jones et al., 1993).
Nitric oxide (NO) is involved in a wide range of biological functions, such as smooth muscle relaxation, neurotransmission, and inflammation (Moncada et al., 1991; Nathan, 1992; Schmidt & Walter, 1994). One of the recognized targets of NO is the soluble form of guanylate cyclase, a haeme-protein whose activity is increased by binding of NO to the haeme moiety of the enzyme (Bredt & Snyder, 1994; Knowles & Moncada, 1994; Moncada et al., 1991). The subsequent increase in intracellular cyclic GMP can then modulate cellular functions including platelet activation/adhesion and vascular tone (Busse et al., 1987). NO is also able to react with oxygen, transition metals, and superoxide anions to generate various redox forms responsible for secondary oxidative stress. Certain redox-activated forms of NO can react with thiol moieties or protein and modify their activity (Stamler, 1994; Stamler et al., 1992). COX is a haeme-protein (Dewitt et al., 1990) that possesses two biochemically and structurally distinct active sites, a cyclo-oxygenase and a peroxidase. The haeme prosthetic group is absolutely required for both activities (Ogino et al., 1978). NO has also been found to modulate prostaglandin (PG) production in different experimental models and cells in culture, although contradictory results have been reported. Salvemini et al. (1993) reported that NO activates PG production in LPS-treated Raw 264.7, a mouse macrophage cell line. This positive modulation was subsequently found in other cellular models (Davidge et al., 1995; Franchi et al., 1994; Rettori et al., 1992; Salvemini et al., 1994; Sautebin et al., 1995; Watkins et al., 1997).
At the same time, NO has been shown both to inhibit or to have no effect on PG formation and COX-2 expression (Curtis et al., 1996; Honda et al., 2000; Minghetti et al., 1996; Stadler et al., 1993; Swierkosz et al., 1995). Moreover, data obtained with purified or recombinant COX-1 or COX-2 are equally conflicting (Salvemini et al., 1995a; 1996; Tsai et al., 1994; Upmacis et al., 1999).
In the present study, we have explored the effects of SIN-1 on the expression of COX and PG production by endothelial cells. SIN-1 induced a rapid expression of COX-2 protein similarly to IL-1α. COX-2 protein induced by SIN-1 was devoid of enzyme activity in contrast to the one expressed in the presence of IL-1α.
Methods
Cell culture
HUVEC were prepared according to the method of Jaffe with some modifications (Jaffe et al., 1973). Passaged cells were subcultured into gelatin-coated plates (Corning, NY, U.S.A.); they were seeded at 6×103 cm−2 and grown to subconfluence in M199 culture medium containing 10% normal human serum and supplemented with heparin and endothelial cell growth supplement (ECGS), as described previously (Habib et al., 1993). Heparin and ECGS were removed 48 h prior to stimulation.
Cell incubations and prostaglandin production
HUVEC at first passage were incubated with different stimuli for 6 h in M199 culture medium containing 0.75% albumax I and 1% foetal bovine serum (FBS). To evaluate COX activity, cells were washed in Hanks' buffer pH 7.4 containing 1 mg ml−1 bovine serum albumin (BSA) and incubated for 30 min in the same buffer with 10 μM arachidonic acid. In some experiments, PGl2 synthase activity was evaluated by incubating the cells with 1 μM PGH2. The supernatants were collected and the stable metabolite of prostacyclin, 6-keto-PGF1α, was measured using enzyme immunoassay (EIA) with acetylcholinesterase-labelled 6-keto-PGF1α as tracer (Pradelles et al., 1985).
Western blot analysis
After incubation, monolayers of HUVEC in 35-mm well plates (2–3×105 cells well−1) were washed twice in PBS, lysed and scraped in Tris/HCl buffer pH 6.8 (4% SDS 20 mM, DTT 0.5 mM, 20% glycerol, EDTA 1 mM, leupeptin 1 μg ml−1, soybean trypsin inhibitor 10 μg ml−1, pefabloc 0.5 mM and benzamidine hydrochloride 1 mM).
Protein content was determined by a micro-bicinchoninic acid assay (Pierce, Rockford, IL, U.S.A.) with BSA as standard. SDS–PAGE analysis was performed using 30 μg proteins of cell lysate (Créminon et al., 1995; Habib et al., 1993). One monoclonal antibody mAb (COX-229) and two specific mAbs (COX-110 and COX-111) were used for COX-2 and COX-1 analysis, respectively (Créminon et al., 1995). MAb directed against β-actin was used to check equal loading of protein. Blots were further incubated with donkey anti-mouse IgG conjugated to horseradish peroxidase (Jackson Immunoresearch, West Grove, PA, U.S.A.) at 1/5000 for 1 hour at room temperature. ECL-chemiluminescence substrates were used to reveal positive bands visualized after exposure to Hyperfilm™ ECL (Amersham Pharmacia Biotech., Les Ulis, France). Densitometric analyses of COX-2 protein bands were performed using an Agfa Arcus II® scanner coupled to Sigma Gel software® (SPSS, Chicago, IL, U.S.A.).
Immunoprecipitation of COX-2
HUVEC in a 100-mm culture dish (approximately 4.5×106 cells) were incubated for 6 h in the presence or absence of SIN-1 and IL-1α and then lysed in solubilization buffer (Hecameg® 20 mM, Tris/HCl pH 8 50 mM containing EDTA 1 mM and benzamidine 1 mM). The lysates were immediately frozen and kept at −70°C until processing. COX activity was assessed after selective immunoprecipitation of COX-2 using specific monoclonal anti-COX-2 peptide antibody coupled to sepharose-CL4B, as described previously with some modifications (Habib et al., 1997). Briefly, after selective immunoprecipitation of COX-2, the immunoprecipitates were resuspended in Tris HCl pH 8.0 50 mM containing phenol 1 mM and BSA 1 mg ml−1. After 1 min pretreatment at 37°C with 1 μM haematin, the reaction was initiated by the addition of 25 μM arachidonic acid. After 10 min incubation, the reaction was stopped by adding 1 vol of ice-cold EIA buffer and the mixture was centrifuged for 1 min at 5000 × g. PGE2, the breakdown product of PGH2 in this reaction, was measured in the supernatant by EIA. The immunoprecipitates were washed five times with 0.5 ml lysis buffer and mixed with Laemmli reagent under reducing conditions. Western blotting was then performed using mAb for COX-2 as described above.
