

Abstract
It was supposed that inhibitors of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG CoA) reductase (statins) might inhibit the expression of the fibrosis-related factor CTGF (connective tissue growth factor) by interfering with the isoprenylation of Rho proteins.
The human renal fibroblast cell line TK173 was used as an in vitro model system to study the statin-mediated modulation of the structure of the actin cytoskeleton and of the expression of CTGF mRNA.
Incubation of the cells with simvastatin or lovastatin time-dependently and reversibly changed cell morphology and the actin cytoskeleton with maximal effects observed after about 18 h.
Within the same time period, statins reduced the basal expression of CTGF and interfered with CTGF induction by lysophosphatidic acid (LPA) or transforming growth factor beta. Simvastatin and lovastatin proved to be much more potent than pravastatin (IC50 1–3 μM compared to 500 μM).
The inhibition of CTGF expression was prevented when the cells were incubated with mevalonate or geranylgeranylpyrophosphate (GGPP) but not by farnesylpyrophosphate (FPP). Specific inhibition of geranylgeranyltransferase-I by GTI-286 inhibited LPA-mediated CTGF expression whereas an inhibitor of farnesyltransferases FTI-276 was ineffective.
Simvastatin reduced the binding of the small GTPase RhoA to cellular membranes. The effect was prevented by mevalonate and GGPP, but not FPP.
These data are in agreement with the hypothesis that interference of statins with the expression of CTGF mRNA is primarily due to interference with the isoprenylation of RhoA, in line with previous studies, which have shown that RhoA is an essential mediator of CTGF induction.
The direct interference of statins with the synthesis of CTGF, a protein functionally related to the development of fibrosis, may thus be a novel mechanism underlying the beneficial effects of statins observed in renal diseases.
Keywords: Lysophosphatidic acid, connective tissue growth factor, transforming growth factor, lovastatin, simvastatin, Rho protein, HMG CoA reductase
Introduction
Inhibitors of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG CoA) reductase (statins) are widely used in the treatment of hypercholesterolaemia, because they interfere with the rate limiting step of cholesterol biosynthesis and thus effectively lower the serum levels of cholesterol and low density lipoproteins (LDL) (Maron et al., 2000). There is increasing evidence that the beneficial effects of statins go beyond the inhibition of cholesterol biosynthesis. The isoprenoids farnesylpyrophosphate (FPP) and geranylgeranylpyrophosphate (GGPP), intermediates of cholesterol biosynthesis, are important lipid attachments for the posttranslational modification of proteins (Zang & Casey, 1996). Modified proteins include the γ subunit of heterotrimeric G proteins, haeme a, nuclear lamins, and small GTPases of the Rho and Ras family.
Dyslipidaemia, hypertension, and proteinuria have been identified as independent risk factors of progressive renal disease (summarized by Keane, 2000). In different animal models of chronic renal glomerular disease, statins have been shown to significantly retard the progression of the disease. Beneficial effects of statins have also been reported in humans with different types of glomerulonephritis or renal impairment (Johnson et al., 1999, and citations therein). In most cases, the beneficial effect was attributed to the lipid lowering capacity of statins. However, in some studies, the degree of vascular and tubulointerstitial injury was also reduced, when the cholesterol levels were not profoundly elevated. This suggested that statins might interfere more directly with cellular processes involved in renal injury. In a recent study Kim et al. (2000) showed that lovastatin ameliorated the development and progression of diabetic nephropathy in streptozotocin-induced diabetic rats. They related this effect to the interference of lovastatin with the glucose-induced upregulation of transforming growth factor beta (TGF-β) and fibronectin mRNA. These data are in line with data obtained in vascular smooth muscle cells, where statins inhibited the synthesis of thrombospondin-1, an activator of TGF-β (Riessen et al., 1999). In human mesangial cells, statins also interfere with the induction of certain inflammatory mediators such as LDL-induced expression of interleukin-6 (Massy et al., 2000) or induction of monocyte chemoattractant protein-1 (Kim et al., 1995; Park et al., 1998). The effects were reversible by mevalonate and thus related to the inhibition of HMG-CoA reductase, but the molecular mechanism of the statin-mediated inhibition was not further investigated.
Whereas most of the renal effects of statins have been related to glomerular diseases and glomerular cells such as mesangial cells, there is some evidence that statins might also be of therapeutic value in the treatment of tubulointerstitial fibrosis, which is characterized by initial proximal tubular cell hypertrophy and hyperplasia followed by proliferation of cortical fibroblasts and excessive extracellular matrix deposition (Müller et al., 1996). The final stage of this tubulointerstitial pathology, atrophy of proximal tubular cells and impaired renal transport function, leads to renal impairment with up to now no satisfactory therapy.
In rat proximal tubular cells, statins inhibited cell proliferation and activated the plasminogen activator/plasmin system in a RhoA-dependent manner (Essig et al., 1998). Using cells from human renal cortex, Johnson et al. (1999) observed a direct effect of statins on the induced collagen synthesis in fibroblasts independent of their effect on cholesterol synthesis. One of the major factors to induce components of the extracellular matrix is TGF-β, which has been shown to play an essential role in fibrotic diseases of the kidney (Bitzer et al., 1998; Piek et al., 1999). Some of the effects of TGF-β on extracellular matrix molecules are not direct effects, but are mediated by connective tissue growth factor (CTGF), which by itself is induced by TGF-β (Grotendorst, 1997). CTGF is a cysteine-rich secreted polypeptide, which belongs to the family of CCN (CYR61, CTGF and NOV) proteins (Bork, 1993; Lau & Lam, 1999). These proteins share structural homologies and function as modulators of complex cellular functions such as growth, differentiation, adhesion and migration (Lau & Lam, 1999). In fibroblasts, CTGF is potently induced by TGF-β (Grotendorst et al., 1996), but may also be induced by lysophosphatidic acid, an activator of heptahelical receptors (Heusinger-Ribeiro et al., 2001). It stimulates fibroblast cell proliferation and mediates TGF-β-induced anchorage-independent growth (Kothapalli et al., 1997). Furthermore, CTGF is a potent stimulator of extracellular matrix synthesis in these cells (Frazier et al., 1996; Duncan et al., 1999).
Elevated levels of CTGF are found in fibrotic lesions (e.g. Igarashi et al., 1996; Oemar et al., 1997; Dammeier et al., 1998) and are suggested to be functionally involved in the development and progression of fibrotic diseases (Gupta et al., 2000). In the kidney, CTGF mRNA levels were elevated in the majority of biopsies obtained from patients with various types of renal diseases characterized by glomerulosclerosis and tubulointerstitial fibrosis (Ito et al., 1998).
We have shown previously that RhoA is critically involved in the regulation of CTGF expression in rat mesangial cells (Hahn et al., 2000) and also in human renal fibroblasts (Heusinger-Ribeiro et al., 2001). It was thus tempting to speculate that the expression of CTGF might be regulated by statins, which modify the isoprenylation of the small GTPases of the Rho and Ras family.
