

Abstract
Beta amyloid (Aβ) is implicated in Alzheimer's disease (AD). Aβ1–42 (5, 10, or 20 μM) was able to increase NO release and decrease cellular viability in primary rat cortical mixed cultures.
L-NOARG and SMTC (both at 10 or 100 μM) – type I NOS inhibitors – reduced cellular NO release in the absence of Aβ1–42. At 100 μM, both drugs decreased cell viability.
L-NIL (10 or 100 μM), and 1400W (1 or 5 μM) – type II NOS inhibitors – reduced NO release and improved viability when either drug was administered up to 4 h post Aβ1–42 (10 μM) treatment. L-NOARG and SMTC (both at 10 or 100 μM) were only able to decrease NO release. Carboxy-PTIO or Trolox (both at 10 or 100 μM) – a NO scavenger and an antioxidant, respectively–increased viability when administered up to 1 h post Aβ1–42 treatment. Either L-NIL (50 μM) or 1400W (3 μM) and Trolox (50 μM) showed synergistic actions.
Peroxynitrite (100 or 200 μM) reduced cell viability. Viabilities were improved by L-NIL (100 μM), 1400W (5 μM), carboxy-PTIO (10 or 100 μM), and Trolox (10 or 100 μM).
Hence, the data show that Aβ1–42 induced NO release in neurons and glial cells, and that Aβ neurotoxicity is, at least in part, mediated by NO. NO concentration modulating compounds and antioxidant may have therapeutic importance in neurological disorders where oxidative stress is likely involved such as in AD.
Keywords: 1400W, Alzheimer's disease, N-iminoethyl-L-lysine, NG-nitro-L-arginine, oxidative stress, S-methyl-L-thiocitulline, Trolox, carboxy-PTIO
Introduction
The immunologic and vascular properties of nitric oxide (NO) has long been recognized (Ignarro et al., 1987; Stuehr & Marletta, 1985). The roles of NO in neurobiology has been a research focus since the discoveries of the inhibitory effects of NO in the peripheral nervous system (Ceccatelli et al., 1994; de man et al., 1991) and NO production in the brain (Bredt et al., 1990; Schmidt et al., 1989). NO is an enzymatic product of nitric oxide synthase (NOS), which exists in three isoforms. In the brain, all three isoforms are present. Type I NOS (neuronal NOS, nNOS) is expressed in neurons in diverse locations with different densities, including the neocortex (Bredt et al., 1990). Type II NOS (inducible NOS, iNOS) is found in astrocytes and microglia (Kröncke et al., 1995). Type III NOS (endothelial NOS, eNOS) has been identified most abundantly in endothelial cells but also in some neurons (Dinerman et al., 1994). The extensive distribution of these NOS in the brain suggests that NO is likely to have important neurophysiological functions. Research thus far has shown that in addition to the regulation of cerebral vasoactivity, NO is likely to participate in learning and memory by acting as the retrograde messenger in long-term potentiation and depression (Haley et al., 1992; Linden & Connor, 1993), as well as participating in axonal remodelling during development (Hess et al., 1993). NO can also be neurotoxic primarily due to its oxidative properties, which includes its ability to induce the production of peroxynitrite, which is a highly destructive reactive oxygen species (ROS) (Beckman et al., 1994a). NO has been implicated in cerebral ischaemia (Dawson, 1994; Eliasson et al., 1999) and in a number of neurodegenerative disorders, including Huntington's disease (Meldrum & Garthwaite, 1990), AIDS-associated dementia (Adamson et al., 1996), multiple sclerosis (Giovannoni et al., 1998), amyotrophic lateral sclerosis (Almer et al., 1999), as well as in Alzheimer's disease (AD) (Smith et al., 1997). These apparent paradoxical effects of NO may be dependent on the NOS isoform involved (Hara et al., 1996; Huang et al., 1996). Despite the possible importance of NO in the aetiology of various neurodegenerative diseases, existing data on the effects of isoform-specific NOS inhibitors on neurological functioning are limited.
Beta-Amyloid (Aβ) is a 40 or 42-amino acids peptide fragment derived from the much larger amyloid precursor protein (APP) secondary to β- and γ-secretase cleavage (Haass & Selkoe, 1993). In the AD brain, significant extraneuronal Aβ deposits have been noted (Selkoe, 1994), and Aβ has been implicated in AD pathogenesis via a number of distinct but intertwined mechanisms, including excitotoxicity, Ca2+ homeostatic disruption, free radical production, neuro-inflammation, and apoptosis (Cotman & Anderson, 1995; Gahtan & Overmier, 1999; Good et al., 1996; Mattson et al., 1992). Moreover, the 42-amino acid peptide fragment (Aβ1–42) appears to be more critically involved in the aetiology of AD (El Agnaf et al., 2000). Previous studies have shown that Aβ stimulated NO production in the brain. However, the data are inconsistent. Several studies demonstrated synergy between Aβ and cytokines to induce microglial NO release (Bonaiuto et al., 1997; Goodwin et al., 1997; Ii et al., 1996; Meda et al., 1995), whereas others argued that the increased NO release was the result of astrocytic activation (Akama et al., 1998; Forloni et al., 1997; Hu et al., 1998; Wallace et al., 1997). Moreover, whether Aβ can induce neuronal NO production (Yang et al., 1998) remains controversial.
Since NO and Aβ have been implicated in neurotoxicity and putative links between these two potentially neurotoxic substances appear to exist, the aim of this study was to examine the effects of Aβ on NO release and the potential protective and rescuing ability of various NOS inhibitors in primary rat cortical neuronal and glial cultures. Our data suggest that Aβ-induced neurotoxicity is NO-mediated, and that isoform-specific NOS inhibition provides beneficial effects.
