

Abstract
Coronary responses to adenosine agonists were assessed in perfused mouse and rat hearts. The roles of nitric oxide (NO) and ATP-dependent K+ channels (KATP) were studied in the mouse.
Resting coronary resistance was lower in mouse vs rat, as was minimal resistance (2.2±0.1 vs 3.8±0.2 mmHg ml−1 min−1 g−1). Peak hyperaemic flow after 20–60 s occlusion was greater in mouse.
Adenosine agonists induced coronary dilation in mouse, with pEC50s of 9.4±0.1 for 2-[p-(2-carboxyethyl)phenethylamino]-5′-N-ethyl carboxamidoadenosine (CGS21680, A2A-selective agonist), 9.3±0.1 for 5′-N-ethylcarboxamidoadenosine (NECA, A1/A2 agonist), 8.4±0.1 for 2-chloroadenosine (A1/A2 agonist), 7.7±0.1 for N6-(R)-(phenylisopropyl)adenosine (R-PIA, A1/A2B selective), and 6.8±0.2 for adenosine. The potency order (CGS21680=NECA>2-chloroadenosine>R-PIA>adenosine) supports A2A adenosine receptor-mediated dilation in mouse coronary vessels. 0.2–2 μM of the A2B-selective antagonist alloxazine failed to alter CGS21680 or 2-chloroadenosine responses.
pEC50s in rat were 6.7±0.2 for CGS21680, 7.3±0.1 for NECA, 7.6±0.1 for 2-chloroadenosine, 7.2±0.1 for R-PIA, and 6.2±0.1 for adenosine (2-chloroadenosine>NECA=R-PIA>CGS21680> adenosine), supporting an A2B adenosine receptor response.
NO-synthase antagonism with 50 μM NG-nitro L-arginine (L-NOARG) increased resistance by ∼25%, and inhibited responses to CGS21680 (pEC50=9.0±0.1), 2-chloroadenosine (pEC50=7.3±0.2) and endothelial-dependent ADP, but not sodium nitroprusside (SNP). KATP channel blockade with 5 μM glibenclamide increased resistance by ∼80% and inhibited responses to CGS21680 in control (pEC50=8.3±0.1) and L-NOARG-treated hearts (pEC50=7.3±0.1), and to 2-chloroadenosine in control (pEC50=6.7±0.1) and L-NOARG-treated hearts (pEC50=5.9±0.2).
In summary, mouse coronary vessels are more sensitive to adenosine than rat vessels. A2A adenosine receptors mediate dilation in mouse coronary vessels vs A2B receptors in rat. Responses in the mouse involve a sensitive NO-dependent response and KATP-dependent dilation.
Keywords: Adenosine, coronary, mouse, rat, receptor, vasodilatation
Introduction
Adenosine receptor sub-types mediating coronary vasodilation appear to differ between species, and controversy exists regarding mechanisms contributing to adenosine-mediated dilation in different (and also within) vascular beds. For example, while there is evidence of NO-dependent components to adenosine responses in coronary vessels (Newmann et al., 1988; Leipert et al., 1992; Vials & Burnstock, 1993; Zanzinger & Bassenge, 1993; Abebe et al., 1994; Kuo & Chancellor, 1995; Hein et al., 1999; 2000), this is not universally observed (Sabouni et al., 1990; Kirkeboen et al., 1994; Lewis & Hourani, 1997; Kemp & Cocks, 1999; Lew & Kao, 1999). There is only preliminary data available regarding the receptor sub-type mediating coronary dilation in the mouse (Morrison et al., 2001), and there are no data regarding mechanisms involved. Therefore, the primary goal of the present study was to functionally characterize the adenosine receptor sub-type mediating coronary dilation in mouse heart. To identify the receptor, coronary responses to adenosine (the endogenous and non-selective agonist), CGS 21680 (A2A selective agonist), NECA (A1/A2 agonist), 2-chloroadenosine (A1/A2 agonist), and R-PIA (A1/A2B agonist) were examined, together with the effects of A2B-selective antagonism with alloxazine. The potential contributions of NO and KATP channel activation to adenosine receptor responses were assessed via competitive inhibition of NO-synthase with L-NOARG and inhibition of KATP channels with glibenclamide. Coronary vascular function in mouse was also compared to that in the more thoroughly characterized rat heart.
Methods
Langendorff perfused heart model
Hearts were isolated from mice and rats as described by us in detail previously (Headrick, 1996; Headrick et al., 2000). Specifically, adult male C57/B16 mice (7–12 weeks old, 26.3±0.3 g body weight, 118±8 mg blotted heart weight) or male Wistar rats (12–16 weeks old, 337±15 g body weight, 1.22±0.19 g blotted heart weight) were anaesthetized with 50 mg kg−1 sodium pentobarbitone, a thoracotomy performed and hearts rapidly excised into ice-cold perfusion fluid. The aorta was cannulated and hearts initially perfused in a retrograde fashion at a hydrostatic pressure of 100 mmHg with modified Krebs bicarbonate buffer containing (in mM): NaCl, 120; NaHCO3, 25; KCl, 4.7; KH2PO4, 1.2; CaCl2, 2.5; Mg2SO4 1.2; glucose, 15; and EDTA, 0.6. Perfusate was equilibrated with 95% O2, 5% CO2 at 37°C, giving a pH of 7.4. Perfusate temperature was maintained at 37°C and hearts were constantly bathed in perfusate within a small water jacketed chamber maintained at 37°C. The left ventricle was vented with a polyethylene drain to prevent accumulation of Thebesian drainage. Coronary perfusion was constantly monitored via a cannulating ultrasonic flow-probe (Transonic Systems Inc., Ithaca, NY, U.S.A.) in the aortic perfusion line and aortic perfusion pressure was monitored using a P23XL pressure transducer (Viggo-Spectramed, Oxnard, CA, U.S.A.) connected to a MacLab data acquisition unit (ADInstruments, Castle Hill, Australia). Aortic pressure was held constant at 100 mmHg. After an initial 20 min of stabilization at intrinsic heart rate all hearts were electrically paced via silver electrodes using a Grass stimulator (Grass S9 stimulator, Quincy, MA, U.S.A.). Mouse hearts were paced at a rate of 400 beats min−1 while rat hearts were paced at 300 beats min−1 (0.5 ms square pulses 20% above threshold, typically 1–4 V). Hearts were allowed to stabilize for an additional 10 min before experimentation. Hearts were then switched to constant flow perfusion at a rate giving an aortic pressure of 100 mmHg prior to drug infusion (13.2±0.7 ml min−1 g−1 in mice, 10.6±0.8 ml min−1 g−1 in rats). This permitted analysis of coronary dilation without the complication of changes in coronary flow rate and drug delivery. Flow was delivered by a Gilson MiniPuls 2 peristaltic pump (Gilson Inc., Middleton WI, U.S.A.).