Immunofluorescence studies
Cells grown in 35-mm culture dishes were incubated for 6 h in the presence or absence of SIN-1 or IL-1α or both. After two washings with PBS, cells were fixed in 2% paraformaldehyde in PBS and permeabilized with saponin (0.1%) for 5 min at room temperature. Subsequent washings and antibody incubations were done in PBS containing 0.1% saponin. After 30 min blocking of non-specific binding with PBS containing 5% BSA, cells were incubated for 1 h at 37°C with 1/50 dilution of the mAb COX-229 followed by two brief washes. Cells were further incubated for 30 min at room temperature with donkey anti-mouse IgG-coupled to Texas Red (1 : 20) (Amersham Pharmacia Biotech.). For negative controls, the same procedure was performed either without adding the primary antibody or after reaction of the primary anti-COX-2 antibody with 10 μg of its cognate peptide. Under these conditions, no significant signals were detected. Microscopy was conducted on a Leica fluorescence microscope (Buffalo, NY, U.S.A.).
RNA extraction and Northern blot analysis
HUVEC in 100-mm culture dishes (approximately 4.5×106 cells) were incubated for 6 h under the indicated conditions and washed once with PBS. Total RNA was extracted with Trizol according to the manufacturer's instruction. Twenty μg of total RNA was fractionated on a formaldehyde/MOPS/EDTA/agarose gel (1%). RNA was transferred to reinforced nitrocellulose and cross-linked by means of u.v. irradiation. The cDNA probes used were a 2-Kb human COX-2 cDNA fragment (Jones et al., 1993) and a β-actin cDNA fragment (Clontech Laboratories Inc, Palo Alto, CA, U.S.A.) and were labelled using a Ready-to-Go kit (Amersham Pharmacia Biotech.). Membranes were first pre-hybridized for 4 h and then hybridized for 18 h with the COX-2 probe (106 c.p.m. ml−1) in 50 mM Tris/HCl buffer, pH 7, containing 50% formamide, 10×Dernhardt's solution, 1 M NaCl, 1% SDS, 5% dextran sulphate, 0.1% pyrophosphate and 100 μg ml−1 salmon sperm DNA. Blots were hybridized overnight at 42°C with the labelled cDNA in the same buffer. Membranes were washed twice for 10 min with 2×SSC/0.1 SDS at room temperature, twice in 1×SSC/0.1 SDS at 60°C and once in 0.1× SSC/0.1 SDS at 60°C. For β-actin detection, membranes were hybridized with 0.5×106 c.p.m. ml−1 of the β-actin probe. Signals were quantified using a Fuji Bio-imaging analyser (Fuji, Tokyo, Japan) and the results were expressed as the ratio of COX-2/β-actin.
Statistics
Data are expressed as means±s.e.mean. Different parameters were evaluated by one-way ANOVA or an unpaired t-test.
Materials
Arachidonic acid, bovine serum albumin (fraction V), cycloheximide, haematin, leupeptin, β-mercaptoethanol, monoclonal anti-β-actin antibody, PMA and 8-bromo-cyclic GMP were from Sigma Aldrich (St Louis, MO, U.S.A.). SB 203580 and PD 98059 were from Calbiochem (La Jolla CA, U.S.A.). PGH2 was from Cayman Chem. (Ann Arbor MI, U.S.A.). SIN-1 was a kind gift of Dr J. Winicki (Hoechst, France). Molsidomine was from Biomol Research Laboratories Inc. (Plymouth Meeting, PA, U.S.A.). 8-pCPT-cyclic GMP [8-(-4-chlorophenylthio) guanosine-3′,5′-cyclic monophosphate] was from Biolog Science Institute (Bremen, Germany). Recombinant human IL-1α and TNFα were obtained from Genzyme (Cambridge, MA, U.S.A.). Albumax I, Trizol and cell culture reagents were from Life Technologies Inc. Ltd (Paisley, Scotland). Hecameg was from Vegatec (Villejuif, France). Peroxynitrite and mAb specific for nitrotyrosine were from Upstate Biotech. Inc (Lake Placid, NY, U.S.A.). Electrophoresis reagents were from Bio-Rad (Richmond, CA, U.S.A.) and chemical products from Prolabo (Paris, France).
Results
Induction of COX-2 by Sin-1
SIN-1 dose-dependently induced the expression of COX-2 in HUVEC, as detected by a specific monoclonal antibody (Figure 1a). COX-2 induction was observed at concentrations as low as 0.1 mM with maximal expression at 1 mM. COX-1 expression, instead, was not modified by SIN-1 (Figure 1a). Molsidomine, the inactive precursor drug of SIN-1, did not induce COX-2 (Figure 1a). Induction of COX-2 by SIN-1 was rapid and was detected after 1 h of incubation, with a maximum at 4–6 h (Figure 1b).
Figure 1.
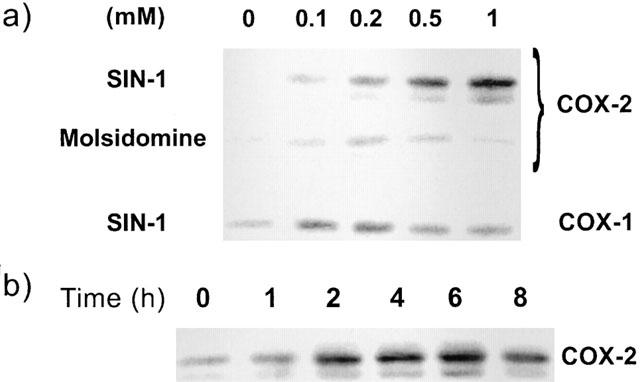
Dose and time-dependent expression of COX-2 by SIN-1. HUVEC were incubated for 6 h with different concentrations of SIN-1 or Molsidomine, the inactive precursor of SIN-1. COX-2 and COX-1 expression was evaluated by Western blot analysis using specific antibody for each isoform. (a) Effect of increasing concentrations of SIN-1 on COX-2 and COX-1 protein. Molsidomine, the inactive precursor of SIN-1, was tested on COX-2 expression. (b) Time-course of COX-2 induction by SIN-1. Results are representative of three different experiments.
COX-2 induction by SIN-1 was also detected by immunofluoresence analysis using the COX-2 antibody. When compared to unstimulated cells (Figure 2a), COX-2 was significantly increased in HUVEC incubated with SIN-1 for 6 h (Figure 2b) and this increase was comparable to that obtained in the presence of IL-1α (Figure 2c). Treatment of HUVEC with IL-1α+SIN-1 further induced COX-2 staining (Figure 2d). Figure 3a shows that SIN-1 alone and in combination with IL-1α increases COX-2 expression, as assessed by Western blot analysis, compared with unstimulated cells or cells treated with IL-1α alone. Densitometric analysis of the autoradiograms was achieved as described in Methods and the results are illustrated in Figure 3b, which shows that SIN-1 alone induced a 5.7±1.0 (n=14) times increase in COX-2 compared with unstimulated cells. SIN-1+IL-1α increased COX-2 expression by 25.3±5.3 times (n=14) (Figure 3b). SIN-1 (0.1 and 1 mM) increased also COX-2 expression induced by TNFα or PMA (Figure 4).
Figure 2.