Methods
Primary cortical mixed cell cultures
Solutions used for primary neuronal and glial cell cultures were obtained from Gibco BRL (Burlington, Ontario, Canada) unless otherwise stated. Culture medium was prepared by adding 10% (v v−1) foetal bovine serum (FBS) and 1% (v v−1) PSN to high glucose, phenol red-free Dulbecco's modified Eagle's medium (D-MEM).
Mixed cortical glial and neuronal cultured cells were prepared from E15 to E18 embryos obtained from Spargue-Dawley rats (Charles River Canada, St-Constant, Québec, Canada). Animal care was according to protocols and guidelines of McGill University Animal Care Committee and the Canadian Council for Animal Care. The embryo brains were dissected, cerebral cortices were separated, and the meninges were removed in Ca2+ and Mg2+-free Hank's balanced salt solution (HBSS). Cortical tissues were collected and washed three times using HBSS; following by digestion in 0.25% trypsin (Sigma, St. Louis, MO, U.S.A.) for 10 min at 37°C. The cortical tissues were rinsed once using HBSS and FBS was added to stop the reaction. The cortical tissue suspension was then triturated 15 times using a Pasteur pippette, followed by 10 min centrifugation at 1000×g. The supernatant was removed and resuspended by adding the prepared culture medium. The suspension was triturated twice. The cortical cells were then plated at day 0 at a density of approximately 15×104 viable cells per well in 24-well plates coated with poly-D-lysine (Sigma). Cells were grown and maintained in the culture medium described above at 37°C and 5% CO2 humidified environment. The culture medium was changed on days 1 and 4.
Experimental treatments
All experimental drugs were purchased from Calbiochem (San Diego, CA, U.S.A.) unless otherwise stated. All drugs were freshly prepared on the day of the experiment. Each experimental condition was tested in triplicated wells, and at least three independent experiments were carried out for each condition.
On day 7 after plating, the culture medium was removed and replaced with freshly prepared culture medium in the presence of either Aβ1–42 (1, 5, 10, or 20 μM; U.S. Peptide, Fullerton, CA, U.S.A.), Aβ42–1 (Bachem, Torrance, CA, U.S.A.), or peroxynitrite (100 or 200 μM) with or without either NG-nitro-L-arginine (L-NOARG, a type I (and III) NOS inhibitor (Furfine et al., 1993); 10 or 100 μM), S-methyl-L-thiocitrulline (SMTC; a type I NOS inhibitor (Furfine et al., 1994); 10 or 100 μM), N-iminoethyl-L-lysine (L-NIL, a type II NOS inhibitor (Moore et al., 1994); 10 or 100 μM), N-(3-(aminomethyl)benzyl)acetamidine (1400W, a type II NOS inhibitor (Garvey et al., 1997); 1 or 5 μM), 2-(4-carboxyphenyl)-4, 4, 5, 5-tetramethylimidazoline-1-oxyl-3-oxide (carboxy-PTIO, a NO scavenger (Hogg et al., 1995); 10 or 100 μM), or 6-hydorxy-2,5,7,8-tetramethylchroman-2-carboxylic acid (Trolox, a vitamin E analogue with antioxidative properties against peroxynitrite-mediated oxidative stress (Salgo & Pryor, 1996); 10 or 100 μM) alone or in combination. The cultured cells were then incubated for 20 h at the conditions described above. For the time-course studies, the cultured cells were pre-treated with the described culture medium containing Aβ1–42 (10 μM). Either L-NIL (100 μM), L-NOARG (100 μM), 1400W (5 μM), SMTC (100 μM), carboxy-PTIO (100 μM) or Trolox (100 μM) were administered at 1, 4, and 8 h later. Assessments were carried out 20 h after Aβ1–42 administration. To examine the combining effects of these drugs, they were paired with each other at half of the maximum concentrations used, except for 1400W, where 3 μM was used.
Assessment of NO release
NO is rapidly converted to nitrate and nitrite in aqueous solutions. NO released by cultured cells after Aβ1–42 and specific NOS inhibitor treatments were inferred by converting the nitrate produced into nitrite by nitrate reductase, followed by the addition of the Griess reagent (NO colorimetric assay kit; Calbiochem), which measured total nitrite production (Nims et al., 1996).
Assessment of cellular viability
The viability of cultured cells was evaluated by using 3-4(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrzolium bromide (MTT) and neutral red (3-amino-7-dimrthyl-amino-2-methylphenazine, NR) colorimetric assays. MTT is an indicator of mitochondrial respiration, which has commonly been used to infer cellular viability (Bastianetto et al., 1999). Since free radicals have been shown to alter the activities of mitochondrial enzymes (Sims, 1996), NR, a dye which is taken up by lysosomes of living cells (Bastianetto et al., 1999), was also used as an additional cellular integrity marker in some experiments to confirm the results obtained with the MTT assay. MTT reduction and NR uptake were quantified at 570 and 540 nm, respectively, by using a micro-plate reader (Bio-Tek Instruments Inc., Ville St-Laurent, Québec, Canada).
Statistical analysis
All data are represented as means±s.e.mean. One-way analysis of variance (ANOVA) with Bonferroni's multiple comparison tests and unpaired Student t-tests were used in all studies to compare controls and treatment groups, with P<0.05 being considered as significant. For cellular viability studies, the viability measures of vehicle-treated control groups were established as 100%.
Results
Effects of Aβ1–42 on NO release
Various concentrations of Aβ1–42 were added to cortical culture cells, and the resultant nitrite concentrations were measured (Figure 1). At 1 μM, Aβ1–42 did not increase nitrite concentration (23.8±1.6 μM). At 5, 10, and 20 μM, Aβ1–42 caused significantly higher nitrite productions (31.9±1.9, 44.3±2.7, and 46.7±3.1 μM, respectively) than control (no Aβ exposure; 21.6±2.2 μM). No significant difference was observed between the 10 and 20 μM concentrations. The reverse peptide sequence Aβ42–1 (10 and 20 μM) failed to induce nitrite production.