Methods
Materials
Recombinant human TGF-β was obtained from TEBU, Frankfurt, Germany. Cell culture reagents were from Biochrom, Berlin, Germany, FCS was from Gibco, Eggenstein, Germany. Geranylgeranylpyrophosphate (GGPP), farnesylpyrophosphate (FPP), mevalonic acid lactone, lysophosphatidic acid (LPA), Hoechst 33258 were from Sigma, Muenchen, Germany. GGPP and FPP were dissolved in methanol/10 mM NH4OH (v v−1, 7/3). The respective solvent control did not affect any of the parameters measured. Mevalonic acid lactone was converted to sodium mevalonate as described by Essig et al. (1998). FTI-276 and GGTI-286 were obtained from Calbiochem, Bad Soden, Germany. Both compounds were dissolved in DMSO at 10 mM. DMSO in the final concentrations used did not affect the expression of CTGF.
Drugs
Simvastatin and Lovastatin were kindly provided by MSD, Sharp and Dohme, München, Germany. Both compounds were dissolved in ethanol and activated as described by Jakobisiak et al. (1991). Pravastatin was kindly provided by Bristol-Myers-Squibb, GmbH, Muenchen, Germany.
Staining of actin filaments
Cells were cultured and growth arrested on glass 8-well multitest slides (ICN, OH, U.S.A.) placed in a Petri dish. Further treatment with different toxins and stimuli was carried out in wet chambers. After treatment cells were fixed with 3% paraformaldehyde in PBS (phosphate buffered saline) for 10 min and then permeabilized with 0.2% Triton X-100 in PBS for 7 min at room temperature. To examine DNA-fragmentation cells were incubated with Hoechst 33258 (8 μg ml−1) for 5 min. After washing, the actin cytoskeleton was stained with rhodamine-phalloidin (Molecular Probes, Leiden, The Netherlands) for 20 min.
Cell culture
Immortalized human renal fibroblasts were kindly provided by G.A. Müller, Göttingen, Germany (Müller et al., 1995). The cells were grown in DMEM supplemented with 2 mM L-glutamine, 4.5 g l−1 glucose, 100 u ml−1 penicillin and 100 μg ml−1 streptomycin containing 10% FCS. Renal fibroblasts (0.8–1.0×106 cells per 10 ml) were plated in 100 mm Petri dishes in medium with 10% FCS. At subconfluency (after 2–3 days) cells were serum starved in DMEM containing 0.5% FCS for 1 day.
Northern blot analysis
Northern blot analysis was performed as described previously (Stroebel & Goppelt-Struebe, 1994). After stimulation for the indicated times, total RNA was extracted according to the protocol of Chomczynski & Sacchi (1987) with minor alterations. Usually, RNA yield was about 60–80 μg per 10 cm Petri dish. Separation of total RNA (20 μg per lane) was achieved by use of 1.2% agarose gels containing 1.9% formaldehyde with 1×MOPS as gel running buffer. Separated RNA was transferred to nylon membranes by capillary blotting and fixed by baking at 80°C for 1–2 h. 18 and 28S rRNA were stained with methylene blue (0.04% in 500 mM sodium acetate, pH 5.2) and directly quantitated by densitometry.
Hybridization was performed with cDNA probes labelled with [32P]-dCTP using the NonaPrimer kit from Appligene, Heidelberg, Germany. A cDNA specific for CTGF was kindly provided by N. Abdel-Wahab, London, U.K. (Mason et al., 1997).
Blots were prehybridized for at least 1 h at 40°C. The probes were allowed to bind overnight at the same temperature. Washing at 40°C was carried out 2×10 min at high salt (2×SSC, 0.2% SDS) and 2×15 min at low salt (0.2×SSC, 0.2% SDS) conditions. DNA/RNA hybrids were detected by autoradiography using Kodak X-Omat AR film. Quantitative analysis was performed by densitometric scanning of the autoradiographs (Bioprofil, Froebel, Wasserburg, Germany). All values were corrected for differences in RNA loading by calculating the ratio of CTGF to 18S rRNA expression.
Western blot analysis
Cellular proteins were isolated using RIPA buffer (50 mM Tris/HCl, pH 7.5, 1% (v v−1) Triton X-100, 0.1% (w v−1) deoxycholic acid, 0.1% (w v−1) SDS, 150 mM NaCl, 1 mM phenylmethylsulfonyl fluoride, 1 mM sodium vanadate, 14 μg ml−1 aprotinin). For Western blot analysis, 10–30 μg protein were separated by SDS–PAGE (10% polyacrylamide), transferred onto PVDF membrane (Pall Biosupport Division Dreieich, Germany) and probed with specific polyclonal antibodies. Polyclonal rabbit antibodies directed against specific peptides of p44 MAP kinase or its phosphorylated form, P-p44 MAP kinase, were obtained from New England Biolabs. The antibody directed against phosphotyrosine was obtained from Santa Cruz, Heidelberg, Germany. The peroxidase-conjugated anti-rabbit secondary antibody was obtained from Amersham (Braunschweig, Germany). Protein-antibody complexes were visualized by the enhanced chemiluminescence detection system (ECL, Amersham).
To obtain cellular membranes, the cells were rinsed twice with ice-cold phosphate-buffered saline (PBS); then they were scraped into 600 μl lysis buffer (250 mM sucrose, 10 mM EGTA, 2 mM EDTA, 20 mM Tris-HCl, pH 7.5; 1 mM PMSF, 0.1% leupeptin and 0.1% aprotinin). The samples were sonified (three times for 10 s each sample) and then centrifuged for 45 min at 100,000×g, 4°C. The high speed pellet, which comprises all membrane subfractions, was resuspended in 50 μl lysis buffer. Proteins were separated on 15% SDS–PAGE and tested for RhoA immunoreactivity as described above. The antibody against RhoA (26C4) was from Santa Cruz. The peroxidase-conjugated anti-mouse secondary antibody was obtained from Amersham (Braunschweig, Germany) and detected with the enhanced chemiluminescence detection system (ECL, Amersham). Improved detection was obtained, when anti-mouse biotinylated IgG was combined with avidin-D-coupled horseradish peroxidase (Vector Laboratories, Burlingame, U.S.A.). To control for equal loading and blotting, the blots were stained with amido black.