Experimental protocol
Stabilized hearts were treated with a dilatory agonist (2-chloroadenosine, R-PIA, CGS21680, NECA, adenosine, SNP, or ADP) infused in incremental concentrations. Agonists were infused at each concentration for 1–3 min period during which vascular responses stabilized. Only one concentration-response curve was acquired per heart. Changes in aortic pressure were measured, and coronary flow was constantly monitored to verify a constant flow rate.
To examine the effects of A2B antagonism in mouse hearts, responses to CGS21680 and 2-chloroadenosine were acquired in the presence of 200 nM alloxazine (n=7 for both groups). Responses to 2-chloroadenosine were also acquired in a sub-set of hearts treated with a higher 2 μm concentration of alloxazine (n=4). Alloxazine infusion was initiated 10 min prior to acquisition of agonist concentration-response curves.
For NO-synthase inhibition in mouse hearts, infusion of 50 μM L-NAME (final concentration) was initiated after stabilization. After a further 10 min period concentration-response curves for CGS21680 (n=10), 2-chloroadenosine (n=8), ADP (n=6), or SNP (n=7) were acquired, as described above. To further examine mechanisms of adenosine-mediated dilation, KATP channel inhibition was studied in mouse hearts. Infusion of 5 μM glibenclamide was initiated alone or in conjunction with 50 μM L-NAME. After a further 10 min period, concentration-response curves for CGS21680 and 2-chloroadenosine were acquired (n=6 in all groups). In preliminary experiments 5 μM glibenclamide was shown to fully block dilatory responses to 0.1–3.0 μM of the KATP opener minoxidil (data not shown).
Reactive hyperaemic responses were assessed in a separate group of mouse (n=7) and rat hearts (n=8) which were perfused at a constant aortic pressure of 100 mmHg after pacing was initiated. After a further 10 min stabilization hearts were subjected to 20 s of total coronary occlusion followed by reperfusion for 15 min, before studying the response to 60 s of coronary occlusion and reperfusion. Flow responses were monitored and the peak hyperaemic response determined (in absolute units and as per cent of baseline resistance), together with per cent flow-debt repayment. Flow-debt repayment was calculated as:
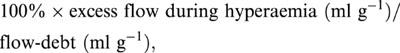 |
where flow-debt is the product of baseline flow (ml min−1 g−1)×period of occlusion (min), and excess flow during hyperaemia was calculated as the volume of flow during the period of the hyperaemic response minus the baseline flow. The period of the hyperaemic response was defined as the time from onset of reperfusion to the point where flow fell to the pre-occlusion level.
Data analysis
Responses to agonists were compared between treatment groups using a multi-way analysis of variance for repeated measures, followed by Tukeys HSD post-hoc test for multiple comparisons when differences were detected. pEC50 values were obtained from concentration-response data (expressed as absolute units) by fitting the following four-parameter logistic equation to data for individual experiments:
where A is the response at zero dose (i.e. the pre-infusion value), and B is the response at infinite dose. The equation was fit to data using the Statistica program (Statsoft, Tulsa, OK, U.S.A.), and individual pEC50 values derived from each fit. pEC50s were compared between treatment groups by one-way analysis of variance with Tukeys HSD post-hoc test. In all statistical tests P<0.05 was considered indicative of statistical significance. Dilatory responses to agonists were calculated as a per cent of resting resistance, or were scaled (as a per cent) to the maximal dilatory response observed. All values are reported as mean±s.e.mean.
Chemicals
2-Chloroadenosine, adenosine, CGS21680, R-PIA, NECA, ADP, SNP, alloxazine, L-NOARG, glibenclamide, and minoxidil were all purchased from Sigma/RBI (Sigma Chemical, Castle Hill, Australia). All other chemicals used were of analytical grade or better.
Results
Coronary vascular function in mouse vs rat heart
Baseline coronary tone, peak reactive hyperaemic responses, and minimal vascular tone observed during infusion of the relatively non-selective A2 adenosine receptor agonist 2-chloroadenosine are shown in Table 1. Resting coronary resistance was lower in mouse vs rat hearts (∼7.5 vs 9.5 mmHg ml−1 min−1 g−1). The minimal resistance achieved during dilation with 2-chloroadenosine was also lower in mouse (2.2 mmHg ml−1 min−1 g−1) vs rat heart (3.8 ml−1 min−1 g−1) (Table 1). The peak reactive hyperaemic response and extent of flow repayment following 60 s occlusion was significantly greater in mouse vs rat (Table 1). Similarly, the peak response following a shorter 20 s occlusion was greater in mouse (35.8±2.3 ml−1 min−1 g−1 at ∼5 s of reperfusion) vs rat (24.3±1.2 ml−1 min−1 g−1 at ∼15 s), as was repayment (136±9% in mouse vs 98±6% in rat). Collectively, data show that resting and minimum coronary resistances are lower in mouse. However, the dynamic range over which coronary vessels dilate is similar in both species (∼5.5 mmHg ml−1 min−1 g−1).
Table 1.
Coronary vascular functional parameters in mouse and rat hearts

Effects of adenosine receptor agonists in mouse vs rat heart
All adenosine receptor agonists concentration-dependently dilated the coronary circulation in mouse and rat hearts (Figure 1). Response magnitude, expressed as per cent change in resistance, was similar in both species. CGS21680 was the most potent agonist in mouse, whereas it was one of the less potent in rat. The rank order of potencies was CGS21680=NECA>2-chloroadenosine>R-PIA>adenosine for mouse. Statistical analysis of pEC50s yielded P values of 0.879 for NECA vs CGS21680, and less than 0.001 for 2-chloroadenosine vs NECA and CGS21680, R-PIA vs 2-chloroadenosine, and adenosine vs R-PIA. This rank order of potencies supports A2A-mediated dilation, and contrasted with that in the rat: 2-chloroadenosine>NECA=R-PIA>CGS21680>adenosine (Table 2). Statistical analysis of pEC50s yielded P values of 0.001 for NECA vs 2-chloroadenosine, 0.835 for R-PIA vs NECA, and less than 0.005 for CGS21680 vs R-PIA and adenosine vs CGS21680. This rank order of potencies supports an A2B-mediated response in rat. In addition, sensitivity to all adenosine agonists was higher in mouse vs rat. Furthermore, as shown in Table 2 (and apparent in curves depicted in Figure 1), the steepness or slope of concentration-response curves is greater in mouse vs rat. Slope factors for all adenosine agonists were significantly higher in mouse (Table 2).
Figure 1.