Figure 1.
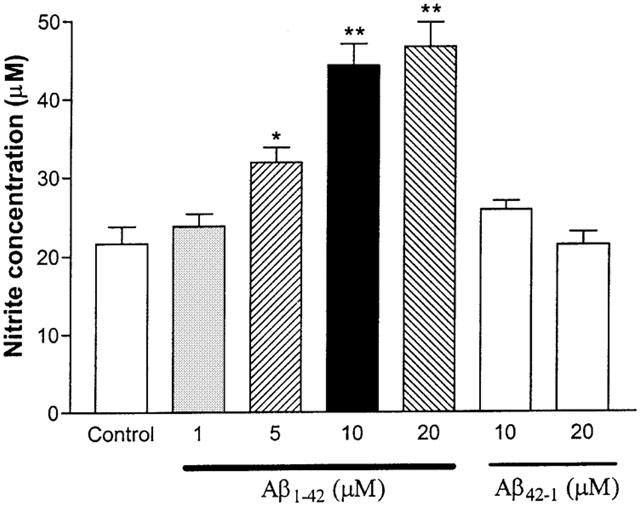
Effects of Aβ1–42 on nitrite production in primary cortical mixed cell cultures. At 5, 10, and 20 μM, Aβ1–42 significantly increased nitrite production as compared to control (no Aβ peptide exposure). The reverse peptide Aβ42–1 (10 and 20 μM) did not increase nitrite production significantly in the cultured cells. Data represent mean±s.e.mean of 6–9 wells from seven independent experiments. One-way ANOVA with Bonferroni's multiple comparison test and Student t-test were used to analyse statistical significance. *P<001; **P<0.001.
Effects of NOS inhibitors on NO release
The effects of two specific type I NOS inhibitors (L-NOARG and SMTC) and two type II NOS inhibitors (L-NIL and 1400W) on nitrite production by the cultured cells were assessed next (Figure 2). L-NOARG and SMTC exposures at 100 μM significantly lowered nitrite production (9.5±1.2 and 11.2±1.1 μM, respectively) when compared to control (no NOS inhibitor exposure; 19.6±1.2 μM). In contrast, both L-NIL (10 and 100 μM) and 1400W (1 and 5 μM) did not show significant effects on nitrite production.
Figure 2.
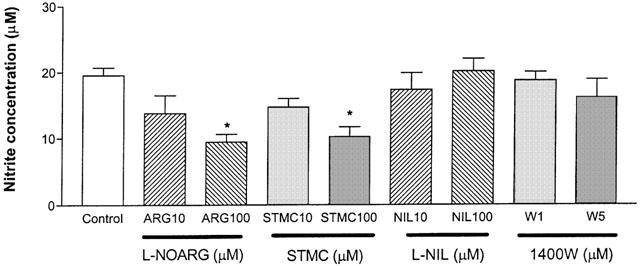
Effects of specific NOS inhibitors on nitrite production in primary cortical mixed cell cultures. L-NOARG and STMC caused significantly lower nitrite release than control (no NOS inhibitor exposure) at 100 μM. L-NIL and 1400W did not show significant effect on nitrite production at their respective concentrations. Data represent mean±s.e.mean of 4–6 wells from five independent experiments. One-way ANOVA with Bonferroni's multiple comparison test and Student t-test were used to analyse statistical significance. *P<0.05.
Effects of NOS inhibitors on NO release in the presence of Aβ1–42
Nitrite productions by the cultured cells after co-administration of Aβ1–42 (10 μM) and various specific NOS inhibitors at different concentrations were measured next (Figure 3). Aβ1–42 by itself induced a significant elevation in nitrite production (46.3±2.7 μM) when compared to the control (without Aβ or NOS inhibitor exposure; 18.6±2.2 μM). Nitrite productions after Aβ1–42 and L-NOARG (100 μM) or Aβ1–42 and SMTC (100 μM) treatments were significantly lower than Aβ1–42 alone (33.5±2.0 and 34.5±1.6 μM, respectively). The co-administration of Aβ1–42 and L-NIL (100 μM) or 1400W (5 μM) induced similar but more significant effects in reducing nitrite production (23.9±1.9 and 27.8±1.0 μM, respectively).
Figure 3.
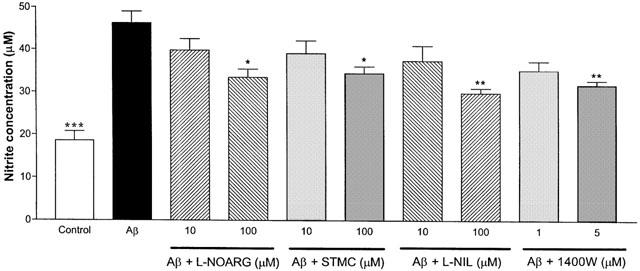
Effects of specific NOS inhibitors on nitrite production in primary cortical mixed cell cultures in the presence of Aβ1–42 (10 μM). Aβ1–42 caused significant increase in nitrite production when compared to control (no Aβ or NOS inhibitor exposure). When L-NOARG (100 μM), STMC (100 μM), L-NIL (100 μM), or 1400W (5 μM), were co-administered with Aβ1–42, nitrite productions were significantly lower than Aβ1–42 alone. Data represent mean±s.e.mean of 8–9 wells from seven independent experiments. One-way ANOVA with Bonferroni's multiple comparison test and Student t-test were used to analyse statistical significance. *P<0.05, **P<0.01; ***P<0.001 compared to Aβ1–42.