Results
Statins affect the actin cytoskeleton
The human renal fibroblast cell line TK173, which has many characteristics in common with primary renal fibroblasts (Müller et al., 1995), was used as a model system to study the effects of statins on cell morphology and gene expression of CTGF. Incubation of the cells with simvastatin or lovastatin led to a time and concentration dependent change in cell morphology. Within 18 h of treatment with 10 μM simvastatin or lovastatin, the originally flat and confluent cells retracted their cytoplasm and developed filopodia (Figure 1A). This was due to a disruption of the actin cytoskeleton as shown in Figure 1B, C. Resting fibroblasts show a distinct pattern of actin stress fibres. Treatment with statins reduced the actin stress fibres, especially those spanning the cytosol. As examples the effects of simvastatin are shown in Figure 1B, C. Concomitant treatment of the cells with mevalonate not only prevented the dissolution of the stress fibres by simvastatin, but rather increased the actin fibres (Figure 1B), indicating the simvastatin-mediated effects on cell morphology to be caused by inhibition of HMG-CoA reductase. More specifically, the morphological changes were prevented by geranylgeranylpyrophosphate (GGPP), but not by farnesylpyrophosphate (FPP), suggesting Rho proteins as targets of statin action (Figure 1A). Staining of the nuclei with the dye Hoechst 33258 did not show any signs of apoptosis when the cells were treated with simvastatin (Figure 1C) or lovastatin (not shown) for 18 h. The changes in cell morphology were fully reversible, when the cells were washed after treatment with statins and then further incubated in cell culture medium without statin for another 18 h (Figure 1A).
Figure 1.
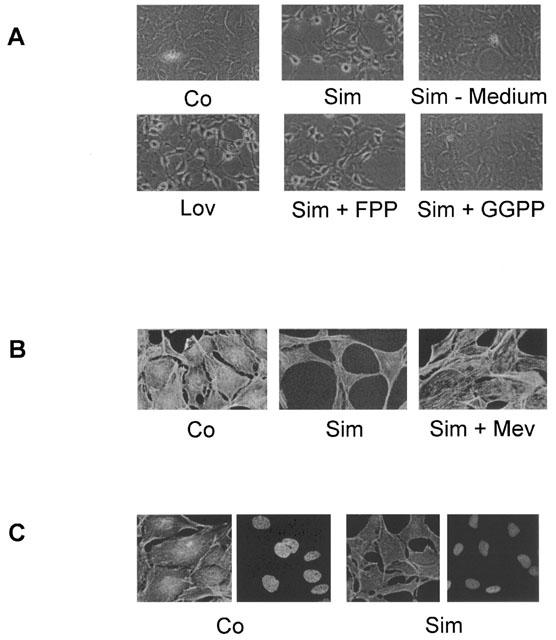
Modulation of cell morphology and the actin cytoskeleton by statins. Fibroblasts were incubated with simvastatin (Sim, 10 μM) or lovastatin (Lov, 10 μM) with or without FPP (10 μM), GGPP (10 μM) or mevalonate (Mev, 500 μM). Sim-Medium: Cells were incubated with simvastatin for 18 h and subsequently cultured in medium with 10% serum without simvastatin for another 18 h. (A) Light microscopy of cells cultured as indicated. Photographs were taken under identical conditions. Original magnification:×200. (B) The actin cytoskeleton was stained with rhodamine-phalloidin. Photographs were taken under identical conditions. Original magnification:×400. (C) The actin cytoskeleton was stained with rhodamine-phalloidin and the nuclei were stained with Hoechst. Photographs were taken under identical conditions. Original magnification:×400. Data shown are representative of at least three experiments for each condition.
Interference of statins with CTGF expression
Previous studies have shown that disruption of the actin cytoskeleton by cytochalasin D or by interference with RhoA signalling by C3 exotoxin interferes with the expression of CTGF (Hahn et al., 2000; Heusinger-Ribeiro et al., 2001). Preincubation of the fibroblasts with simvastatin (10 μM) for 6–18 h led to a time-dependent decrease of basal CTGF expression (Figure 2). More prominently, the strong induction of CTGF by LPA was completely abolished when the cells were pre-treated with the statin for 18 h.
Figure 2.
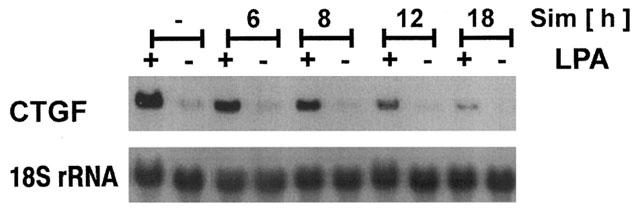
Time dependent interference of simvastatin with CTGF mRNA expression. Fibroblasts were preincubated with simvastatin (Sim; 10 μM) for the times indicated. Cells were stimulated with LPA (10 μM) for 90 min. CTGF mRNA was detected by Northern blot analysis.
Interference with CTGF expression was concentration-dependent. At 10–30 μM simvastatin or lovastatin completely inhibited the LPA-induced expression of CTGF with an IC50 of 1–3 μM (Figure 3A, B). Simvastatin proved to be more potent than lovastatin in all experiments. Pravastatin, a water soluble statin, was much less potent: even at a concentration of 500 μM, only a partial inhibition of CTGF expression was observed (Figure 3C). Interference of statins with CTGF expression was not restricted to LPA as stimulus. TGF-β is a potent stimulus of CTGF expression although with a slower kinetic (Heusinger-Ribeiro et al., 2001). After a 6 h incubation period, CTGF mRNA expression was increased 4.7±1.4 fold, means±s.d., n=10. Preincubation with lovastatin or simvastatin (10 μM; Figure 3D) for 18 h reduced the TGF-β-mediated increased expression of CTGF to near background levels. Pravastatin (500 μM) partially reduced the induction of CTGF by TGF-β (Figure 3C).
Figure 3.
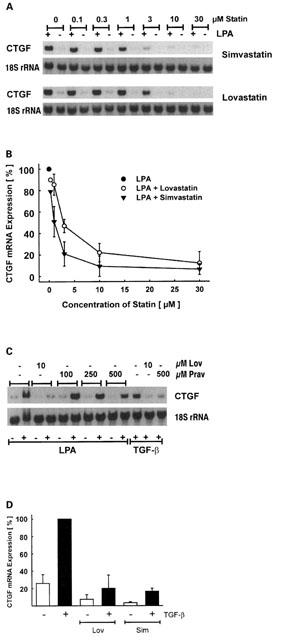
Concentration dependent inhibition of CTGF mRNA expression by statins. Fibroblasts were preincubated with simvastatin or lovastatin for 18 h as indicated. Cells were stimulated with LPA (10 μM) for 90 min. (A) Typical Northern blot analysis of CTGF mRNA expression. (B) To compare different experiments, stimulation with LPA was set to 100%. Data are means±s.d. or±half range of 2–3 experiments. (C) Fibroblasts were preincubated with lovastatin (Lov) or pravastatin (Prav) for 18 h as indicated. Stimulation with LPA (10 μM) was for 90 min, with TGF-β (5 ng ml−1) for 6 h. (D) To compare different experiments, stimulation with TGF-β (5 ng ml−1) was set to 100%. Simvastatin and lovastatin were used at 10 μM. Data are means±s.d. of three experiments.