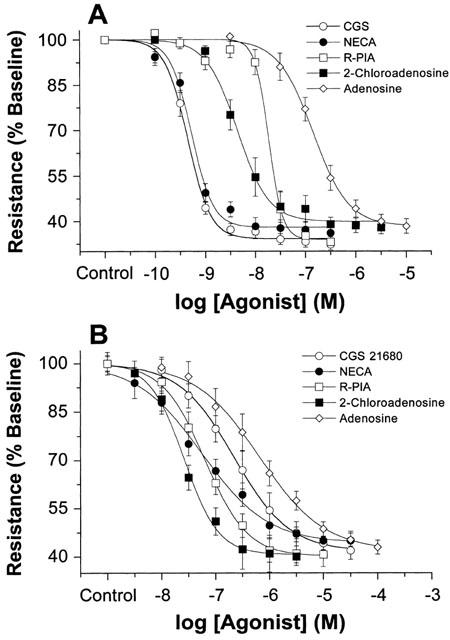
Concentration-response curves for adenosine agonist mediated vasodilation in (A) mouse, and (B) rat hearts. Responses to adenosine (n=8 for mouse, n=7 for rat), 2-chloroadenosine (n=7 for mouse, n=7 for rat), R-PIA (n=6 for mouse, n=8 for rat), NECA (n=7 for mouse, n=7 for rat), and CGS 21680 (n=8 for mouse, n=7 for rat) were studied. Responses are shown as per cent of baseline coronary resistance. All values are means±s.e.mean.
Table 2.
Concentration-response data for adenosine agonists in mouse and rat hearts
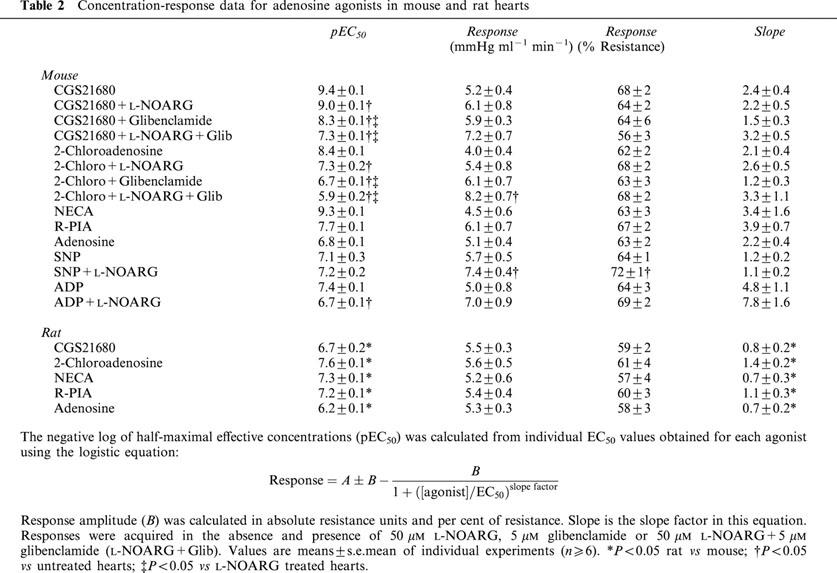
A2B-selective antagonist alloxazine, when infused at 200 nM concentration, failed to alter responses to either A2A-selective CGS21680 (pEC50=9.2±0.1) or non-selective 2-chloroadenosine (pEC50=8.2±0.1) (Figure 2). A 10 fold higher concentration of alloxazine also failed to alter responses to 2-chloroadenosine in mouse heart (Figure 2B).
Figure 2.
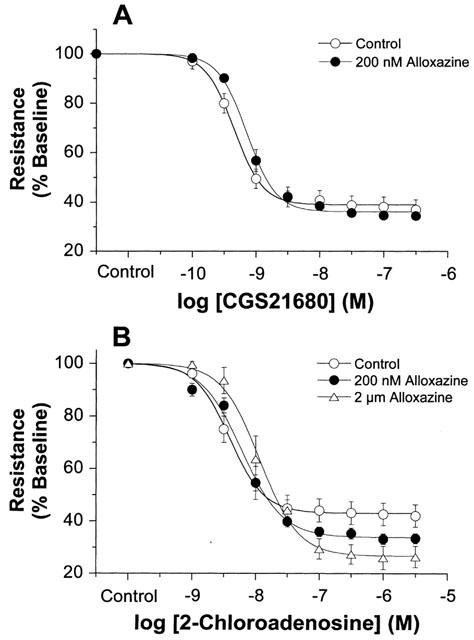
Concentration-response curves for (A) CGS21680 and (B) 2-chloroadenosine in mouse hearts in the presence and absence of alloxazine. Responses were obtained for CGS21680 alone (n=8) or in the presence of 200 nM alloxazine (n=7), and for 2-chloroadenosine alone (n=7) or in the presence of 200 nM (n=7) or 2 μm (n=4) alloxazine. Control data for CGS21680 and 2-chloroadenosine are taken from Figure 1. Responses are shown as per cent coronary resistance. All values are means±s.e.mean. *P<0.05 vs values in untreated hearts (P<0.05).
Effects of NO-synthase inhibition in mouse heart
Treatment of mouse hearts with 50 μM L-NOARG significantly increased baseline coronary resistance by ∼25% to 9.5±0.6 mmHg ml−1 min−1 g−1 (P<0.05). Responses to the adenosine receptor agonists CGS21680 and 2-chloroadenosine were significantly reduced by L-NOARG (Figures 3 and 4). Inhibitory effects were present whether responses were expressed relative to resting resistance (Figure 3) or scaled to maximal dilation (data not shown). Effects of L-NOARG were primarily observed at low to moderate agonist concentrations, with no depression of responses to high agonist levels (i.e. above 10−8 M CGS21680 and 10−7 M 2-chloroadenosine) (Figure 3). Coronary sensitivities to both CGS21680 and 2-chloroadenosine (reflected by pEC50s) were significantly depressed by L-NOARG treatment, with a greater shift in 2-chloroadenosine sensitivity (13 fold) vs CGS21680 sensitivity (2.5 fold) (Table 2). Representative traces from hearts treated with 2-chloroadenosine and CGS21680±L-NOARG, are shown in Figure 4.
Figure 3.
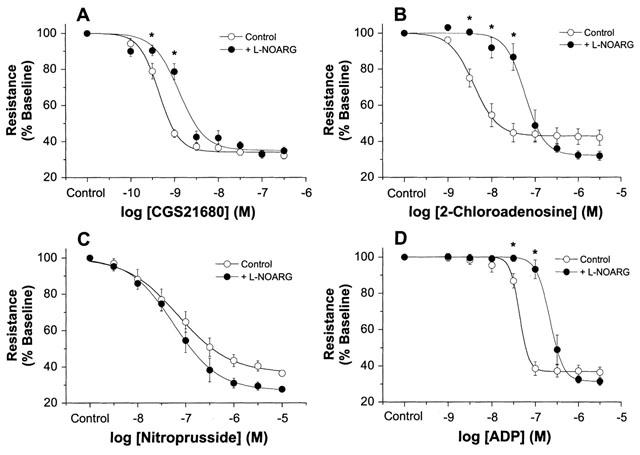
Concentration-response curves from mouse hearts in the presence and absence of 50 μM L-NOARG. Responses were obtained for (A) CGS21680 (n=10), (B) 2-chloroadenosine (n=8), (C) SNP (n=6 in both groups), and (D) ADP (n=7 in both groups). Control responses for CGS21680 and 2-chloroadenosine are taken from the data shown in Figure 1. Responses are shown as per cent of baseline coronary resistance. All values are means±s.e.mean. * P<0.05 vs values in untreated hearts (P<0.05).