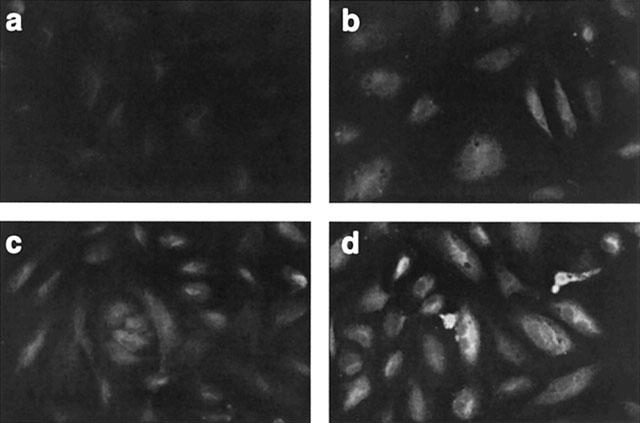
Immunofluorescence analyses of COX-2 in HUVEC exposed to SIN-1 and/or IL-1α. Cells were treated as described in the legend of Figure 1 and further processed as described in the Methods section. (a) Unstimulated cells, (b) 1 mM SIN-1, (c) 25 u ml−1 IL-1α, (d) 1 mM SIN-1+25 u ml−1 IL-1α. Magnification×250.
Figure 3.
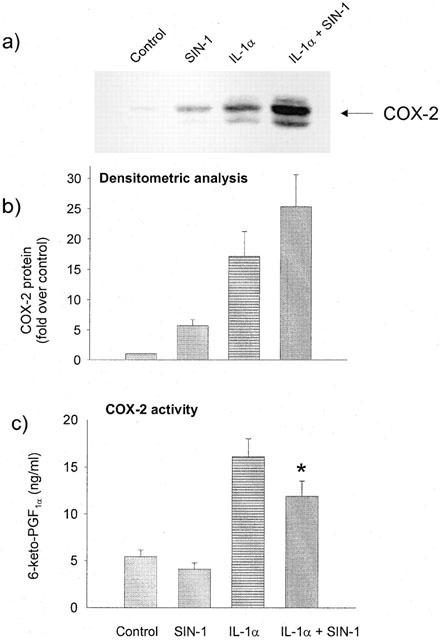
Effect of SIN-1 and IL-1α on COX-2 expression and COX activity in HUVEC. (a) COX-2 expression was analysed after 6 h incubation in the absence (control) or presence of 1 mM SIN-1 and/or 25 u ml−1 IL-1α as indicated in the legend of Figure 1. This Western blot represents 14 similar experiments. (b) The COX-2 signal was quantitated by densitometric analysis and results are expressed as ×-fold increase over unstimulated cells (control). (c) COX activity was assayed in HUVEC after 6 h incubation with 1 mM SIN-1 and/or 25 u ml−1 IL-1α. Cells were incubated in the presence of 10 μM arachidonic acid for 30 min. 6-keto-PGF1α concentration was determined in the supernatant by EIA. Data are expressed as mean±s.e. of 14 experiments. Statistical analysis was performed by means of Student's unpaired t-test (*P<0.05 with respect to IL-1α).
Figure 4.
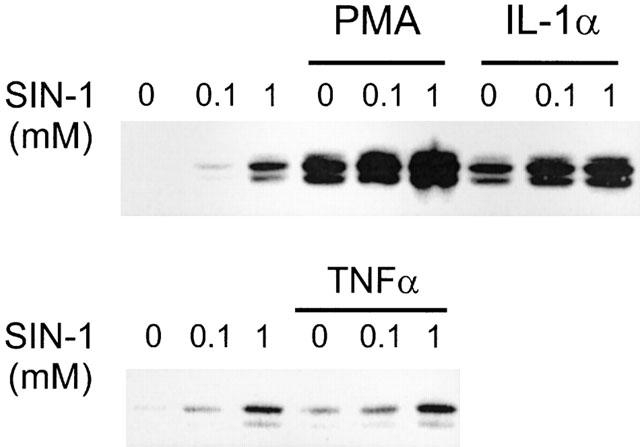
Effect of SIN-1 on COX-2 expression in the presence of different endothelial cell activators. HUVEC were incubated for 6 h with two different concentrations of SIN-1 in the absence or presence of 5 nM PMA, 25 u ml−1 IL-1α or 100 u ml−1 TNFα. COX-2 expression was evaluated as indicated in the legend of Figure 1. The effect of TNFα was studied in a different set of experiments. Results are representative of three different experiments.
The upregulation of COX-2 by SIN-1 alone or in combination with IL-1α was compared with PG formation and COX activity. No statistically significant difference was observed between SIN-1 treated and untreated cells (1.74±0.22 ng ml−1 for SIN-1 treated versus 1.05±0.16 ng ml−1 for untreated, mean±s.e.mean, n=14) in the release of 6-keto-PGF1α. IL-1α, instead, markedly increased the levels of 6-keto-PGF1α accumulated in the medium (4.64±0.95 ng ml−1), but SIN-1 did not exert additional effects (5.2±0.75 ng ml−1).
Moreover, SIN-1 failed to increase COX-2 activity also when cells were incubated with excess exogenously added arachidonate, both in the absence or presence of IL-1α (Figure 3c). Thus the modifications in the expression of COX-2 induced by SIN-1 contrasted with the absence of modifications of the cyclo-oxygenase activity.
Since NO donors have been reported to affect the activity of prostacyclin synthase, we assessed the PGI2 synthase activity by adding to the cells exogenous PGH2, the substrate of PGI2 synthase, and measuring the amount of 6-keto-PGF1α. Neither SIN-1 alone nor SIN-1+IL-1α modified prostacyclin synthase activity (90±16, 80±17 and 92±17 ng ml−1, for unstimulated cells, SIN-1 and SIN-1+IL-1α respectively, n=3).
These data suggest that in HUVEC the SIN-1 induced expression of COX-2 devoid of enzyme activity, as inferred from the absence of modifications in the production of PG.
Recovery of enzyme activity in immunoprecipitates
We then addressed the question whether COX activity could be recovered after isolation of the enzyme by immunoprecipitation. To this end, the enzyme activity was assessed after immunoprecipitation of COX-2 from cells incubated with SIN-1 alone or with IL-1α. In this assay, arachidonic acid was added in the presence of haematin to restore the prosthetic group. The levels of PGE2, which reflect the formation of PGH2, the product of COX activity, were measured. As shown in Figure 5a, PGE2 levels were significantly greater when cells were treated with SIN-1. Similarly, SIN-1+IL-1α elicited higher levels of PGE2 compared to IL-1α alone. Western blot analysis of the immunoprecipitated COX-2 showed the induction of COX-2 by SIN-1 (Figure 5b), as described to occur in total cell lysates (Figure 3a).
Figure 5.