Effects of NOS inhibitors on NO release after Aβ1–42 pre-treatment
Various specific NOS inhibitors were administered at 1, 4 and 8 h after Aβ1–42 treatment, and the resultant nitrite concentrations were assessed (Figure 4a–d). L-NOARG at 100 μM (Figure 4a) and SMTC at 100 μM (Figure 4b) were able to significantly reduce nitrite production (26.5±2.0 and 25.2±1.1 μM, respectively) as compared to Aβ1–42 treatment alone (38.3±2.7 μM), when administered after Aβ1–42 at the 1 h time point. When L-NIL at 100 μM (Figure 4c) was administered 1 or 4 h after Aβ1–42 pre-treatment, the resultant nitrite levels (26.5±2.1 and 28.0±1.9 μM, respectively) were significantly lower than Aβ1–42 treatment alone. No significant difference was observed between Aβ1–42 and Aβ1–42 with L-NIL if added at 8 h after Aβ1–42 treatment. Similar results were obtained with 1400W (5 μM; 27.4±2.5 μM at 1 h and 28.7±1.3 μM at 4 h; Figure 4d). All conditions that contained either of the type I NOS inhibitors showed higher nitrate release than those with type II NOS inhibitors.
Figure 4.
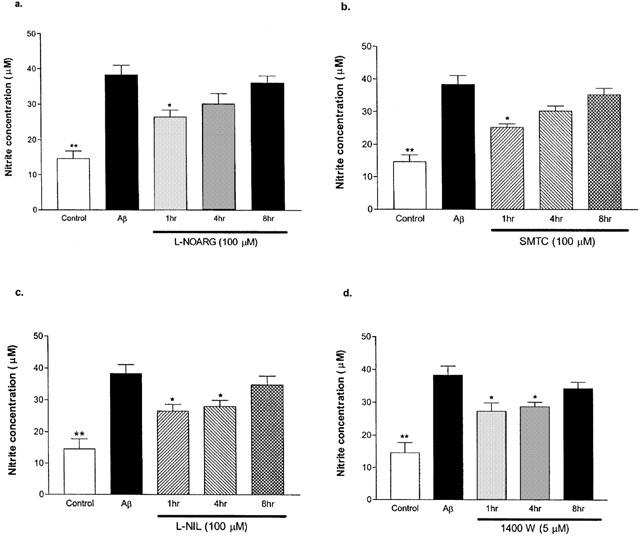
Effects of specific NOS inhibitors on nitrite production in primary cortical mixed cell cultures at 1, 4, and 8 h post Aβ1–42 (10 μM) treatment. Aβ1–42 caused significantly higher nitrite production than control (no Aβ or NOS inhibitor). L-NOARG (a) and SMTC (b) significantly decreased nitrite release when administered at 1 h post Aβ1–42 treatment. L-NIL (c) and 1400W (d) caused significant reductions in nitrite release when administered up to 4 h after Aβ1–42 treatment. Data represent mean±s.e.mean of 4–6 wells from four independent experiments. One-way ANOVA with Bonferroni's multiple comparison test and Student t-test were used to analyse statistical significance. *P<001;**P<0.001 compared to Aβ1–42.
Effects of Aβ1–42 on cellular viability
Various concentrations of Aβ1–42 were administered to the cultured cells, and viabilities were measured by assessing MTT reductions (Figure 5a) and NR uptakes (Figure 5b). At 5, 10 and 20 μM, Aβ1–42 significantly lowered MTT levels (78±2, 62±3 and 58±4% of control, respectively). No significant difference on MTT reductions were observed between Aβ1–42 at 10 and 20 μM. Moreover, all concentrations tested here significantly reduced NR uptakes when compared to control (82±2, 74±3, 62±3 and 55±4% of control for 1, 5, 10 and 20 μM, respectively). The reverse control peptide Aβ42–1 (10 and 20 μM) had no effect on MTT or NR activities.
Figure 5.
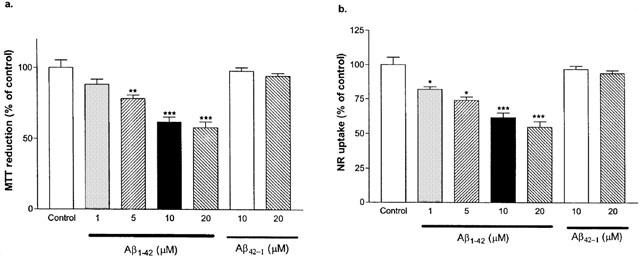
Effects of Aβ1–42 and Aβ42–1 on MTT reduction and neutral red (NR) uptake by primary cortical mixed cell cultures. Aβ1–42 (5, 10 and 20 μM) caused significant decreases in MTT reductions (a) as compared to the control (no Aβ peptide exposure). Aβ42–1, at 10 or 20 μM, showed no significant effect on MTT reduction. Aβ1–42, (1, 5, 10 and 20 μM) caused significant decreases in NR uptakes (b) as compared to the control. Aβ42–1, at 10 or 20 μM, had no significant effect on NR uptake by the cultured cells. Data represent mean±s.e.mean of 6–9 wells from six independent experiments. One-way ANOVA with Bonferroni's multiple comparison test and Student t-test were used to analyse statistical significance. *P<0.05; **P<0.01; ***P<0.001.
Effects of NOS inhibitors on cellular viability
MTT reductions (Figure 6a) and NR uptakes (Figure 6b) by cultured cells after exposures to various specific NOS inhibitors were measured next. L-NOARG and SMTC at 100 μM concentration decreased both MTT (84±2 and 87±1% of control, respectively) and NR (81±2 and 80±1% of control, respectively) levels. L-NIL and 1400W had no significant effect on either viability markers with the concentration tested.