RhoA as potential mediator of the inhibitory effect of statins
In order to investigate the molecular mechanism of statin-mediated inhibition of CTGF induction, cells were incubated with statins in the presence of mevalonate, to functionally overcome the inhibition of HMG CoA reductase. Mevalonate completely prevented the inhibitory action of lovastatin or simvastatin. In the presence of mevalonate, the basal levels of CTGF expression remained unaltered (Figure 4A) and the stimulation by LPA (Figure 4A, D) and TGF-β (Figure 4C, E) was not affected by the statins. As RhoA has previously been shown to be critically involved in the regulation of CTGF expression, it was supposed to be the target of statins interfering with CTGF expression. To test this hypothesis, fibroblasts were incubated with statins in the presence of GGPP to allow geranylgeranylation of Rho proteins or with FPP to substitute for the farnesylation of Ras proteins. As can be seen from Figure 4A–D, only GGPP was able to overcome the simvastatin- or lovastatin-mediated inhibition whereas FPP was without effect. This indicated that Rho proteins, but not Ras proteins were involved in the statin-mediated inhibition of CTGF. In the absence of statins, neither GGPP nor FPP affected basal or stimulated expression of CTGF (Figure 4B and data not shown).
Figure 4.
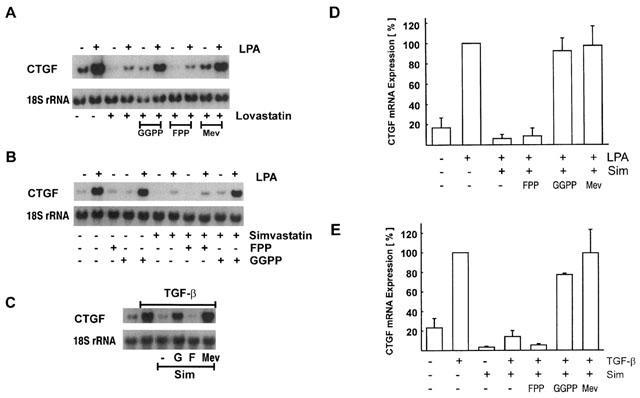
Interference with statin action by GGPP and mevalonate but not FPP. Fibroblasts were preincubated with lovastatin (10 μM; A) or simvastatin (10 μM; B,C) in the presence or absence of GGPP (G; 10 μM), FPP (F; 10 μM) or mevalonate (M; 500 μM). Stimulation time was 90 min for LPA (10 μM) and 6 h for TGF-β (5 ng ml−1). To compare different experiments, stimulation with LPA (A,B,D) or TGF-β (C,E) was set to 100%. Data are means±half range or±s.d. of 2–3 experiments.
The importance of isoprenylation of Rho proteins for the induction of CTGF was further substantiated by GGTI-286, an inhibitor of the geranylgeranyltransferase-I (Lerner et al., 1997). Pre-treatment of the fibroblasts with GGTI-286 for 18 h substantially reduced LPA-mediated induction of CTGF, while FTI-276, an inhibitor of the farnesyltransferase, was without significant effect (Figure 5). A direct effect of simvastatin on the modification of RhoA was shown by Western blot analysis of membrane-bound RhoA (Figure 6A). Treatment of the fibroblasts with increasing concentrations of simvastatin (2–50 μM) for 18 h reduced the membrane-bound RhoA. Cytosolic RhoA was detectable on gels loaded with 30 μg protein (Figure 6B). This corresponded to only 1/40 of the cellular protein used for detection in the membrane fraction, indicating that most of the RhoA protein is located in the cytosol in an inactive state. Upon treatment with simvastatin, the cytosolic RhoA concentration increased even beyond the portion attributable to the release from membranes suggesting induced RhoA synthesis by simvastatin. Binding of RhoA to cellular membranes was dependent on geranylgeranylation, because mevalonate and GGPP were able to prevent the effect of simvastatin, whereas FPP was without effect (Figure 6C).
Figure 5.
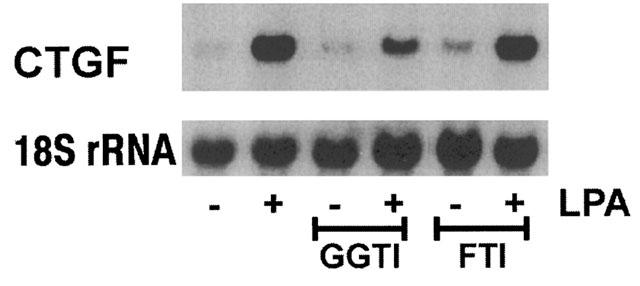
Inhibition of geranylgeranyltransferase-I inhibits CTGF mRNA induction. Fibroblasts were preincubated with GGTI-286 (10 μM) or FTI-276 (10 μM) for 18 h and then incubated with LPA for 90 min. The blot is representative of two identical experiments.
Figure 6.
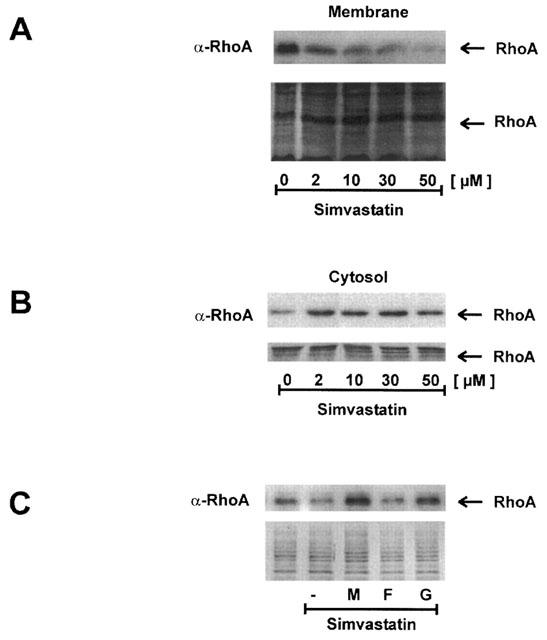
Reduced membrane binding of RhoA by simvastatin is dependent on interference with geranylgeranylation. Fibroblasts were incubated with simvastatin for 18 h. Membranes (A) and cytosol (B) were separated by differential centrifugation. One hundred and fifty micrograms of membrane protein and 30 μg of cytosolic protein were separated by SDS–PAGE and analysed by Western blotting using an antibody directed against RhoA. Blots were stained with amido black to confirm equal loading and blotting. To avoid side effects, the blots were loaded with controls in the middle of the blot and rearranged for the presentation. (C) Fibroblasts were incubated with or without simvastatin for 18 h. Part of the cells was incubated with simvastatin plus mevalonate (M, 500 μM), FPP (F, 10 μM) or GGPP (G, 10 μM) as indicated. Membrane protein was analysed using a biotin-avidin system as described in Methods. To confirm equal loading, the upper part of the blot stained with amido black is shown.