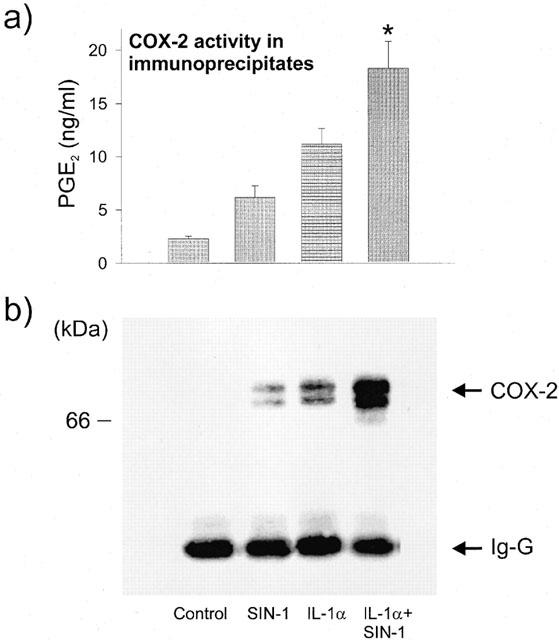
Recovery of COX-2 activity in immunoprecipitates. Cells were incubated for 6 h in the absence (control) or presence of 1 mM SIN-1 and/or 25 u ml−1 IL-1α as indicated in the legend of Figure 1. (a) COX-2 activity. COX-2 protein was immunoprecipitated using an immunoaffinity column of monoclonal antibody selective for COX-2 and COX activity was assayed by incubating the immunoprecipitated proteins with 25 μM of arachidonic acid and haematin as described in the Methods section. After 15 min of incubation, the reaction was stopped and the PGE2, one of the metabolites of PGH2 was measured in the supernatant by EIA. Results are expressed as mean±s.e.mean of seven experiments. *P<0.05 with respect to IL-1α, unpaired t-test. (b) Detection of COX-2 protein after immunoprecipitation. The immunoprecipitates were recovered in Laemmli buffer and assayed for COX-2 expression by Western blot analysis. IgG corresponds to immunoglobulin recovered by immunoprecipitation.
Nitrosylation of protein on the tyrosine residues has been proposed as one of the mechanisms of action of NO and its derivatives. Therefore, we assessed whether the COX-2 induced by SIN-1 was tyrosine nitrated. Using specific antibodies raised against nitrotyrosine, we were unable, in our experimental conditions and within the limit of detection of the antibodies, to show that immunoprecipitated COX-2 was tyrosine nitrated (data not shown).
Characterization of the SIN-1-dependent COX-2 induction
Cycloheximide, an inhibitor of protein synthesis, suppressed the induction of COX-2 by SIN-1 in the absence or presence of IL-1α, PMA or TNFα (data not shown), suggesting that SIN-1 induced de novo synthesis of the protein. Moreover, Northern blot analysis showed that SIN-1 increased basal and IL-1α-dependent COX-2 mRNA levels (Figure 6).
Figure 6.
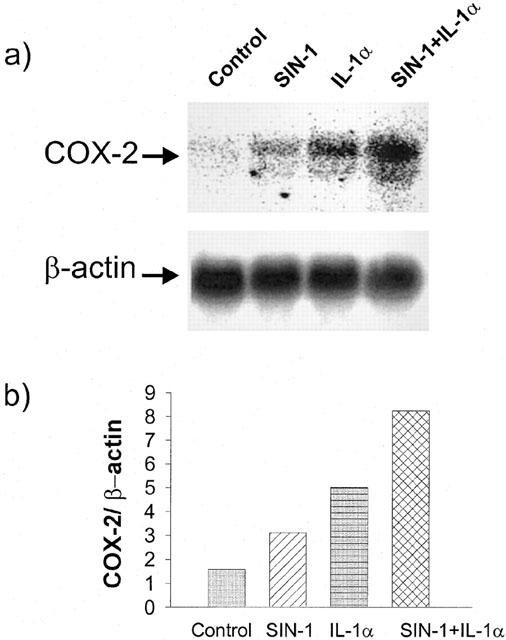
Northern blot analysis of COX-2. Cells were incubated with 1 mM SIN-1, and/or 25 u ml−1 IL-1α for 6 h. COX-2 mRNA levels were evaluated by Northern analysis as indicated in the Methods section. Signals were evaluated by phosphoimager using Fuji imaging-plate. (a) Northern blot analysis; (b) COX-2 signals were quantified relative to β-actin. Results are representative of four different experiments.
To determine whether activation of guanylate cyclase and cyclic GMP by NO played a role in the induction of COX-2, we evaluated the effect of 8-bromo-cyclic GMP and 8-pCT-cyclic GMP, a PDE-resistant cyclic GMP analogue, on COX-2 expression. Both analogues failed to mimic the effect of SIN-1 (Figure 7).
Figure 7.
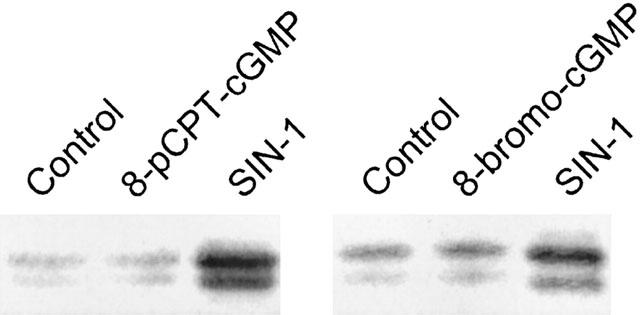
Effect of cyclic GMP analogues on COX-2 expression. HUVEC were incubated with 1 mM of 8-pCPT-cyclic GMP or 8-bromo-cyclic GMP for 6 h. COX-2 expression was evaluated as indicated in the legend of Figure 1. Results are representative of two different experiments.
Next, we tested the effect of peroxynitrite on the induction of COX-2. SIN-1 has been demonstrated to release very rapidly superoxide and NO which react to form the highly oxidant substance, peroxynitrite. Figure 8 shows that 0.5–1 mM peroxynitrite was able to induce COX-2, although to a lesser extent with respect to SIN-1. These data suggest that the formation of peroxynitrite could account at least partially for the observed induction of COX-2 exerted by SIN-1.
Figure 8.
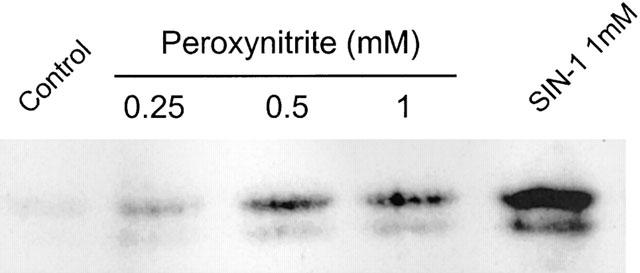
Induction of COX-2 by peroxynitrite. HUVEC were incubated with different concentrations of peroxynitrite or 1 mM SIN-1 for 6 h. COX-2 expression was evaluated as indicated in the legend of Figure 1. Results are representative of three different experiments.