Figure 6.
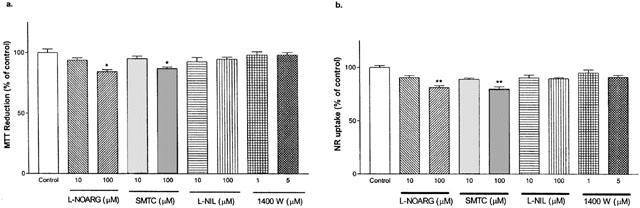
Effect of specific NOS inhibitors on MTT reduction and neutral red (NR) uptake by primary cortical mixed cell cultures. L-NOARG and SMTC caused significant decreases in MTT reduction (a) and NR uptake (b) at 100 μM concentration. L-NIL and 1400W showed no effects on MTT reduction or NR uptake. Data represent mean±s.e.mean of 6–9 well from four independent experiments. One-way ANOVA with Bonferroni's multiple comparison test and Student t-test were used to analyse statistical significance. *P<0.05;** P<0.01.
Effects of NOS inhibitors, carboxy-PTIO, and Trolox on cellular viability in the presence of Aβ1–42
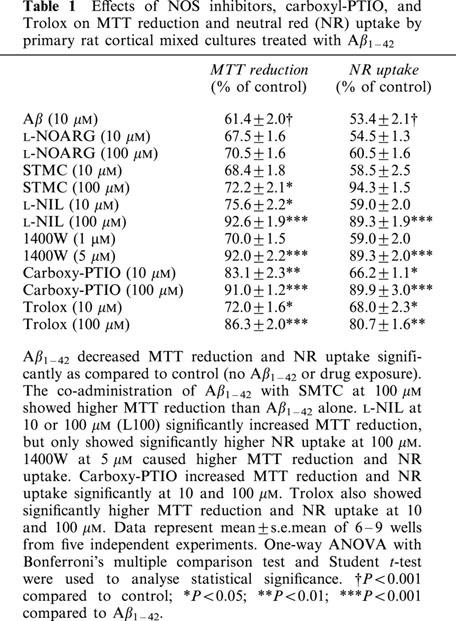
Carboxy-PTIO and Trolox possess antioxidative effects against NO-mediated oxidative stress. Their potential effects against Aβ toxicity were investigated here along with NOS inhibitors (Table 1). The co-administration of SMTC (100 μM) and Aβ1–42 significantly reversed the effects of Aβ1–42 alone (72±2% vs 61±2% of control). Similar results were obtained with a co-administration of L-NIL at 10 or 100 μM (76±2 and 93±2% of control, respectively), 1400W at 5 μM (92±2% of control), carboxy-PTIO at 10 and 100 μM (83±2 and 91±1% of control, respectively) or Trolox at 10 or 100 μM (72±2 and 86±2% of control, respectively) and Aβ1–42. Similar results were obtained using NR uptake as viability marker.
Table 1.
Effects of NOS inhibitors, carboxyl-PTIO, and Trolox on MTT reduction and neutral red (NR) uptake by primary rat cortical mixed cultures treated with Aβ1–42
Effects of NOS inhibitors, carboxy-PTIO, and Trolox on cellular viability after Aβ1–42 pre-treatment
Various specific NOS inhibitors, carboxy-PTIO, or Trolox were administered at 1, 4, and 8 h post Aβ1–42 treatment, and MTT reductions (Figure 7a–f) were measured. Both L-NOARG at 100 μM (Figure 7a) and SMTC at 100 μM (Figure 7b) had no effects on MTT levels when compared to Aβ1–42 alone (56±5%). When L-NIL (100 μM; Figure 7c) or 1400W (5 μM; Figure 7d) was administered 1 or 4 h after Aβ1–42 treatment, the resultant MTT reduction values (93±2 and 89±4% of control, 94±4 and 90±2% of control, respectively) were significantly higher than Aβ1–42 treatment alone. No significant difference in MTT reduction was observed when L-NIL or 1400W was added at 8 h later. Carboxy-PTIO (Figure 7e) and Trolox (Figure 7f) at 100 μM reversed the effects of Aβ1–42 when added 1 h post-Aβ1–42 treatment (91±5 and 74±5% of control, respectively).
Figure 7.
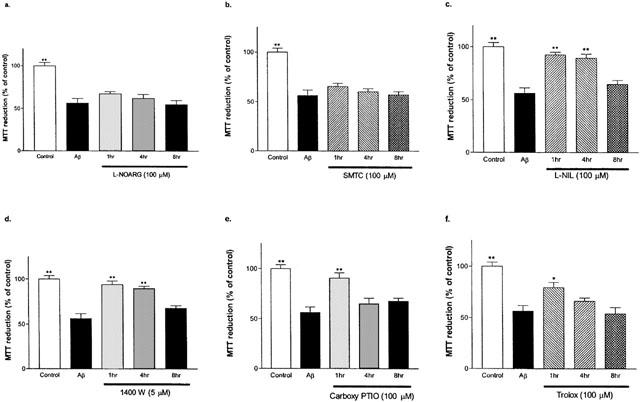
Effects of specific NOS inhibitors, carboxy-PTIO, and Trolox on MTT reduction by primary cortical mixed cell cultures at 1, 4 and 8 h post Aβ1–42 (10 μM) treatment. Aβ1–42 caused significant MTT reduction decrease when compared to the control (no Aβ or drug exposure). L-NOARG (a) or SMTC (b) treatments did not show significant increase in MTT reduction compared Aβ1–42 alone. L-NIL (c) or 1400W (d) at 1 or 4 h, but not at 8 h, caused significant increases in MTT reductions. Carboxyl-PTIO (e) or Trolox (f) treatment showed significantly higher MTT reduction only at 1 h after Aβ1–42 treatment. MTT levels of L-NIL and 1400W administered at 1 and 4 h after Aβ1–42 treatment, as well as carboxy-PTIO at 1 h post Aβ1–42 treatment were similar to control. Data represent mean±s.e.mean of 4–6 wells from four independent experiments. One-way ANOVA with Bonferroni's multiple comparison test and Student t-test were used to analyse statistical significance. *P<0.01; **P<0.001 compared to Aβ1–42.