Figure 4.
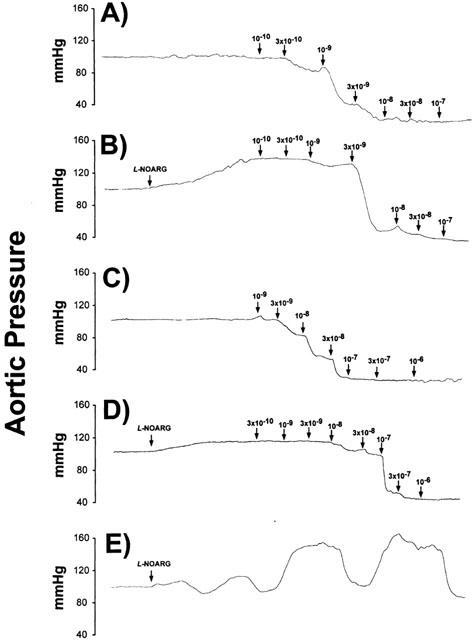
Representative tracings from mouse hearts treated with (A) CGS21680, (B) CGS21680+50 μM L-NOARG, (C) 2-chloroadenosine, and (D) 2-chloroadenosine+50 μM L-NOARG. The trace shown in (E) is from a heart displaying phasic oscillations in coronary tone during infusion of 50 μm L-NOARG.
L-NOARG failed to reduce responses or alter sensitivity to the endothelial-independent dilator SNP (Figure 3C, Table 2). There were no significant inhibitory effects when responses were expressed either as per cent of resting coronary resistance (Figure 3C), or scaled to the maximal dilation observed (data not shown). Indeed, due to slightly increased resting resistance with L-NOARG, there was an insignificant trend towards slightly enhanced responses to higher levels of nitroprusside (when expressed relative to the initial resting resistance). Dilatory responses to the endothelial-dependent dilator ADP were significantly inhibited by L-NOARG (Figure 3D, Table 2).
Curiously, a significant number of hearts displayed phasic oscillations in coronary tone during treatment with L-NOARG, rendering it difficult to acquire concentration-response data in those hearts. Representative traces from various experiments, including the latter hearts displaying oscillations in tone, are shown in Figure 4.
Effects of KATP channel inhibition in mouse heart
Treatment of mouse hearts with 5 μM glibenclamide significantly increased baseline coronary resistance by ∼35% to 9.8±0.5 mmHg ml−1 min−1 g−1 (P<0.05). Treatment with 5 μM glibenclamide+50 μM L-NOARG further increased resting resistance to 12.6±0.7 mmHg ml−1 min−1 g−1 (P<0.05). Responses to CGS21680 and 2-chloroadenosine were significantly inhibited by glibenclamide, and were further inhibited by co-treatment with glibenclamide+L-NOARG (Figure 5). The degree of inhibition observed with glibenclamide was considerably greater than that with L-NOARG alone. The pEC50s for CGS21680 and 2-chloroadenosine were shifted to higher concentrations by an order of magnitude in the presence of glibenclamide (Table 2). The substantial inhibitory effects of glibenclamide were additive to those for L-NOARG. The pEC50s for CGS21680 and 2-chloroadenosine were further shifted by at least an order of magnitude in hearts treated with glibenclamide+L-NOARG together, relative to pEC50s for hearts treated with either glibenclamide or L-NOARG alone (Table 2).
Figure 5.
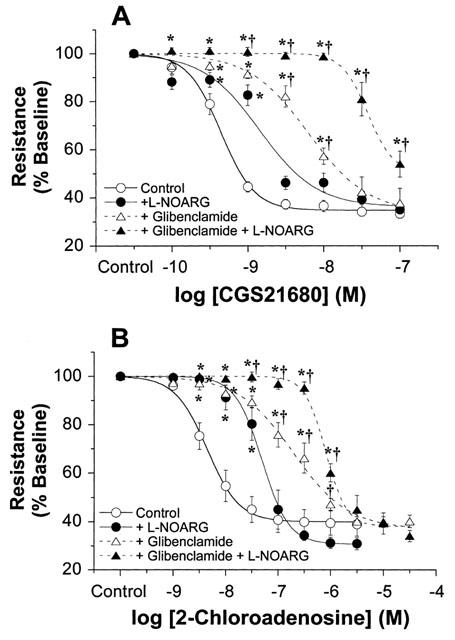
Concentration-response curves from mouse hearts in the presence and absence of 5 μM glibenclamide or 5 μM glibenclamide+50 μM L-NOARG. Responses were obtained for (A) CGS21680 (n=10), and (B) 2-chloroadenosine (n=8). Control responses and responses in the presence of L-NOARG alone are also shown (from Figure 3). Responses are shown as per cent of baseline coronary resistance. All values are means±s.e.mean. *P<0.05 vs values in untreated hearts (P<0.05). †P<0.05 vs values in L-NOARG treated hearts (P<0.05).
Discussion
Little information exists regarding adenosine receptor sub-types mediating coronary dilation in the mouse, or mechanisms of adenosine-mediated coronary dilation in this species. Our data indicate that mouse heart is more sensitive to adenosine agonists than rat heart, and show that different receptor sub-types mediate dilation in these species. Adenosine-mediated coronary dilation in the mouse involves NO-dependent and KATP-dependent components.
Coronary vascular function in mouse vs rat heart
Baseline and maximally dilated coronary resistances were lower in mouse vs rat heart (Table 1). Additionally, reactive hyperaemic responses were higher in the mouse (Table 1). The mouse circulation therefore appears to operate at lower resting and dilated resistances than in the rat, likely reflecting correlations between vascular density, mitochondrial capacity, metabolic rate and body mass (Pietschmann et al., 1982; Rakusan & Tomanek, 1986). Owing to greater metabolic rate in mouse (i.e. mouse heart rate is 550–600 beats min−1 vs ∼350 beats min−1 in the rat), the murine heart requires a much greater O2 delivery. This can be accomplished by limited adaptations in O2 carrying capacity of blood and O2 transport from blood to mitochondria, and/or it can be met by increased coronary vascularization and coronary flow, as shown here. Higher hyperaemic responses and lower coronary resistance in mouse heart permits greater resting and maximal O2 delivery.