Finally we evaluated some aspects of SIN-1 induced signalling relative to COX-2 expression. In particular we assessed the potential role of some protein kinases in this effect. PKC represents an important signalling pathway required for the expression of COX-2 induced by a variety of stimuli. The PKC inhibitor Ro 31-8220 suppressed the induction of COX-2 by SIN-1 alone or in the presence of IL-1α (Figure 9). Moreover, prolonged incubation (18 h) of cells with PMA, described to downregulate PKC, lead to inhibition of the SIN-1- and/or IL-1α-dependent induction of COX-2 (data not shown).
Figure 9.
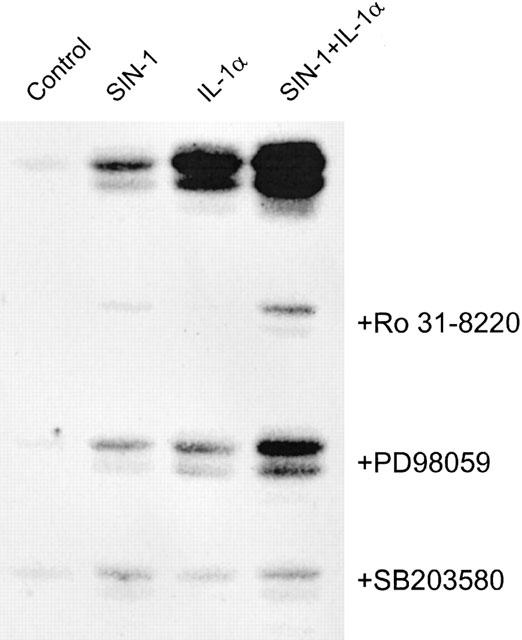
Involvement of kinases in COX-2 induction by SIN-1. Cells were treated in the absence or presence of 3 μM of the PKC inhibitor, Ro 31-8220, 10 μM of the p38MAP kinase inhibitor, SB203580 or 25 μM of the MEK kinase inhibitor, PD98059, for 30 min prior to the addition of 1 mM SIN-1 and/or 25 u ml−1 IL-1α followed by 6 h incubation. COX-2 expression was evaluated by Western blot analysis. Results are representative of three different experiments for Ro 31-8220 and two different experiments for SB203580 and PD98059.
Exposure of cells to SB203580, a specific p38 kinase inhibitor, abolished the expression of COX-2 induced by SIN-1 or IL-1α, whereas the MEK inhibitor PD98059 partially inhibited COX-2 induction by SIN-1 (Figure 9).
Discussion
In a variety of in vivo or cell systems, NO has been reported to modulate expression of COX-2 or the formation of PG. Earlier studies by Salvemini et al. (1993) showed that LPS-treated mouse macrophage cell lines exposed to NO synthase inhibitors produced lower amounts of PG. This type of modulation was found in other cellular (Davidge et al., 1995; Franchi et al., 1994; Rettori et al., 1992; Sautebin et al., 1995; Watkins et al., 1997) and animal models (Salvemini et al., 1994; 1995b). Opposite effects, however, have been recently reported in rat mesangial cells (Tetsuka et al., 1996), vascular cells (Kosonen et al., 1998) or chondrocytes (Notoya et al., 2000). We have recently shown that in rat macrophages activated by lipopolysaccharide inhibition of NO synthase enhanced the production of PG which paralleled by an increase in COX-2 expression (Habib et al., 1997). These data suggest that endogenous NO down-regulates the induction of COX-2 in macrophages. This effect was also found in an in vivo model of rats chronically treated with a NO synthase inhibitor (Henrion et al., 1997). In in vitro COX assays performed with purified or recombinant COX, NO had no clear effect on the ability of purified COX to oxygenate arachidonic acid (Upmacis et al., 1999), although some studies have suggested the opposite (Tsai et al., 1994). Indeed, interactions between NO and COX are more likely to lead to inhibition than activation of the enzyme. A possible explanation is the differences between experimental conditions aimed to evaluate the direct effect of NO on recombinant enzymes in simplified systems where NO is added to purified COX and whole cell systems where NO intermediates can be formed. Also, the effects of NO may depend upon various factors, including the cell type, the type of NO donor and the concentration of NO formed in the cell.
In the present study, we assessed the effect of SIN-1, a potent NO donor, on cyclo-oxygenase activity and expression in cultured HUVEC. Previous studies have shown that incubation of HUVEC with two other NO donors, GEA 3175 and S-nitroso-N-acetylpenicillamine (SNAP), leads to an inhibition of prostacyclin formation without modification of the expression of COX (Kosonen et al., 1998).
The most intriguing finding of the present study is the absence of an increase in PG release or activity which can be expected following the induction of COX-2 by SIN-1. The enzyme activity of COX-2 was partially recovered following immunoprecipitation of the enzyme. These results, together with the demonstration by Kosonen et al. (1998) that COX-2 is the enzyme responsible for the majority of the prostacyclin produced in HUVEC, indicate that SIN-1 induces the synthesis of an inactive form of COX-2. This inactive form is, however, not irreversibly modified, since the enzyme was active after addition of haematin, a Fe-protoporphyrin source. Some non-steroid anti-inflammatory drugs have been reported to induce inactive COX-2 since the newly synthesized enzyme is inactivated by the drug (Meade et al., 1999). In the case of NO donors, one possibility is that SIN-1 modifies the reserves of haeme in the cell as described in endothelial cells by Juckett et al. (1998) who have recently demonstrated that NO nitrosylates intracellular free haeme in endothelial cells in culture and forms NO complexes. This might result in an impaired accessibility of the newly synthesized enzyme to haeme. Also, NO can directly bind the haeme moieties of haeme proteins, thereby causing the displacement or the disruption of the axial ligand. The observation that the production of 6-keto-PGF1α was identical when PGH2 was added to cells ruled out any effect of SIN-1 on prostacyclin synthase, although Wade & Fitzpatrick (1997) have described a modulation of the recombinant PGI2 synthase by different concentrations of NO. Recent results have shown that induction of COX-2 may not always be accompanied by an increased capacity of the cells to generate PG. Intracellular localization of COX or availability of AA released by the different phospholipases, secreted or cyctoplasmic, have been suggested to play an additional role in the regulation of the formation of PG (Reddy & Herschman, 1997). Although SIN-1 is by no means an inducer of physiological relevance, it may represent a useful tool in investigating the mechanisms by which an increase in COX-2 content is not accompanied by a concomitant variation in activity.