Effects of NOS inhibitors, carboxy-PTIO, and Trolox on cellular viability in the presence of peroxynitrite
Since NO and peroxynitrite are closely related ROS, the toxic effect of peroxynitrite was also examined. Peroxynitrite was administered alone or in combination with various specific NOS inhibitors or antioxidants, and the resultant MTT reductions were measured (Figure 8). Administration of peroxynitrite (100 or 200 μM) significantly decreased MTT values in the cultured cells (31±2 and 5±3%, respectively). The co-administration of carboxy-PTIO (10 or 100 μM) and peroxynitrite (100 μM) significantly reversed the effects of the peroxynitrite treatment (57±2 and 65±3%, respectively). Similar results were obtained with co-administration of peroxynitrite (100 μM) and Trolox (10 or 100 μM) L-NIL (100 μM), 1400W (5 μM), with Trolox being the most potent of all drugs. Both L-NOARG and SMTC at 100 μM failed to reverse the effects of peroxynitrite.
Figure 8.
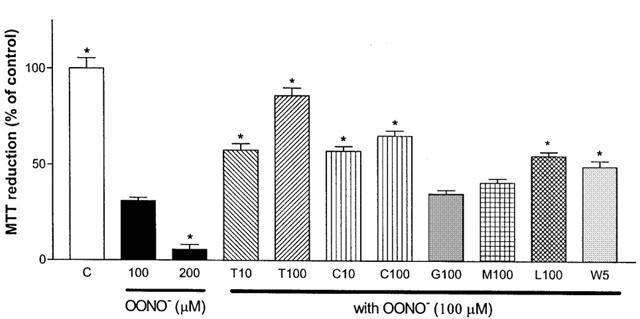
Effects of NOS inhibitors, Carboxy-PTIO, and Trolox on MTT reductions by primary cortical mixed cell cultures in the presence of peroxynitrite. Peroxynitrite (OONO; 100 and 200 μM) caused significant MTT reduction decreases as compared to the control (no peroxynitrite or drug exposure). The co-administration of peroxynitrite (100 μM) with Trolox at 10 (T10) or 100 μM (T100), carboxy-PTIO at 10 (C10) or 100 μM (C100), L-NIL at 100 μM (L100), or 1400W at 5 μM (W5) showed significantly higher MTT reductions than peroxynitrite treatment alone. L-NOARG at 100 μM (G100) or SMTC at 100 μM (M100) showed no significant difference in MTT reductions. Data represent mean±s.e.mean of 6–9 wells from five independent experiments. One-way ANOVA with Bonferroni's multiple comparison test and Student t-test were used to analyse statistical significance. *P<0.001 compared to peroxynitrite at 100 μM.
Effects of NOS inhibitors, carboxy-PTIO and Trolox on cellular viability in the presence of Aβ1–42 when administered in combinations
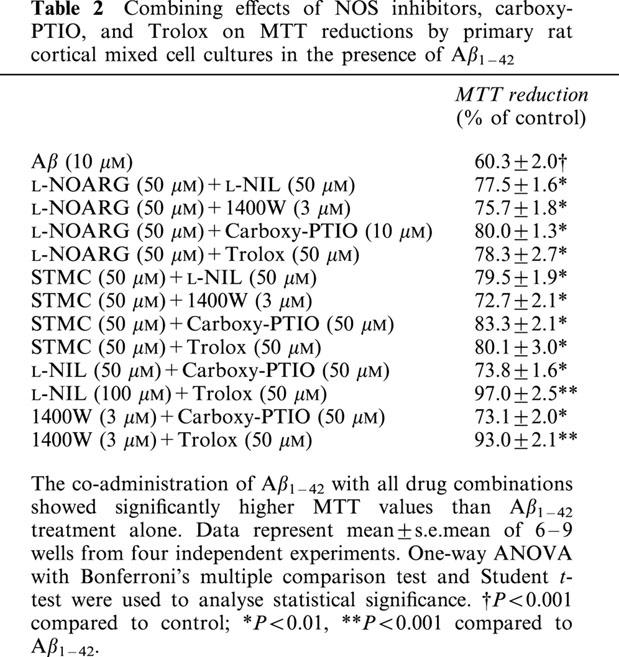
The effects of multiple drug treatments on Aβ1–42 (10 μM)-induced loss in cell viability were investigated next (Table 2). Aβ1–42 showed significantly lower MTT reduction (58±2% of control). All drug combinations were able to reverse the effect of Aβ1–42 on MTT reduction but to different extents. The co-administration of L-NIL (50 μM) and Trolox (50 μM) in the presence of Aβ1–42 fully reversed the effects of Aβ1–42 on MTT level (97±2%). Similar results were seen with 1400W (3 μM) and Trolox co-administration (89±1%). On the other hand, the co-administrations of L-NIL or 1400W with carboxyl-PTIO or type I NOS inhibitors failed to show additional effects on MTT values when compared to L-NIL or 1400W alone (data not shown). Similarly, the co-administration of carboxyl-PTIO (50 μM) or Trolox with either of the type I NOS inhibitors did not show significantly different MTT values to those of carboxyl-PTIO or Trolox alone.
Table 2.