LPA binds to seven transmembrane receptors which couple to trimeric G-proteins (Goetzl & An, 1998; Contos et al., 2000). One of the subunits of trimeric G-proteins, Gγ, is modified by geranylgeranyl moieties. Therefore, the question was addressed, whether early signalling events of LPA receptor activation were modulated by statins. Fibroblasts were preincubated with simvastatin (10 μM) for 20 h and then stimulated with LPA. Protein tyrosine phosphorylation was assessed after 5 min. The tyrosine phosphorylation pattern changed upon incubation with LPA (Figure 7A). Incubation with simvastatin modified the pattern of tyrosine phosphorylation, but did not abrogate LPA signalling. The phosphorylated bands observed at about 42 kDa were identified as phosphorylated p42/44 MAP kinase (Figure 7B). Activation of these enzymes represents a very early activation step of signal transduction after LPA stimulation (e.g. Goppelt-Struebe et al., 2000).
Figure 7.
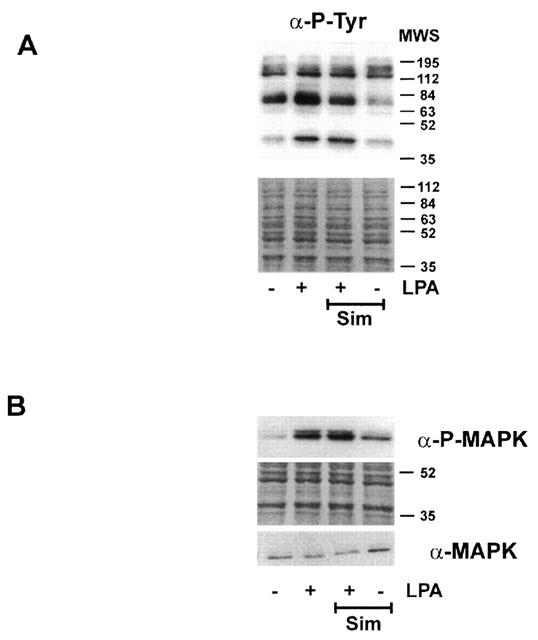
Modulation of early LPA signalling by statins. Fibroblasts were preincubated with simvastatin (Sim, 10 μM) for 18 h and then stimulated with LPA for 5 min. Cellular protein was separated by SDS–PAGE and phosphotyrosine proteins detected by Western blot analysis (A). The phosphorylated form of p42/44 MAP kinase was detected by a phospho-specific antibody (B). To detect MAP kinase, samples were run in parallel on a separate gel. The position of the molecular weight standard (MWS) is indicated. Blots were stained with amido black to confirm equal loading and blotting. Blots shown are representative of three experiments.
Discussion
Rho and Ras proteins as targets of statins have been described in various cell types, most often related to modulation of cell growth and apoptosis (e.g. Negre-Aminou et al., 1997). Growth-arrested cells were used in this study and during the time period of the experiments (up to 24 h), no signs of apoptosis were detectable. The changes in cell morphology were fully reversible indicating that under these conditions, the statins were not toxic for the cells. Susceptibility towards the growth inhibitory and pro-apoptotic effects of statins seems to be dependent on the cell type (Negre-Aminou et al., 1997), with fibroblasts being less responsive than e.g. tubular epithelial cells (Vrtovsnik et al., 1999).
The changes in cell morphology were related to the disruption of the actin cytoskeleton similarly as observed after treatment of the cells with cytochalasin D or toxin B, an inhibitor of proteins of the Rho family. The dissolution of the actin stress fibres was prevented by mevalonate, indicating that the changes were caused by inhibition of HMG CoA reductase. Products of the mevalonate pathway, the isoprenoid lipids farnesyl- and geranylgeranylpyrophosphate, are critically involved in the regulation of the activity of the small GTPases of the Ras and Rho family (Zhang & Casey, 1996). In the course of posttranslational processing these lipids are added by the enzymes farnesyltransferase and geranylgeranyltransferase-1, respectively. Isoprenylation is a prerequisite of membrane association of the small GTPases, which determines the GTP binding activity of these enzymes (van aelst & D'souza-Schorey, 1997). Interference with the isoprenoid synthesis by statins strongly reduced the membrane-bound fraction of RhoA in fibroblasts, in line with data obtained in other cells, e.g. Chinese hamster ovary cells (Gnad et al., 2000) or endothelial cells (Laufs et al., 2000). It was confirmed that binding of RhoA to cellular membranes is dependent on geranylgeranylation, because mevalonate and GGPP were able to prevent the effect of simvastatin, whereas in the presence of FPP, simvastatin reduced the binding of RhoA. The portion of membrane-bound RhoA was small compared to the total cellular RhoA protein. As described recently in endothelial cells (Laufs et al., 2000), statins upregulated the synthesis of RhoA, which accumulated in the cytosolic fraction, but due to the missing posttranslational modification did not increase the pool of RhoA available for signalling.
Treatment of fibroblasts with statins interfered with the induction of the expression of CTGF. In line with the requirement of posttranslational processing of the GTPases, a preincubation period of several hours was required to obtain full inhibition. Lovastatin and simvastatin almost completely prevented the expression of CTGF at a concentration of 10–30 μM, the IC50 was determined to be in the range of 1–3 μM. These concentrations are in the same range as those needed to interfere with cellular proliferation (Negre-Aminou et al., 1997; Vrtovsnik et al., 1997), with the integrity of the actin cytoskeleton (Koch et al., 1997) or with the induction of matrix proteins (Riessen et al., 1999). In all experiments, simvastatin proved to be about two times more potent than lovastatin, which is in line with the clinical experience, where two times higher doses of lovastatin compared to simvastatin are required to obtain a comparable reduction in LDL cholesterol (Illingworth et al., 1994; Desager & Horsmans, 1996; Maron et al., 2000). Pravastatin, in contrast, which is clinically considered to be equally potent as lovastatin, interfered with CTGF induction only at much higher concentrations (0.5–1 mM). Most likely because of its hydrophobicity, pravastatin is poorly taken up by cells in culture.
Basal levels of CTGF mRNA were observed in the fibroblast cell line, which were also reduced by treatment with statins. This reflected the dynamic regulation of CTGF mRNA which has a half life less than 2 h (Pereira et al., 2000, and unpublished results). In previous studies, inhibition of Rho proteins by toxinB or RhoA by C3 exotoxin similarly reduced the basal CTGF expression, indicating that RhoA is involved in the regulation of the basal levels of CTGF (Hahn et al., 2000; Heusinger-Ribeiro et al., 2001).