The induction of COX-2 by SIN-1 alone or in the presence of IL-1α occurred in a time- and dose-dependent manner. This induction was cyclic GMP-independent since the non-hydrolyzed analogue of cyclic GMP, 8-pCPT-cyclic GMP, did not induce COX-2 in these cells. In this respect, both cyclic GMP-dependent and -independent expression of COX-2 has been described and these apparent discrepancies might be mainly related to differences in regulation of the redox status according to cell type (Davidge et al., 1995; Salvemini et al., 1993; Tetsuka et al., 1996).
Spontaneous decomposition of SIN-1, is accompanied by virtually simultaneous release of superoxide and NO, which immediately react with each other to generate the highly reactive peroxynitrite (Goodwin et al., 1999; Hogg et al., 1992). Also, superoxide is formed in cells after activation with cytokines under some conditions and can interact with NO to form peroxynitrite. In our cellular model, exogenous peroxynitrite induced COX-2, albeit to a lesser extent than SIN-1, suggesting a direct involvement of this compound in COX-2 induction. Peroxynitrite has been reported to react directly with enzymes including COX (Landino et al., 1996) and to modulate vascular tone (Upmacis et al., 1999).
The effect of SIN-1 was the consequence of a signalling event as it was suppressed by PKC and MAP kinase inhibitors. The activation of PKC induces COX-2 expression in many cell types such as Swiss 3T3 fibroblasts and mouse mast cells (Herschman, 1996; Kujubu et al., 1991; Okada et al., 2000). Recently, Yoshida et al. (1999) have demonstrated that NO donors such as SIN-1 or SNAP activated purified PKC in vitro. Moreover, NO generated during reperfusion following ischaemia activated PKC isoforms in rat heart.
In this study we have observed that the increase in COX-2 levels after incubation with SIN-1 was dependent on PKC activation, since Ro 31-8220, a specific inhibitor of PKC, reduced the expression of COX-2. PKC is not the only kinase involved in COX-2 induction by SIN-1 in HUVEC. Other protein kinases such as the three MAP kinase subfamilies, ERK, p38, and JNK/SAPK, can be involved in the COX-2 induction (Herschman, 1996). Using selective inhibitors of MAP kinases, we have demonstrated that p38 MAP kinase, and to a lesser extent ERK kinase, play a role in COX-2 induction by SIN-1. NO and related chemical species have been demonstrated to activate MAP kinases (Lander et al., 1996). However, different modulation was demonstrated in other experimental models where exogenous free radicals mainly activated p38 MAP kinase and less often ERK, suggesting that activation of MAP kinases is cell type-specific (Jun et al., 1999; Lander et al., 1996).
In summary, we have shown that SIN-1 induces COX-2 expression in HUVEC through a signalling pathway which involves PKC and p38 MAP kinase. The newly synthesized protein is devoid of enzyme activity which suggests that NO donors differently affect the expression of COX and its activity.
Acknowledgments
This paper is dedicated to the late Jacques Maclouf who initiated and supported this study. He will always be remembered as a great mentor, colleague and friend. We thank Prof Elena Tremoli (Univ Milan) for the helpful discussion. This work was supported by the Institut National de la Recherche sur la Santé et la Médecine (INSERM) and grants from the Association pour la Recherche sur le Cancer (ARC □num;2076). S. Eligini was supported by scholarships from the Fondation pour la Recherche Médicale and the Fondation Sanofi-Thrombose pour la Recherche.
Abbreviations
- COX
cyclo-oxygenase
- EIA
enzyme immunoassay
- HUVEC
human umbilical vein endothelial cells
- IL
interleukine
- MAP
mitogen-activated protein
- PG
prostaglandin
- PGI2
prostacyclin
- PKC
protein kinase C
- SIN-1
3-morpholinosydnonimine
References
- BREDT D.S., SNYDER S.H. Nitric oxide: a physiologic messenger molecule. Annu. Rev. Biochem. 1994;63:175–195. doi: 10.1146/annurev.bi.63.070194.001135. [DOI] [PubMed] [Google Scholar]
- BUSSE R., LUCKHOFF A., BASSENGE E. Endothelium-derived relaxant factor inhibits platelet activation. Naunyn Schmiedebergs Arch. Pharmacol. 1987;336:566–571. doi: 10.1007/BF00169315. [DOI] [PubMed] [Google Scholar]
- CRÉMINON C., HABIB A., MACLOUF J., PRADELLES P., GRASSI J., FROBERT Y. Differential measurement of constitutive (COX-1) and inducible (COX-2) cyclooxygenase expression in human umbilical vein endothelial cells using specific immunometric enzyme immunoassays. Biochim. Biophys. Acta. 1995;1254:341–348. doi: 10.1016/0005-2760(94)00197-7. [DOI] [PubMed] [Google Scholar]
- CURTIS J.F., REDDY N.G., MASON R.P., KALYANARAMAN B., ELING T.E. Nitric oxide: a prostaglandin H synthase 1 and 2 reducing cosubstrate that does not stimulate cyclooxygenase activity or prostaglandin H synthase expression in murine macrophages. Arch. Biochem. Biophys. 1996;335:369–376. doi: 10.1006/abbi.1996.0518. [DOI] [PubMed] [Google Scholar]
- DAVIDGE S.T., BAKER P.N., LAUGHLIN M.K., ROBERTS J.M. Nitric oxide produced by endothelial cells increases production of eicosanoids through activation of prostaglandin H synthase. Circ. Res. 1995;77:274–283. doi: 10.1161/01.res.77.2.274. [DOI] [PubMed] [Google Scholar]
- DEWITT D.L., EL-HARITH E.A., KRAEMER S.A., ANDREWS M.J., YAO E.F., ARMSTRONG R.L., SMITH W.L. The aspirin and heme-binding sites of ovine and murine prostaglandin endoperoxide synthases. J. Biol. Chem. 1990;265:5192–5198. [PubMed] [Google Scholar]
- FRANCHI A.M., CHAUD M., RETTORI V., SUBURO A., MCCANN S.M., GIMENO M. Role of nitric oxide in eicosanoid synthesis and uterine motility in estrogen-treated rat uteri. Proc. Natl. Acad. Sci. U.S.A. 1994;91:539–543. doi: 10.1073/pnas.91.2.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOODWIN D.C., LANDINO L.M., MARNETT L.J. Effects of nitric oxide and nitric oxide-derived species on prostaglandin endoperoxide synthase and prostaglandin biosynthesis. Faseb. J. 1999;13:1121–1136. doi: 10.1096/fasebj.13.10.1121. [DOI] [PubMed] [Google Scholar]