Combining effects of NOS inhibitors, carboxy-PTIO, and Trolox on MTT reductions by primary rat cortical mixed cell cultures in the presence of Aβ1–42
Discussion
The current study examined the effects of Aβ on NO release, and the possible protective and rescuing abilities of NO/ROS modulating agents. Our results showed that Aβ1–42, but not Aβ42–1, caused a concentration-dependent increase in nitrite production in cultured cortical cells, along with decreases in cellular viability. In the absence of Aβ1–42, L-NIL and 1400W – two type II NOS inhibitors – did not have significant effect on cellular nitrite production nor cellular viability, whereas L-NOARG and SMTC – two type I NOS inhibitors – caused cellular nitrite production and viability to decrease at the higher concentration tested. Both type II NOS inhibitors caused reductions in nitrite productions and maintained cellular viability in the presence of Aβ1–42, whereas type I NOS inhibitors decreased nitrite production – to lesser extents than type II NOS inhibitors – but failed to support viability as assessed using MTT and NR assays. Most interestingly, type I and type II NOS inhibitors decreased cellular nitrite productions even if administered 1 or 4 h post Aβ1–42 treatments, respectively. However, only the type II NOS inhibitors were able to maintain cellular viability when administered 4 h post Aβ1–42 treatments. Peroxynitrite decreased cellular viability significantly, while Trolox, a vitamin E analogue antioxidant, reversed this effect. Both type II NOS inhibitors and carboxy-PTIO had similar, but less effective, actions as Trolox, whereas the type I NOS inhibitors were ineffective. In the presence of Aβ1–42, the co-administration of a type II NOS inhibitor with Trolox showed synergistic effects and fully maintained cellular viability.
The neurotoxicity of Aβ fragments (e.g. Aβ25–35, Aβ1–40, Aβ1–42) is well documented (Selkoe, 1994; Yankner et al., 1989). In the present study, Aβ1–42 was used since it is one of the naturally occurring sequences which is found in higher levels than Aβ1–40 in the AD brain, and possibly participating in the atiology of the disease (Lorenzo & Yankner, 1994) (El Agnaf et al., 2000). Previous studies have provided contradictory evidence regarding the ability of Aβ to induce NO production in neurons and glial cells (Forloni et al., 1997; Ii et al., 1996; Yang et al., 1998). In this study, Aβ, in a concentration-dependent manner, caused significant NO release – inferred by increased nitrite production – in primary rat cortical mixed cell cultures.
In the brain, NO is produced by three isoforms of NOS. Both type I and type III NOS – expressed primarily in neurons and endothelial cells, respectively – are constitutive (Bredt et al., 1990; Dinerman et al., 1994). Type II NOS – expressed in glial cells – is activated by many agents, including lipopolysaccarides and a number of cytokines (Dinerman et al., 1994), and is minimally expressed under basal conditions. The current study shows that under basal conditions, type II NOS inhibitors did not affect NO release, whereas type I NOS inhibitors were clearly able to inhibit it. These results indicate that the constitutive NOS isoform(s) are responsible for the production of NO under basal conditions. Moreover, L-NOARG is shown to inhibit both type I and type III NOS (Furfine et al., 1993), whereas SMTC is selective for type I NOS only. The lack of difference in the inhibition of NO release between L-NOARG and SMTC suggests that type III NOS does not contribute significantly to NO release in our model. Although the Griess reaction is not an absolute measure of NO production, this does not interfere with the overall interpretation of results in regard to the respective role of type I or type II NOS inhibition and the effects of Aβ1–42.
In the presence of Aβ1–42, both type I and type II NOS inhibitors showed significant effects in reducing NO release. These data suggest that Aβ-induced NO release was mediated by both type I and type II NOS. In other words, the origin of NO appeared to be both neuronal and glial, rather than solely glial as proposed earlier (Ii et al., 1996). Documented evidence shows that Aβ peptides and cytokines can act in synergy to induce type II NOS-mediated NO release from activated microglial cells and/or reactive astrocytes. The current results are in accordance with these studies, even if it does not establish whether astrocytes or microglia, or both, are responsible for the type-II NOS-mediated NO production. It is worthwhile to mention that the micromolar Aβ concentration ranges have been used repeatedly in many in vitro and in vivo toxicity studies (Doré et al., 1997; Huang et al., 2000; Nitta et al., 1993; Tang et al., 2000; Wei et al., 2000). These concentrations are often greater than that of the documented endogenous Aβ load necessary to cause neurotoxicity (Callahan et al., 2001; Hulstaert et al., 1999). However, the extrapolation between experimental and endogenous Aβ burden is difficult since these in vitro and in vivo intracerebroventricular infusion experiments represent acute toxicity, whereas endogenous Aβ toxicity is most likely a chronic phenomenon related to long-term exposure to low but constant levels of the peptide.
The observation that Aβ1–42 caused significant increase in NO release while decreasing cellular viability suggests that NO is likely to be neurotoxic. This hypothesis is supported by the findings that type II NOS inhibitors were able to decrease NO production while improving or maintaining cellular viability. The time-course also provided further evidence that Aβ1–42-induced NO release is neurotoxic. Moreover, the ability of type II NOS inhibitors to maintain cellular viability even up to 4 h post Aβ1–42-treatments demonstrates the neuroresecuing properties of these agents. Interestingly, the observed NO-induced neurotoxicity appeared to be NOS-isoform specific, since type I NOS inhibitors were able to reduce NO release in the presence of Aβ1–42 but failed to improve cellular viability under these conditions. Alternatively, the apparent lack of effect for type I NOS inhibitors on Aβ1–42-induced MTT reduction could possibly be explained by the fact that Aβ1–42 appeared to show greater effects on type II than type I NOS. Further investigation of NOS isoform-specific neurotoxicity is certainly worthwhile since in animal models of cerebral ischaemia, the resultant infarct damage is apparently dependent on type I and type III NOS, with the former being neurotoxic while the latter may be neuroprotective (Hara et al., 1996; Huang et al., 1996).