The adenosine receptor mediating coronary dilation in mouse and rat hearts
Endogenous adenosine may play an important role in modulating coronary vascular resistance during and following pathophysiological stimuli (Berne, 1980; Olsson & Pearson, 1990), although its role in regulation of coronary tone under more physiological conditions in vivo is questionable (Tune et al., 2001). We show that murine coronary vessels are highly sensitive to adenosine agonists. Sensitivities to all agonists, including adenosine, are higher in mouse than in rat (Figure 1, Table 2). Importantly, the rank order of agonist potencies differs between mouse and rat (Table 2). As noted initially by Gurden et al. (1993), two differing orders of agonist potencies are observed for A2 receptor mediated vascular responses. They observed an order of potency of CGS21680⩾NECA>R-PIA in canine coronary vessels, reflecting an A2A receptor-mediated response, and NECA>R-PIA>CGS21680 in guinea-pig aorta, reflecting an A2B receptor-mediated response. Kull et al. (1999) documented a functional potency profile of NECA⩾CGS21680>R-PIA⩾2-chloroadenosine>adenosine for rat and human A2A receptors. We obtained an order of CGS21680=NECA>2-chloroadenosine>R-PIA>adenosine in the mouse which is comparable to that for canine (Gurden et al., 1993), human and rat A2A receptors (Kull et al., 1999). In contrast we obtained a profile of 2-chloroadenosine > NECA=R-PIA> CGS21680>adenosine in rat, supporting a primarily A2B response.
To test for A2B receptors we also infused the A2B selective antagonist alloxazine (Figure 2). While there is a lack of potent and highly selective A2B antagonists, alloxazine exhibits ∼10 fold selectivity for murine A2B receptors vs A2A receptors (Brackett & Daly, 1994) and completely inhibits A2B receptors at 2 μM. We chose a lower concentration in order to minimize antagonism at the A2A receptors which appear to be present. At 200 nM alloxazine failed to alter responses to A2A selective CGS21680 and non-selective 2-chloroadenosine (Figure 2). Hearts receiving the maximally effective 2 μM concentration also displayed unaltered responses to 2-chloroadenosine (Figure 2B). These data verify A2A mediated responses to both receptor agonists, and indicate an absence of functional A2B receptor in mouse coronary vessels.
Previous studies indicate that A2A receptors mediate coronary dilation in dog (Glover et al., 1996), pig (Lew & Kao, 1999; Hein et al., 1999), and guinea-pig (Belardinelli et al., 1998). Functional identification of A2A receptors in the pig (Lew & Kao, 1999; Hein et al., 1999) has recently been supported by molecular analysis (Hein et al., 2001). In contrast, A2B receptors are of primary importance in mediating coronary dilation in humans (Kemp & Cocks, 1999) and in the rat (Lewis & Hourani, 1997; Rose'meyer et al., 1999). A preliminary study by Morrison et al. (2001) indicates that gene deletion of the A2A receptor eliminates CGS21680-mediated coronary dilation in the mouse. While findings from genetically modified knock-out animals are not directly applicable to wild-type tissues, and although we have only assessed receptor identity in a single strain of mice, and rodents strains can show variability in cardiovascular responses, our data together with the findings of Morrison et al. (2001) support A2A-mediated coronary dilation in the murine heart.
It is worth noting that the slope of concentration-response curves is greater for all agonists in rat vs mouse (Table 2). This suggests that the efficacy of adenosine agonists at murine A2A receptors may be greater than at rat A2B receptors, and/or supports heterogeneity in adenosine responses in rat. Lewis & Hourani (1997) provide evidence that both A2A and A2B receptors may mediate dilation in rat heart, and there is support for an unidentified, potentially intracellular receptor in rat vessels (Prentice & Hourani, 1996; Prentice et al., 1997) in addition to mouse aorta (Prentice et al., 2001). This multiplicity of effector mechanisms may contribute to a broader concentration range over which agonists act in rat. The narrow range and steep slope in mouse is consistent with a single coronary receptor sub-type.
In assessing the identity of the adenosine receptor we chose to assess the rank order of agonist potencies, and study effects of a selective antagonist. This conventional pharmacological approach remains a key method for identification of adenosine receptors (Belardinelli et al., 1998; Brackett & Daly, 1994; Gurden et al., 1993; Hein et al., 1999; Kemp & Cocks, 1999; Kull et al., 1999; Lew & Kao, 1999; Prentice et al., 1996; 1997; Rose'meyer et al., 1999), and was adopted despite alternate methodologies, including epigenetic approaches (targeted gene knock-out and anti-sense oligonucleotides) (Nyce, 1999). Despite selectivity of epigenetic techniques, these methods possess limitations. Anti-sense studies in intact organs are largely restricted to brain and lungs due to difficulties in selectively introducing oligonucleotides, and these studies are limited by poor penetration into cells and nuclei. With respect to gene knock-out, an A2A knockout mouse has been assessed in terms of neurophysiological responses (Ledent et al., 1997), and there are preliminary data regarding adenosine and CGS21680 mediated coronary dilation in this model (Morrison et al., 2001). While these findings support an important A2A response in murine vessels, interpretation of gene knock-out data are limited since they assess effects of a genes long-term absence rather than the normal role of the gene product itself. Absence of the gene may lead to unknown developmental, morphological and functional changes, together with unpredicted compensatory responses. Receptor identification remains best achieved via complimentary pharmacological and molecular approaches.
Role of NO in adenosine receptor-mediated coronary dilation in mouse
There is evidence that adenosine is a mixed endothelial-dependent/independent dilator, activating both endothelial and smooth muscle receptors within the same vessel (Headrick & Berne, 1990; Moritoki et al., 1990; Rose'meyer & Hope, 1990; Headrick et al., 1992; Vials & Burnstock, 1993; Hein et al., 1999). To test the potential involvement of NO in adenosine responses, we studied effects of NO-synthase inhibition with L-NOARG. L-NOARG significantly increased baseline resistance and inhibited responses to endothelial-dependent ADP but not SNP, verifying selective inhibition of NO-dependent responses, and supporting a role for endogenous NO in control of resting tone in murine coronary vessels (Table 1, Figure 3). Curiously, a number of hearts displayed phasic oscillations in coronary resistance during L-NOARG treatment (Figure 4). Though the mechanism of these oscillations is unclear, it may reflect competition between constriction due to reduced NO release and relaxation due to locally released dilators. Importantly, while L-NOARG did not alter responses to SNP, it significantly attenuated responses to 2-chloroadenosine and CGS 21680, substantially increasing EC50s and threshold concentrations at which agonists induced dilation (Figure 3, Table 2).
While our data agree with studies supporting partial NO- or endothelial-dependent coronary responses to adenosine in guinea-pig (Newmann et al., 1988; Leipert et al., 1992; Vials & Burnstock, 1993), dog (Rubanyi & Vanhoutte, 1985; Zanzinger & Bassenge, 1993), and pig (Abebe et al., 1994; Kuo & Chancellor, 1995; Hein et al., 1999; 2000), they contrast with studies in human and porcine coronary vessels (Sabouni et al., 1990; Kirkebeon et al., 1994; Kemp & Cocks, 1999; Lew & Kao, 1999), and in rat heart (Lewis & Hourani, 1997). Varying observations from different species supports pronounced species differences in the mechanisms of adenosine-mediated coronary dilation. Contradictory observations made within a single species (e.g. Abebe et al., 1994; Kirkebeon et al., 1994; Kuo & Chancellor, 1995; Hein et al., 1999; Lew & Kao, 1999) demonstrates a need for further research. In this respect, both A2A and A2B receptors may mediate coronary dilation in the same species and there appear to be differences in transduction mechanisms for these sub-types. There is a greater weight of unequivocal data supporting NO-dependence of adenosine A2A responses (Newmann et al., 1988; Leipert et al., 1992; Vials & Burnstock, 1993) whereas there is controversy regarding NO-dependence of coronary A2B responses (Kirkebeon et al., 1994; Kemp & Cocks, 1999; Lewis & Hourani, 1997; Lew & Kao, 1999).