- HABIB A., BERNARD C., LEBRET M., CREMINON C., ESPOSITO B., TEDGUI A., MACLOUF J. Regulation of the expression of cyclooxygenase-2 by nitric oxide in rat peritoneal macrophages. J. Immunol. 1997;158:3845–3851. [PubMed] [Google Scholar]
- HABIB A., CREMINON C., FROBERT Y., GRASSI J., PRADELLES P., MACLOUF J. Demonstration of an inducible cyclooxygenase in human endothelial cells using antibodies raised against the carboxyl-terminal region of the cyclooxygenase-2. J. Biol. Chem. 1993;268:23448–23454. [PubMed] [Google Scholar]
- HENRION D., DECHAUX E., DOWELL F.J., MACLOUF J., SAMUEL J.L., LEVY B.I., MICHEL J.B. Alteration of flow-induced dilatation in mesenteric resistance arteries of L-NAME treated rats and its partial association with induction of cyclo-oxygenase-2. Br. J. Pharmacol. 1997;121:83–90. doi: 10.1038/sj.bjp.0701109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HERSCHMAN H.R. Prostaglandin synthase 2. Biochim. Biophys. Acta. 1996;1299:125–140. doi: 10.1016/0005-2760(95)00194-8. [DOI] [PubMed] [Google Scholar]
- HOGG N., DARLEY-USMAR V.M., WILSON M.T., MONCADA S. Production of hydroxyl radicals from the simultaneous generation of superoxide and nitric oxide. Biochem. J. 1992;281:419–424. doi: 10.1042/bj2810419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HONDA S., MIGITA K., HIRAI Y., UEKI Y., YAMASAKI S., URAYAMA S., KAWABE Y., FUKUDA T., KAWAKAMI A., KAMACHI M., KITA M., IDA H., AOYAGI T., EGUCHI K. Induction of COX-2 expression by nitric oxide in rheumatoid synovial cells. Biochem. Biophys. Res. Commun. 2000;268:928–931. doi: 10.1006/bbrc.2000.2228. [DOI] [PubMed] [Google Scholar]
- JAFFE E.A., NACHMAN R.L., BECKER C.G., MINICK C.R. Culture of human endothelial cells derived from umbilical veins. Identification by morphologic and immunologic criteria. J. Clin. Invest. 1973;52:2745–2756. doi: 10.1172/JCI107470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JONES D.A., CARLTON D.P., MCINTYRE T.M., ZIMMERMAN G.A., PRESCOTT S.M. Molecular cloning of human prostaglandin endoperoxide synthase type II and demonstration of expression in response to cytokines. J. Biol. Chem. 1993;268:9049–9054. [PubMed] [Google Scholar]
- JUCKETT M., ZHENG Y., YUAN H., PASTOR T., ANTHOLINE W., WEBER M., VERCELLOTTI G. Heme and the endothelium. Effects of nitric oxide on catalytic iron and heme degradation by heme oxygenase. J. Biol. Chem. 1998;273:23388–23397. doi: 10.1074/jbc.273.36.23388. [DOI] [PubMed] [Google Scholar]
- JUN C.D., OH C.D., KWAK H.J., PAE H.O., YOO J.C., CHOI B.M., CHUN J.S., PARK R.K., CHUNG H.T. Overexpression of protein kinase C isoforms protects RAW 264.7 macrophages from nitric oxide-induced apoptosis: involvement of c-Jun N-terminal kinase/stress-activated protein kinase, p38 kinase, and CPP-32 protease pathways. J. Immunol. 1999;162:3395–3401. [PubMed] [Google Scholar]
- KNOWLES R.G., MONCADA S. Nitric oxide synthases in mammals. Biochem. J. 1994;298:249–258. doi: 10.1042/bj2980249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOSONEN O., KANKAANRANTA H., MALO-RANTA U., RISTIMAKI A., MOILANEN E. Inhibition by nitric oxide-releasing compounds of prostacyclin production in human endothelial cells. Br. J. Pharmacol. 1998;125:247–254. doi: 10.1038/sj.bjp.0702042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KUJUBU D.A., FLETCHER B.S., VARNUM B.C., LIM R.W., HERSCHMAN H.R. TIS10, a phorbol ester tumor promoter-inducible mRNA from Swiss 3T3 cells, encodes a novel prostaglandin synthase/cyclooxygenase homologue. J. Biol. Chem. 1991;266:12866–12872. [PubMed] [Google Scholar]
- LANDER H.M., JACOVINA A.T., DAVIS R.J., TAURAS J.M. Differential activation of mitogen-activated protein kinases by nitric oxide-related species. J. Biol. Chem. 1996;271:19705–19709. doi: 10.1074/jbc.271.33.19705. [DOI] [PubMed] [Google Scholar]
- LANDINO L.M., CREWS B.C., TIMMONS M.D., MORROW J.D., MARNETT L.J. Peroxynitrite, the coupling product of nitric oxide and superoxide, activates prostaglandin biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 1996;93:15069–15074. doi: 10.1073/pnas.93.26.15069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MEADE E.A., MCINTYRE T.M., ZIMMERMAN G.A., PRESCOTT S.M. Peroxisome proliferators enhance cyclooxygenase-2 expression in epithelial cells. J. Biol. Chem. 1999;274:8328–8334. doi: 10.1074/jbc.274.12.8328. [DOI] [PubMed] [Google Scholar]
- MINGHETTI L., POLAZZI E., NICOLINI A., CREMINON C., LEVI G. Interferon-gamma and nitric oxide down-regulate lipopolysaccharide-induced prostanoid production in cultured rat microglial cells by inhibiting cyclooxygenase-2 expression. J. Neurochem. 1996;66:1963–1970. doi: 10.1046/j.1471-4159.1996.66051963.x. [DOI] [PubMed] [Google Scholar]
- MONCADA S., PALMER R.M., HIGGS E.A. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- NATHAN C. Nitric oxide as a secretory product of mammalian cells. FASEB. J. 1992;6:3051–3064. [PubMed] [Google Scholar]
- NOTOYA K., JOVANOVIC D.V., REBOUL P., MARTEL-PELLETIER J., MINEAU F., PELLETIER J.P. The induction of cell death in human osteoarthritis chondrocytes by nitric oxide is related to the production of prostaglandin E2 via the induction of cyclooxygenase-2. J. Immunol. 2000;165:3402–3410. doi: 10.4049/jimmunol.165.6.3402. [DOI] [PubMed] [Google Scholar]
- O'BANION M., SADOWSKI H.B., WINN V., YOUNG D.A. A serum- and glucocorticoid-regulated 4-kilobase mRNA encodes a cyclooxygenase-related protein. J. Biol. Chem. 1991;266:23261–23267. [PubMed] [Google Scholar]