Statin-mediated interference with the induced expression of CTGF was prevented by mevalonate and more specifically by GGPP, whereas FPP was without effect. Furthermore, specific inhibition of geranylgeranyltransferase-I reduced CTGF expression, while no effect was observed when farnesyltransferase was inhibited. These data indicate that induction of CTGF is critically dependent on proteins which are geranylgeranylated. The gamma subunit of trimeric G-proteins is geranylgeranylated (e.g. Pomerantz et al., 1997) and statins might thus affect very early steps in LPA signalling, the receptor of which couples to trimeric G-proteins. Tyrosine phosphorylation and activation of the p42/44 MAP kinase pathway were taken as measures of early cellular activation. The phosphorylation pattern was modified indicating distinct interference with LPA signalling. Activation of p42/44 MAP kinases was not significantly affected by pre-treatment with concentrations of simvastatin, which completely prevented CTGF induction. These data cannot exclude modification of a certain subtype of G-protein subunit gamma, but argue against the possibility that the effects of statins on CTGF expression have to be attributed solely to impaired LPA signalling or to down-regulation of LPA receptors, as has been shown for the angiotensin II receptor ATI (Wassmann et al., 2001). The most likely isoprenylated protein to be responsible for the reduced induction of CTGF is thus RhoA, which has been previously shown to be critically involved in the regulation of the expression of CTGF and the organization of actin stress fibres (Hahn et al., 2000; Heusinger-Ribeiro et al., 2001). Disruption of the actin cytoskeleton by cytochalasin D or by inhibition of RhoA signalling by C3 exotoxin or inhibition of Rho kinases interfered with LPA- as well as TGF-β-mediated induction of CTGF. RhoA as signalling module of LPA-mediated cellular activation is well established, whereas much less is known about a connection between TGF-β signalling and RhoA-mediated events. In recent reports, Kucich et al. (2000) and Rosenbloom et al. (2000) showed that inhibitors of geranylgeranyltransferase-I inhibited the TGF-β-mediated induction of fibronectin and collagen I expression, respectively. It is interesting to note that CTGF is an inductor of both of these matrix molecules (Murphy et al., 1999). Furthermore, CTGF has been recognized as mediator of TGF-β-induced collagen synthesis (Duncan et al., 1999). One might thus speculate that inhibition of CTGF synthesis may be the reason for at least part of the effects of geranylgeranyltransferase inhibitors on matrix synthesis.
In vivo, CTGF is associated with renal interstitial fibrosis shown in animal models (Frazier et al., 2000) and in human biopsies (Ito et al., 1998). Interference with CTGF expression may therefore be involved in the beneficial effects of statins in experimental kidney disease (Johnson et al., 1999).
We could thus show that statins, simvastatin and lovastatin, by interference with protein geranylgeranylation inhibit the induction of CTGF in renal fibroblasts. The effect was independent of the stimulus used, LPA- and TGF-β-mediated induction were likewise reduced. As CTGF is associated with the development of fibrotic lesions, these findings might imply statins as potentially beneficial in the prevention or treatment of fibrotic diseases.
Acknowledgments
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (SFB 423, B3). The expert technical assistance of Mrs M. Rehm is gratefully acknowledged.
Abbreviations
- CTGF
connective tissue growth factor
- FPP
farnesylpyrophosphate
- GGPP
geranylgeranylpyrophosphate
- HMG CoA
3-hydroxy-3-methylglutaryl-coenzyme A
- LDL
low density lipoproteins
- LPA
lysophosphatidic acid
- TGF
transforming growth factor
References
- BITZER M., STERZEL R.B., BÖTTINGER E.P. Transforming growth factor-beta in renal disease. Kidney Blood Press. Res. 1998;21:1–12. doi: 10.1159/000025837. [DOI] [PubMed] [Google Scholar]
- BORK P. The modular architecture of a new family of growth regulators related to connective tissue growth factor. FEBS Lett. 1993;327:125–130. doi: 10.1016/0014-5793(93)80155-n. [DOI] [PubMed] [Google Scholar]
- CHOMCZYNSKI P., SACCHI N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- CONTOS J.J., ISHII I., CHUN J. Lysophosphatidic acid receptors. Mol. Pharmacol. 2000;58:1188–1196. doi: 10.1124/mol.58.6.1188. [DOI] [PubMed] [Google Scholar]
- DAMMEIER J., BRAUCHLE M., FALK W., GROTENDORST G.R., WERNER S. Connective tissue growth factor: a novel regulator of mucosal repair and fibrosis in inflammatory bowel disease. Int. J. Biochem. Cell Biol. 1998;30:909–922. doi: 10.1016/s1357-2725(98)00046-6. [DOI] [PubMed] [Google Scholar]
- DESAGER J.P., HORSMANS Y. Clinical pharmacokinetics of 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors. Clin. Pharmacokinet. 1996;31:348–371. doi: 10.2165/00003088-199631050-00003. [DOI] [PubMed] [Google Scholar]
- DUNCAN M.R., FRAZIER K.S., ABRAMSON S., WILLIAMS S., KLAPPER H., HUANG X., GROTENDORST G.R. Connective tissue growth factor mediates transforming growth factor beta-induced collagen synthesis: down-regulation by cAMP. FASEB J. 1999;13:1774–1786. [PubMed] [Google Scholar]
- ESSIG M., VRTOVSNIK F., NGUYEN G., SRAER J.D., FRIEDLANDER G. Lovastatin modulates in vivo and in vitro the plasminogen activator/plasmin system of rat proximal tubular cells: role of geranylgeranylation and Rho proteins. J. Am. Soc. Nephrol. 1998;9:1377–1388. doi: 10.1681/ASN.V981377. [DOI] [PubMed] [Google Scholar]
- FRAZIER K.S., PAREDES A., DUBE P., STYER E. Connective tissue growth factor expression in the rat remnant kidney model and association with tubular epithelial cells undergoing transdifferentiation. Vet. Pathol. 2000;37:328–335. doi: 10.1354/vp.37-4-328. [DOI] [PubMed] [Google Scholar]