Functional antagonism vs NO-synthase inhibition
There may be two effects of L-NOARG in coronary vessels–enhanced functional antagonism increasing the resistance over which dilatory agonists can induce relaxation, and direct antagonism of NO-synthase dependent responses. Lewis & Hourani (1997) recently concluded that apparent antagonism of adenosine via NO-synthase inhibitors may reflect functional antagonism (i.e. vasoconstriction). They found that L-NOARG reduced adenosine responses in rat, but also reduced responses to NO-synthase independent SNP. However, responses to SNP were quite low in their study and L-NOARG only modestly reduced these in contrast with 2–10 fold differences in adenosine-response magnitude (Lewis & Hourani, 1997). We show that L-NOARG increases resting coronary resistance (Table 1), and this should alter dilatory response amplitude. If ‘pre-constrictors' do not directly inhibit mechanisms of action of a dilator, enhanced functional antagonism and resting tone simply increases the amplitude (but not sensitivity) of dilatory responses, as noted by Lew (1995). Functional antagonism is useful in increasing response amplitude for dilators, removing constraints imposed by degree of pre-constriction, and permitting analysis of agonist efficacies (Broadley & Nicholson, 1979; Lew, 1995). These factors are exemplified by responses to SNP: L-NOARG tends to enhance response amplitude to high levels of SNP when responses are expressed relative to the higher initial tone (Figure 3C). It is necessary to normalize such responses to make meaningful comparisons, and to remove differences which are not relevant (Lew, 1995). These minor differences are absent when SNP responses are scaled to maximal dilation (data not shown). Importantly, in direct contrast to SNP, responses to low levels of adenosine agonists are substantially reduced by L-NOARG while maximal responses are unaltered (Figure 3), the threshold effective concentration of 2-chloroadenosine is increased ∼10 fold (Figure 3), and coronary sensitivity to 2-chloroadenosine and CGS21680 is significantly reduced (Table 2). Selective effects of L-NOARG on A2-mediated responses (and on ADP responses) supports significant NO-dependence of adenosine-mediated dilation in mouse heart.
Assuming that responses to low levels of 2-chloroadenosine in the presence of L-NOARG can be considered largely NO-synthase independent, we can estimate NO-synthase dependent dilation by subtraction of NO-synthase independent responses from control (mixed) responses (Headrick & Berne, 1990; Headrick et al., 1992). As shown in Figure 6 the NO-synthase dependent L-NOARG sensitive response is important at low levels of A2 receptor activation. Dilation with 1–30 nM 2-chloroadenosine is almost entirely NO-dependent (Figure 6). At higher levels of activation NO-synthase independent dilation predominates. This is consistent with previous observations indicating that NO-dependent adenosine responses are sensitive yet small in amplitude (Headrick & Berne, 1990; Moritoki et al., 1990; Rose'meyer & Hope, 1990; Headrick et al., 1992; Vials & Burnstock, 1993). While small amplitude might be considered evidence for a minimal functional role (Vials & Burnstock, 1993), high sensitivity (Figure 6) ensures that it is quantitatively important at low, physiologically relevant levels of receptor activation.
Figure 6.
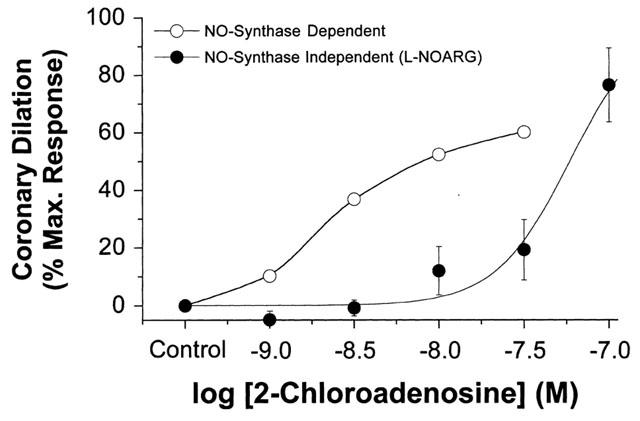
NO-synthase dependent and NO-synthase independent responses to 2-chloroadenosine in perfused mouse heart. The raw data is derived from the results shown in Figure 3. The NO-synthase independent response is represented by the dilatory response in the presence of 50 μM L-NOARG. The NO-synthase dependent response is calculated as the difference between the control response (in the absence of L-NOARG) and the NO-synthase independent response (in the presence of L-NOARG). Responses are scaled to per cent of maximal dilation observed.
Role of KATP channels in adenosine receptor-mediated coronary dilation in mouse
In addition to release of NO, activation of KATP channels is implicated in A2A adenosine receptor-mediated coronary dilation (Kuo & Chancellor, 1995; Randall, 1995; Hein et al., 1999; 2001). In contrast, A2B receptors may mediate coronary dilation in human vessels via NO and KATP-independent mechanisms (Kemp & Cocks, 1999). Our data reveal that KATP blockade with glibenclamide, at a level which fully antagonizes responses to minoxidil, markedly inhibits responses to CGS21680 and 2-chloroadenosine (Figure 5). Importantly, effects of KATP channel inhibition are additive to (and greater than) those for NO-synthase inhibition via L-NOARG. Considering the 120–300 fold reduction in sensitivity to CGS21680 and 2-chloroadenosine with combined NO-synthase and KATP inhibition, these two pathways appear to be the primary mechanisms contributing to A2A-mediated dilation. The more pronounced inhibitory effects of KATP channel blockade and additivity with the effects of L-NOARG suggest that A2A receptors trigger NO-mediated dilation via mechanisms distinct from activation of smooth muscle and/or endothelial KATP channels.
Concluding remarks
In conclusion, the present study demonstrates that peak coronary flows, reactive hyperaemia and adenosine sensitivity are all substantially greater in mouse vs rat heart. Adenosine agonist potency profiles and effects of alloxazine support A2A adenosine receptor-mediated coronary dilation in mouse vs A2B adenosine receptor-mediated dilation in rat. Our data demonstrate a significant NO-dependent and KATP channel-dependent components to adenosine mediated coronary dilation in the mouse. A2A-mediated dilation appears more strongly dependent on KATP channels than NO formation. However, although NO-independent dilation predominates over NO-dependent dilation at moderate to high agonist levels, the high-sensitivity NO-dependent response may play an important role under physiological conditions when adenosine concentrations and the level of A2A receptor activation are relatively low.