- OGINO N., OHKI S., YAMAMOTO S., HAYAISHI O. Prostaglandin endoperoxide synthetase from bovine vesicular gland microsomes. Inactivation and activation by heme and other metalloporphyrins. J. Biol. Chem. 1978;253:5061–5068. [PubMed] [Google Scholar]
- OKADA Y., VOZNESENSKY O., HERSCHMAN H., HARRISON J., PILBEAM C. Identification of multiple cis-acting elements mediating the induction of prostaglandin G/H synthase-2 by phorbol ester in murine osteoblastic cells. J. Cell. Biochem. 2000;78:197–209. [PubMed] [Google Scholar]
- PRADELLES P., GRASSI J., MACLOUF J. Enzyme immunoassays of eicosanoids using acetylcholine esterase as label: an alternative to radioimmunoassay. Anal. Chem. 1985;57:1170–1173. doi: 10.1021/ac00284a003. [DOI] [PubMed] [Google Scholar]
- REDDY S.T., HERSCHMAN H.R. Prostaglandin synthase-1 and prostaglandin synthase-2 are coupled to distinct phospholipases for the generation of prostaglandin D2 in activated mast cells. J. Biol. Chem. 1997;272:3231–3237. doi: 10.1074/jbc.272.6.3231. [DOI] [PubMed] [Google Scholar]
- RETTORI V., GIMENO M., LYSON K., MCCANN S.M. Nitric oxide mediates norepinephrine-induced prostaglandin E2 release from the hypothalamus. Proc. Natl. Acad. Sci. U.S.A. 1992;89:11543–11546. doi: 10.1073/pnas.89.23.11543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SALVEMINI D., CURRIE M.G., MOLLACE V. Nitric oxide-mediated cyclooxygenase activation. A key event in the antiplatelet effects of nitrovasodilators. J. Clin. Invest. 1996;97:2562–2568. doi: 10.1172/JCI118704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SALVEMINI D., MISKO T.P., MASFERRER J.L., SEIBERT K., CURRIE M.G., NEEDLEMAN P. Nitric oxide activates cyclooxygenase enzymes. Proc. Natl. Acad. Sci. U.S.A. 1993;90:7240–7244. doi: 10.1073/pnas.90.15.7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SALVEMINI D., SEIBERT K., MASFERRER J.L., MISKO T.P., CURRIE M.G., NEEDLEMAN P. Endogenous nitric oxide enhances prostaglandin production in a model of renal inflammation. J. Clin. Invest. 1994;93:1940–1947. doi: 10.1172/JCI117185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SALVEMINI D., SEIBERT K., MASFERRER J.L., SETTLE S.L., MISKO T.P., CURRIE M.G., NEEDLEMAN P. Nitric oxide and the cyclooxygenase pathway. Adv. Prostaglandin. Thromboxane. Leukot. Res. 1995a;23:491–493. [PubMed] [Google Scholar]
- SALVEMINI D., SETTLE S.L., MASFERRER J.L., SEIBERT K., CURRIE M.G., NEEDLEMAN P. Regulation of prostaglandin production by nitric oxide; an in vivo analysis. Br. J. Pharmacol. 1995b;114:1171–1178. doi: 10.1111/j.1476-5381.1995.tb13330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAUTEBIN L., IALENTI A., IANARO A., DI ROSA M. Modulation by nitric oxide of prostaglandin biosynthesis in the rat. Br. J. Pharmacol. 1995;114:323–328. doi: 10.1111/j.1476-5381.1995.tb13230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHMIDT H.H., WALTER U. NO at work. Cell. 1994;78:919–925. doi: 10.1016/0092-8674(94)90267-4. [DOI] [PubMed] [Google Scholar]
- SMITH W.L. Prostanoid biosynthesis and mechanisms of action. Am. J. Physiol. 1992;263:F181–191. doi: 10.1152/ajprenal.1992.263.2.F181. [DOI] [PubMed] [Google Scholar]
- SMITH W.L., DEWITT D.L. Prostaglandin endoperoxide H synthases-1 and -2. Adv. Immunol. 1996;62:167–215. doi: 10.1016/s0065-2776(08)60430-7. [DOI] [PubMed] [Google Scholar]
- STADLER J., HARBRECHT B.G., DI SILVIO M., CURRAN R.D., JORDAN M.L., SIMMONS R.L., BILLIAR T.R. Endogenous nitric oxide inhibits the synthesis of cyclooxygenase products and interleukin-6 by rat Kupffer cells. J. Leukoc. Biol. 1993;53:165–172. doi: 10.1002/jlb.53.2.165. [DOI] [PubMed] [Google Scholar]
- STAMLER J.S. Redox signaling: nitrosylation and related target interactions of nitric oxide. Cell. 1994;78:931–936. doi: 10.1016/0092-8674(94)90269-0. [DOI] [PubMed] [Google Scholar]
- STAMLER J.S., SINGEL D.J., LOSCALZO J. Biochemistry of nitric oxide and its redox-activated forms. Science. 1992;258:1898–1902. doi: 10.1126/science.1281928. [DOI] [PubMed] [Google Scholar]
- SWIERKOSZ T.A., MITCHELL J.A., WARNER T.D., BOTTING R.M., VANE J.R. Co-induction of nitric oxide synthase and cyclo-oxygenase: interactions between nitric oxide and prostanoids. Br. J. Pharmacol. 1995;114:1335–1342. doi: 10.1111/j.1476-5381.1995.tb13353.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TETSUKA T., DAPHNA-IKEN D., MILLER B.W., GUAN Z., BAIER L.D., MORRISON A.R. Nitric oxide amplifies interleukin 1-induced cyclooxygenase-2 expression in rat mesangial cells. J. Clin. Invest. 1996;97:2051–2056. doi: 10.1172/JCI118641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TSAI A.L., WEI C., KULMACZ R.J. Interaction between nitric oxide and prostaglandin H synthase. Arch. Biochem. Biophys. 1994;313:367–372. doi: 10.1006/abbi.1994.1400. [DOI] [PubMed] [Google Scholar]
- UPMACIS R.K., DEEB R.S., HAJJAR D.P. Regulation of prostaglandin H2 synthase activity by nitrogen oxides. Biochemistry. 1999;38:12505–12513. doi: 10.1021/bi983049e. [DOI] [PubMed] [Google Scholar]
- WADE M.L., FITZPATRICK F.A. Nitric oxide modulates the activity of the hemoproteins prostaglandin 12 synthase and thromboxane A2 synthase. Arch. Biochem. Biophys. 1997;347:174–180. doi: 10.1006/abbi.1997.0348. [DOI] [PubMed] [Google Scholar]
- WATKINS D.N., GARLEPP M.J., THOMPSON P.J. Regulation of the inducible cyclo-oxygenase pathway in human cultured airway epithelial (A549) cells by nitric oxide. Br. J. Pharmacol. 1997;121:1482–1488. doi: 10.1038/sj.bjp.0701283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YOSHIDA K., MIZUKAMI Y., KITAKAZE M. Nitric oxide mediates protein kinase C isoform translocation in rat heart during postischemic reperfusion. Biochim. Biophys. Acta. 1999;1453:230–238. doi: 10.1016/s0925-4439(98)00105-7. [DOI] [PubMed] [Google Scholar]