- FRAZIER K., WILLIAMS S., KOTHAPALLI D., KLAPPER H., GROTENDORST G.R. Stimulation of fibroblast cell growth, matrix production, and granulation tissue formation by connective tissue growth factor. J. Invest. Dermatol. 1996;107:404–411. doi: 10.1111/1523-1747.ep12363389. [DOI] [PubMed] [Google Scholar]
- GNAD R., AKTORIES K., KAINA B., FRITZ G. Inhibition of protein isoprenylation impairs rho-regulated early cellular response to genotoxic stress. Mol. Pharmacol. 2000;58:1389–1397. doi: 10.1124/mol.58.6.1389. [DOI] [PubMed] [Google Scholar]
- GOETZL E.J., AN S. Diversity of cellular receptors and functions for the lysophospholipid growth factors lysophosphatidic acid and sphingosine 1-phosphate. FASEB J. 1998;12:1589–1598. [PubMed] [Google Scholar]
- GOPPELT-STRUEBE M., FICKEL S., REISER C.O. The platelet-derived-growth-factor receptor, not the epidermal-growth-factor receptor, is used by lysophosphatidic acid to activate p42/44 mitogen-activated protein kinase and to induce prostaglandin G/H synthase-2 in mesangial cells. Biochem. J. 2000;345:217–224. [PMC free article] [PubMed] [Google Scholar]
- GROTENDORST G.R. Connective tissue growth factor: a mediator of TGF-beta action on fibroblasts. Cytokine. Growth Factor. Rev. 1997;8:171–179. doi: 10.1016/s1359-6101(97)00010-5. [DOI] [PubMed] [Google Scholar]
- GROTENDORST G.R., OKOCHI H., HAYASHI N. A novel transforming growth factor beta response element controls the expression of the connective tissue growth factor gene. Cell Growth Differ. 1996;7:469–480. [PubMed] [Google Scholar]
- GUPTA S., CLARKSON M.R., DUGGAN J., BRADY H.R. Connective tissue growth factor: potential role in glomerulosclerosis and tubulointerstitial fibrosis. Kidney Int. 2000;58:1389–1399. doi: 10.1046/j.1523-1755.2000.00301.x. [DOI] [PubMed] [Google Scholar]
- HAHN A., HEUSINGER-RIBEIRO J., LANZ T., ZENKEL S., GOPPELT-STRUEBE M. Induction of connective tissue growth factor by activation of heptahelical receptors. Modulation by rho proteins and the actin cytoskeleton. J. Biol. Chem. 2000;275:37429–37435. doi: 10.1074/jbc.M000976200. [DOI] [PubMed] [Google Scholar]
- HEUSINGER-RIBEIRO J., EBERLEIN M., ABDEL WAHAB N., GOPPELT-STRUEBE M.Lysophosphatidic acid-induced expression of connective tissue growth factor in human renal fibroblasts: Regulatory role of RhoA and cAMP J. Am. Soc. Nephrol. 2001. in press [DOI] [PubMed]
- IGARASHI A., NASHIRO K., KIKUCHI K., SATO S., IHN H., FUJIMOTO M., GROTENDORST G.R., TAKEHARA K. Connective tissue growth factor gene expression in tissue sections from localized scleroderma, keloid, and other fibrotic skin disorders. J. Invest. Dermatol. 1996;106:729–733. doi: 10.1111/1523-1747.ep12345771. [DOI] [PubMed] [Google Scholar]
- ILLINGWORTH D.R., ERKELENS D.W., KELLER U., THOMPSON G.R., TIKKANEN M.J. Defined daily doses in relation to hypolipidaemic efficacy of lovastatin, pravastatin, and simvastatin. The Lancet. 1994;343:1554–1555. doi: 10.1016/s0140-6736(94)92945-9. [DOI] [PubMed] [Google Scholar]
- ITO Y., ATEN J., BENDE R.J., OEMAR B.S., RABELINK T.J., WEENING J.J., GOLDSCHMEDING R. Expression of connective tissue growth factor in human renal fibrosis. Kidney Int. 1998;53:853–861. doi: 10.1111/j.1523-1755.1998.00820.x. [DOI] [PubMed] [Google Scholar]
- JAKOBISIAK M., BRUNO S., SKIERSKI J.S., DARZYNKIEWICZ Z. Cell cycle-specific effects of lovastatin. Proc. Natl. Acad. Sci. U.S.A. 1991;88:3628–3632. doi: 10.1073/pnas.88.9.3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JOHNSON D.W., SAUNDERS H.J., FIELD M.J., POLLOCK C.A. In vitro effects of simvastatin on tubulointerstitial cells in a human model of cyclosporin nephrotoxicity. Am. J. Physiol. 1999;276:F467–F475. doi: 10.1152/ajprenal.1999.276.3.F467. [DOI] [PubMed] [Google Scholar]
- KEANE W.F. The role of lipids in renal disease: Future challenges. Kidney Int. 2000;57:27–31. [PubMed] [Google Scholar]
- KIM S.I., HAN D.C., LEE H.B. Lovastatin inhibits transforming growth factor-beta 1 expression in diabetic rat glomeruli and cultured rat mesangial cells. J. Am. Soc. Nephrol. 2000;11:80–87. doi: 10.1681/ASN.V11180. [DOI] [PubMed] [Google Scholar]
- KIM S.-Y., GUIJARRO C., O'DONNELL M.P., KASISKE B.L., KIM Y., KEANE W.F. Human mesangial cell production of monocyte chemoattractant protein-1: Modulation by lovastatin. Kidney Int. 1995;48:363–371. doi: 10.1038/ki.1995.304. [DOI] [PubMed] [Google Scholar]
- KOCH G., BENZ C., SCHMIDT G., OLENIK C., AKTORIES K. Role of Rho proteins in lovastatin-induced breakdown of actin cytoskeleton. J. Pharmacol. Exp. Ther. 1997;283:901–909. [PubMed] [Google Scholar]
- KOTHAPALLI D., FRAZIER K.S., WELPLY A., SEGARINI P.R., GROTENDORST G.R. Transforming growth factor beta induces anchorage-independent growth of NRK fibroblasts via a connective tissue growth factor- dependent signaling pathway. Cell Growth Differ. 1997;8:61–68. [PubMed] [Google Scholar]
- KUCICH U., ROSENBLOOM J.C., SHEN G., ABRAMS W.R., HAMILTON A.D., SEBTI S.M., ROSENBLOOM J. TGF-beta1 stimulation of fibronectin transcription in cultured human lung fibroblasts requires active geranylgeranyl transferase I, phosphatidylcholine-specific phospholipase C, protein kinase C-delta, and p38, but not erk1/erk2. Arch. Biochem. Biophys. 2000;374:313–324. doi: 10.1006/abbi.1999.1625. [DOI] [PubMed] [Google Scholar]
- LAU L.F., LAM S.C. The CCN family of angiogenic regulators: the integrin connection. Exp. Cell Res. 1999;248:44–57. doi: 10.1006/excr.1999.4456. [DOI] [PubMed] [Google Scholar]