Acknowledgments
This work was supported in part by grants from the National Heart Foundation of Australia (G99B0246), and the National Health and Medical Research Council of Australia (971168).
Abbreviations
- CGS21680
2-[p-(2-carboxyethyl)phenethylamino]-5′-N-ethyl carboxamidoadenosine
- KATP
ATP-dependent K+ channel
- L-NOARG
NG-nitro L-arginine
- NECA
5′-N-ethylcarboxamidoadenosine
- NO
nitric oxide
- R-PIA
N6-(R)-(phenylisopropyl) adenosine
- SNP
sodium nitroprusside
References
- ABEBE W., MAKUJINA S.R., MUSTAFA S.J. Adenosine receptor-mediated relaxation of porcine coronary artery in presence and absence of endothelium. Am. J. Physiol. Heart Circ. Physiol. 1994;266:H2018–H2025. doi: 10.1152/ajpheart.1994.266.5.H2018. [DOI] [PubMed] [Google Scholar]
- BELARDINELLI L., SHRYOCK J.C., SNOWDY S., ZHANG Y., MONOPOLI A., LOZZA G., ONGINI E., OLSSON R.A., DENNIS D.M. The A2A adenosine receptor mediates coronary vasodilation. J. Pharmacol. Exp. Ther. 1998;284:1066–1073. [PubMed] [Google Scholar]
- BERNE R.M. The role of adenosine in regulation of coronary blood flow. Circ. Res. 1980;47:807–813. doi: 10.1161/01.res.47.6.807. [DOI] [PubMed] [Google Scholar]
- BRACKETT L.E., DALY J.W. Functional characterization of the A2B adenosine receptor in NIH 3T3 fibroblasts. Biochem. Pharmacol. 1994;47:801–814. doi: 10.1016/0006-2952(94)90480-4. [DOI] [PubMed] [Google Scholar]
- BROADLEY K.J., NICHOLSON C.D. Functional antagonism as a means of determining dissociation constants and relative efficacies of sympathomimetic amines in guinea-pig isolated atria. Br. J. Pharmacol. 1979;66:397–404. doi: 10.1111/j.1476-5381.1979.tb10844.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GLOVER D.K., RUIZ M., YANG J.Y., KOPLAN B.A., ALLEN T.R., SMITH W.H., WATSON D.D., BARRETT R.J., BELLER G.A. Pharmacological stress thallium scintigraphy with 2-cyclohexylmethylidenehydrazinoadenosine (WRC-0470). A novel, short-acting adenosine A2A receptor agonist. Circulation. 1996;94:1726–1732. doi: 10.1161/01.cir.94.7.1726. [DOI] [PubMed] [Google Scholar]
- GURDEN M.F., COATES J., ELLIS F., EVANS B., FOSTER M., HORNBY E., KENNEDY I., MARTIN D.P., STRONG P., VARDEY C.J. Functional characterization of three adenosine receptor types. Br. J. Pharmacol. 1993;109:693–698. doi: 10.1111/j.1476-5381.1993.tb13629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEADRICK J.P. Impact of aging on adenosine levels, A1/A2 responses, arrhythmogenesis, and energy metabolism in rat heart. Am. J. Physiol. Heart Circ. Physiol. 1996;270:H897–H906. doi: 10.1152/ajpheart.1996.270.3.H897. [DOI] [PubMed] [Google Scholar]
- HEADRICK J.P., BERNE R.M. Endothelium-dependent and independent relaxations to adenosine in guinea pig aorta. Am. J. Physiol. Heart Circ. Physiol. 1990;259:H62–H67. doi: 10.1152/ajpheart.1990.259.1.H62. [DOI] [PubMed] [Google Scholar]
- HEADRICK J.P., GAUTHIER N.S., MORRISON R.R., MATHERNE G.P. Chronotropic and vasodilatory responses to adenosine and b-adrenoceptor activation in mouse heart: effects of adenosine A1 receptor overexpression. Clin. Exp. Pharm. Physiol. 2000;27:185–190. doi: 10.1046/j.1440-1681.2000.03218.x. [DOI] [PubMed] [Google Scholar]
- HEADRICK J.P., NORTHINGTON F.C., HYNES M., MATHERNE G.P., BERNE R.M. Relative responses to luminal and adventitial adenosine in perfused arteries. Am. J. Physiol. Heart Circ. Physiol. 1992;263:H1437–H1446. doi: 10.1152/ajpheart.1992.263.5.H1437. [DOI] [PubMed] [Google Scholar]
- HEIN T.W., BELARDINELLI L., KUO L. Adenosine A2A receptors mediate coronary microvascular dilation to adenosine: role of nitric oxide and ATP-sensitive potassium channels. J. Pharmacol. Exp. Ther. 1999;291:655–664. [PubMed] [Google Scholar]
- HEIN T.W., LIAO J.C., KUO L. oxLDL specifically impairs endothelium-dependent NO-mediated dilation of coronary arterioles. Am. J. Physiol. Heart Circ. Physiol. 2000;278:H175–H183. doi: 10.1152/ajpheart.2000.278.1.H175. [DOI] [PubMed] [Google Scholar]
- HEIN T.W., WANG W., ZOGHI B., MUTHUCHAMY M., KUO L. Functional and molecular characterization of receptor subtypes mediating coronary microvascular dilation to adenosine. J. Mol. Cell. Cardiol. 2001;33:271–282. doi: 10.1006/jmcc.2000.1298. [DOI] [PubMed] [Google Scholar]
- KEMP B.K., COCKS T.M. Adenosine mediates relaxation of human small resistance-like coronary arteries via A2B receptors. Br. J. Pharmacol. 1999;126:1796–1800. doi: 10.1038/sj.bjp.0702462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIRKEBOEN K.A., NAESS P.A., OFFSTAD J., ILEBEKK A. Effects of regional inhibition of nitric oxide synthesis in intact porcine hearts. Am. J. Physiol. Heart Circ. Physiol. 1994;266:H1516–H1527. doi: 10.1152/ajpheart.1994.266.4.H1516. [DOI] [PubMed] [Google Scholar]
- KULL B., ARSLAN G., NILSSON C., OWMAN C., LORENZEN A., SCHWABE U., FREDHOLM B.B. Differences in the order of potency for agonists but not antagonists at human and rat adenosine A2A Receptors. Biochem. Pharmacol. 1999;57:65–75. doi: 10.1016/s0006-2952(98)00298-6. [DOI] [PubMed] [Google Scholar]
- KUO L., CHANCELLOR J.D. Adenosine potentiates flow-induced dilation of coronary arterioles by activating KATP channels in endothelium. Am. J. Physiol. Heart Circ. Physiol. 1995;269:H541–H549. doi: 10.1152/ajpheart.1995.269.2.H541. [DOI] [PubMed] [Google Scholar]
- LEDENT C., VAUGEOIS J.M., SCHIFFMANN S.N., PEDRAZZINI T., EL YACOUBI M., VANDERHAEGHEN J.J., COSTENTIN J., HEATH J.K., VASSART G., PARMENTIER M. Aggressiveness, hypoalgesia and high blood pressure in mice lacking the adenosine A2a receptor. Nature. 1997;388:674–678. doi: 10.1038/41771. [DOI] [PubMed] [Google Scholar]
- LEIPERT B., BECKER B.F., GERLACH E. Different endothelial mechanisms involved in coronary responses to known vasodilators. Am. J. Physiol. Heart Circ. Physiol. 1992;262:H1676–H1683. doi: 10.1152/ajpheart.1992.262.6.H1676. [DOI] [PubMed] [Google Scholar]
- LEW M. Extended concentration-response curves used to reflect full agonist efficacies and receptor occupancy-response coupling ranges. Br. J. Pharmacol. 1995;115:745–752. doi: 10.1111/j.1476-5381.1995.tb14996.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEW M.J., KAO S.W. Examination of adenosine receptor-mediated relaxation of the pig coronary artery. Clin. Exp. Pharmacol. Physiol. 1999;26:438–443. doi: 10.1046/j.1440-1681.1999.03054.x. [DOI] [PubMed] [Google Scholar]
- LEWIS C.D., HOURANI S.M.O. Involvement of function antagonism in the effects of adenosine antagonists and L-NAME in the rat isolated heart. Gen. Pharmacol. 1997;29:421–427. doi: 10.1016/s0306-3623(96)00466-1. [DOI] [PubMed] [Google Scholar]
- MORRISON R.R., HASSAN TALUKDER M.A., LEDENT C., MUSTAFA J.Adenosine-mediated increase in myocardial contractility and coronary flow in adenosine A2A receptor knockout mice FASEB J. 200115A763(Abstract) [Google Scholar]
- MORITOKI H., MATSUGI T., TAKASE H., UEDA H., TANIOKA A. Evidence for the involvement of cyclic GMP in adenosine-induced, age dependent vasodilatation. Br. J. Pharmacol. 1990;100:569–575. doi: 10.1111/j.1476-5381.1990.tb15848.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NEWMANN W.H., BECKER B.F., HEIER M., NEES S., GERLACH E. Endothelium-mediated coronary dilatation by adenosine does not depend on endothelial adenylate cyclase activation: studies in isolated guinea-pig hearts. Pfluegers Arch. 1988;413:1–7. doi: 10.1007/BF00581221. [DOI] [PubMed] [Google Scholar]
- NYCE J.W. Insight into adenosine receptor function using antisense and gene-knockout approaches. TiPS. 1999;20:79–83. doi: 10.1016/s0165-6147(99)01305-x. [DOI] [PubMed] [Google Scholar]
- OLSSON R.A., PEARSON J.D. Cardiovascular purinoceptors. Physiol. Rev. 1990;70:761–845. doi: 10.1152/physrev.1990.70.3.761. [DOI] [PubMed] [Google Scholar]
- PIETSCHMANN M., BARTELS H., FONS R. Capillary supply of heart and skeletal muscle of small bats and non-flying mammals. Respir. Physiol. 1982;50:267–282. doi: 10.1016/0034-5687(82)90023-8. [DOI] [PubMed] [Google Scholar]
- PRENTICE D.J., HOURANI S.M.O. Activation of multiple sites by adenosine in the rat isolated aorta. Br. J. Pharmacol. 1996;118:1509–1517. doi: 10.1111/j.1476-5381.1996.tb15567.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRENTICE D.J., PAYNE S.L., HOURANI S.M.O. Activation of two sites by adenosine receptor agonists to cause relaxation in rat isolated mesenteric artery. Br. J. Pharmacol. 1997;122:1509–1515. doi: 10.1038/sj.bjp.0701524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRENTICE D., BOON K., HOURANI S. Relaxation of mouse isolated aorta to adenosine and its analogues does not involve adenosine A1, A2 or A3 receptors. Eur. J. Pharmacol. 2001;415:251–255. doi: 10.1016/s0014-2999(01)00841-x. [DOI] [PubMed] [Google Scholar]
- RAKUSAN K., TOMANEK R.J. Distribution of mitochondria in normal and hypertrophic myocytes from the rat heart. J. Mol. Cell. Cardiol. 1986;18:299–305. doi: 10.1016/s0022-2828(86)80412-6. [DOI] [PubMed] [Google Scholar]
- RANDALL M.D. The involvement of ATP-sensitive potassium channels and adenosine in the regulation of coronary flow in the isolated perfused rat heart. Br. J. Pharmacol. 1995;116:3068–3074. doi: 10.1111/j.1476-5381.1995.tb15965.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROSE'MEYER R.B., HARDEN F.A., VARELA J.I., HARRISON G.J., WILLIS R.J. Age-related changes in adenosine in rat coronary resistance vessels. Gen. Pharmacol. 1999;32:35–40. doi: 10.1016/s0306-3623(98)00023-8. [DOI] [PubMed] [Google Scholar]
- ROSE'MEYER R.B., HOPE W. Evidence that A2 purinoceptors are involved in endothelium-dependent relaxation of rat thoracic aorta. Br. J. Pharmacol. 1990;100:575–580. doi: 10.1111/j.1476-5381.1990.tb15849.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RUBANYI G., VANHOUTTE P.M. Endothelium-removal decreases relaxations of canine coronary arteries caused by ß-adrenergic agonists and adenosine. J. Cardiovasc. Pharmacol. 1985;7:139–144. doi: 10.1097/00005344-198501000-00023. [DOI] [PubMed] [Google Scholar]
- SABOUNI M.H., RAMAGOPAL M.V., MUSTAFA S.J. Relaxation by adenosine and its analogues of potassium-contracted human coronary arteries. Naunyn Schmiedeberg's Arch. Pharmacol. 1990;341:388–390. doi: 10.1007/BF00180667. [DOI] [PubMed] [Google Scholar]
- TUNE J.D., RICHMOND K.N., GORMAN M.W., FEIGL E.O. KATP channels, nitric oxide, and adenosine are not required for local metabolic coronary vasodilation. Am. J. Physiol. Heart Circ. Physiol. 2001;280:H868–H875. doi: 10.1152/ajpheart.2001.280.2.H868. [DOI] [PubMed] [Google Scholar]
- VIALS A., BURNSTOCK G. A2-purinoceptor-mediated relaxation in the guinea-pig coronary vasculature: a role for nitric oxide. Br. J. Pharmacol. 1993;109:424–429. doi: 10.1111/j.1476-5381.1993.tb13586.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZANZINGER J., BASSENGE E. Coronary vasodilation to acetylcholine, adenosine and bradykinin in dogs: effects of inhibition of NO-synthesis and captopril. Eur. Heart. J. 1993;14:164–168. [PubMed] [Google Scholar]