- LAUFS U., ENDRES M., CUSTODIS F., GERTZ K., NICKENIG G., LIAO J.K., BOHM M. Suppression of endothelial nitric oxide production after withdrawal of statin treatment is mediated by negative feedback regulation of rho GTPase gene transcription. Circulation. 2000;102:3104–3110. doi: 10.1161/01.cir.102.25.3104. [DOI] [PubMed] [Google Scholar]
- LERNER E.C., HAMILTON A.D., SEBTI S.M. Inhibition of Ras prenylation: a signaling target for novel anti-cancer drug design. Anticancer Drug Des. 1997;12:229–238. [PubMed] [Google Scholar]
- MARON D.J., FAZIO S., LINTON M.F. Current perspectives on statins. Circulation. 2000;101:207–213. doi: 10.1161/01.cir.101.2.207. [DOI] [PubMed] [Google Scholar]
- MASON R., LI X.J., WAHAB N.A. High glucose induces the expression of connective tissue growth factor in human mesangial cells. J. Am. Soc. Nephrol. 1997;8:642A. [Google Scholar]
- MASSY Z.A., KIM Y., GUIJARRO C., KASISKE B.L., KEANE W.F., O'DONNELL M.P. Low-density lipoprotein-induced expression of interleukin-6, a marker of human mesangial cell inflammation: effects of oxidation and modulation by lovastatin. Biochem. Biophys. Res. Commun. 2000;267:536–540. doi: 10.1006/bbrc.1999.1992. [DOI] [PubMed] [Google Scholar]
- MULLER G.A., SCHETTLER V., MULLER C.A., STRUTZ F. Prevention of progression of renal fibrosis: how far are we. Kidney Int. Suppl. 1996;54:S75–S82. [PubMed] [Google Scholar]
- MURPHY M., GODSON C., CANNON S., KATO S., MACKENZIE H.S., MARTIN F., BRADY H.R. Suppression subtractive hybridization identifies high glucose levels as a stimulus for expression of connective tissue growth factor and other genes in human mesangial cells. J. Biol. Chem. 1999;274:5830–5834. doi: 10.1074/jbc.274.9.5830. [DOI] [PubMed] [Google Scholar]
- MÜLLER G.A., FRANK J., RODEMANN H.P., ENGLER-BLUM G. Human renal fibroblast cell lines (tFKIF and tNKF) are new tools to investigate pathophysiologic mechanisms of renal interstitial fibrosis. Exp. Nephrol. 1995;3:127–133. [PubMed] [Google Scholar]
- NEGRE-AMINOU P., VAN VLIET A.K., VAN ERCK M., VAN THIEL G.C., VAN LEEUWEN R.E., COHEN L.H. Inhibition of proliferation of human smooth muscle cells by various HMG-CoA reductase inhibitors; comparison with other human cell types. Biochim. Biophys. Acta. 1997;1345:259–268. doi: 10.1016/s0005-2760(96)00184-1. [DOI] [PubMed] [Google Scholar]
- OEMAR B.S., WERNER A., GARNIER J.M., DO D.D., GODOY N., NAUCK M., MARZ W., RUPP J., PECH M., LUSCHER T.F. Human connective tissue growth factor is expressed in advanced atherosclerotic lesions. Circulation. 1997;95:831–839. doi: 10.1161/01.cir.95.4.831. [DOI] [PubMed] [Google Scholar]
- PARK Y.S., GUIJARRO C., KIM Y., MASSY Z.A., KASISKE B.L., KEANE W.F., O'DONNELL M.P. Lovastatin reduces glomerular macrophage influx and expression of monocyte chemoattractant protein-1 mRNA in nephrotic rats. Am. J. Kidney Dis. 1998;31:190–194. doi: 10.1053/ajkd.1998.v31.pm9428473. [DOI] [PubMed] [Google Scholar]
- PEREIRA R.C., DURANT D., CANALIS E. Transcriptional regulation of connective tissue growth factor by cortisol in osteoblasts. Am. J. Physiol Endocrinol. Metab. 2000;279:E570–E576. doi: 10.1152/ajpendo.2000.279.3.E570. [DOI] [PubMed] [Google Scholar]
- PIEK E., HELDIN C.-H., TEN DIJKE P. Specificity, diversity, and regulation in TGF-beta superfamily signaling. FASEB J. 1999;13:2105–2124. [PubMed] [Google Scholar]
- POMERANTZ K.B., LANDER H.M., SUMMERS B., ROBISHAW J.D., BALCUEVA E., HAJJAR D.P. Gprotein-mediated signaling in cholesterol-enriched arterial smooth muscle cells. 1. Reduced membrane-associated G-protein content due to diminished isoprenylation of G-gamma subunits and p21ras. Biochemistry. 1997;36:9253–9531. doi: 10.1021/bi963069l. [DOI] [PubMed] [Google Scholar]
- RIESSEN R., AXEL D.I., FENCHEL M., HERZOG U.U., ROSSMANN H., KARSCH K.R. Effect of HMG-CoA reductase inhibitors on extracellular matrix expression in human vascular smooth muscle cells. Basic. Res. Cardiol. 1999;94:322–332. doi: 10.1007/s003950050158. [DOI] [PubMed] [Google Scholar]
- ROSENBLOOM J., SAITTA B., GAIDAROVA S., SANDORFI N., ROSENBLOOM J.C., ABRAMS W.R., HAMILTON A.D., SEBTI S.M., KUCICH U., JIMENEZ S.A. Inhibition of type I collagen gene expression in normal and systemic sclerosis fibroblasts by a specific inhibitor of geranylgeranyl transferase I. Arthritis Rheum. 2000;43:1624–1632. doi: 10.1002/1529-0131(200007)43:7<1624::AID-ANR28>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- STROEBEL M., GOPPELT-STRUEBE M. Signal transduction pathways responsible for serotonin-mediated prostaglandin G/H synthase expression in rat mesangial cells. J. Biol. Chem. 1994;269:22952–22957. [PubMed] [Google Scholar]
- VAN AELST L., D'SOUZA-SCHOREY C. Rho GTPases and signaling networks. Genes Dev. 1997;11:2295–2322. doi: 10.1101/gad.11.18.2295. [DOI] [PubMed] [Google Scholar]
- VRTOVSNIK F., COUETTE S., PRIE D., LALLEMAND D., FRIEDLANDER G. Lovastatin-induced inhibition of renal epithelial tubular cell proliferation involves a p21ras activated, AP-1-dependent pathway. Kidney Int. 1997;52:1016–1027. doi: 10.1038/ki.1997.423. [DOI] [PubMed] [Google Scholar]
- VRTOVSNIK F., ESSIG M., IIMURA O., FRIEDLANDER G. Effect of lipid-lowering strategies on tubular cell biology. Kidney Int. Suppl. 1999;71:S92–S96. doi: 10.1046/j.1523-1755.1999.07123.x. [DOI] [PubMed] [Google Scholar]
- WASSMANN S., LAUFS U., BAUMER A.T., MULLER K., KONKOL C., SAUER H., BOHM M., NICKENIG G. Inhibition of Geranylgeranylation Reduces Angiotensin II-Mediated Free Radical Production in Vascular Smooth Muscle Cells: Involvement of Angiotensin AT1 Receptor Expression and Rac1 GTPase. Mol. Pharmacol. 2001;59:646–654. doi: 10.1124/mol.59.3.646. [DOI] [PubMed] [Google Scholar]
- ZANG F.L., CASEY P.J. Protein prenylation: Molecular mechanisms and functional consequences. Annu. Rev. Biochem. 1996;65:241–269. doi: 10.1146/annurev.bi.65.070196.001325. [DOI] [PubMed] [Google Scholar]