Peroxynitrite is a radical species generated by a reaction between NO and superoxide anions (Beckman et al., 1994a, 1994b). It leads to necrotic cell death by causing typical free radical damages and energy depletion secondary to glycolytic pathway impairment and polyADP-ribose polymerase (PARP) overactivation, a cellular response occurring as an attempt to repair excessive DNA damage (Beckman et al., 1994b; Ha & Snyder, 1999; Koppal et al., 1999). The current data shows that peroxynitrite treatment significantly reduced cell viability. Trolox has been shown to have protective effect against peroxynitrite toxicity (Salgo & Pryor, 1996) and was able to protect cultured cells in the model used here. Interestingly, type II NOS inhibitors and carboxy-PTIO also provided partial protection against peroxynitrite-induced toxicity. These findings can be taken as an indication that peroxynitrite may induce type II NOS expression and subsequent NO release. Under pathological conditions where type II NOS-mediated NO release is increased, the resultant NO release would lead to peroxynitrite formation, thereby providing a positive feedback mechanism to induce further NO release. Hence, type II NOS inhibitors may be a useful adjunct in attenuating peroxynitrite-induced toxicity.
Taken together, our results suggest that NO may be neurotoxic, and that Aβ1–42-induced toxicity, at least in part, is NO-mediated. Moreover, the fact that Trolox was able to improve cellular viability in the presence of Aβ1–42 suggests that peroxynitrite also played a role in Aβ1–42/NO-mediated cell toxicity. However, Trolox was not able to fully maintain cell viability in the presence of Aβ1–42, thereby revealing that other mechanisms are likely to be involved. Data in the literature suggest that in addition to the production of peroxynitrite, NO, by itself, is a ROS that can cause oxidative damages. It also promotes arachidonic acid inflammatory cascade (Guidarelli et al., 2000; Honda et al., 2000), and is involved in apoptosis (Dimmeler & Zeiher, 1997). Our results also show that lower concentrations of type II NOS inhibitors were able to fully protect against Aβ1–42-induced toxicity when administered concurrently with Trolox, revealing the synergistic actions of type II NOS inhibitors and antioxidants in attenuating the toxic effects of Aβ related peptides.
Although the present data suggest that Aβ peptide-induced neurotoxicity may be due to elevated NO release, the intracellular mechanism(s) which lead to the observed increased in NO production remains to be fully established. Existing data show that Aβ peptide activates several subtypes of mitogen-activated protein (MAP) kinases as well as the transcription factor cyclic AMP response element binding protein (CREB) (Sato et al., 1997). Interestingly, an increase in MAP kinase activity has been shown to induce type II NOS-mediated NO production and that the genes encoding type I and II NOS contain CREB binding sites (Kibbe et al., 2000; Sasaki et al., 2000), thereby suggesting that Aβ peptide may affect type I and II NOS expression. Further studies are needed to elucidate whether Aβ peptide-induced NO release is secondary to an increase of NOS expression, thereby raising the basal level of NO release, or an increase of existing enzyme activities.
AD is a complex syndrome that multiple factors are likely to be involved in its aetiology. Based on the findings of the present study and others, NO may be a key element in AD, and the therapeutic values of type II NOS inhibitors, NO scavengers, and antioxidants certainly deserve to be explored further. Existing data have already shown the beneficial potential of antioxidants such as vitamin E in slowing the progression of AD (Butterfield et al., 1999), the synergistic actions between antioxidants and NO-reducing agents may provide additional benefits in the treatment of AD.
Acknowledgments
The authors would like to thank Dr Wen-Hua Zheng for his excellent technical assistance. This study is supported by Canadian Institutes for Health Research (CIHR), RSMQ-FRSQ, and the Alzheimer Society of Canada.
Abbreviations
- AD
Alzheimer's disease
- Aβ
β-amyloid
- carboxy-PTIO
2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide
- D-MEM
Dulbecco's modified Eagle's medium
- FBS
foetal bovine serum
- HBSS
Hank's balanced salt solution
- L-NIL
N-iminoethyl-L-lysine
- L-NOARG
NG-nitro-L-arginine
- MTT
3-4(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrzolium bromide
- NO
nitric oxide
- NR
neutral red
- NOS
nitric oxide synthase
- ROS
reactive oxygen species
- SMTC
S-methyl-L-thiocitulline
- Trolox
6-hydorxy-2,5,7,8-tetramethylchroman-2-carboxylic acid
- 1400W
N-(3-(aminomethyl)benzyl)acetamidine
References
- ADAMSON D.C., WILDEMANN B., SASAKI M., GLASS J.D., MCARTHUR J.C., CHRISTOV V.I., DAWSON T.M., DAWSON V.L. Immunologic NO synthase: elevation in severe AIDS dementia and induction by HIV-1 gp41. Science. 1996;274:1917–1921. doi: 10.1126/science.274.5294.1917. [DOI] [PubMed] [Google Scholar]
- AKAMA K.T., ALBANESE C., PESTELL R.G., VAN ELDIK L.J. Amyloid beta-peptide stimulates nitric oxide production in astrocytes through an NFkappaB-dependent mechanism. Proc. Natl. Acad. Sci. U.S.A. 1998;95:5795–5800. doi: 10.1073/pnas.95.10.5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALMER G., VUKOSAVIC S., ROMERO N., PRZEDBORSKI S. Inducible nitric oxide synthase up-regulation in a transgenic mouse model of familial amyotrophic lateral sclerosis. J. Neurochem. 1999;72:2415–2425. doi: 10.1046/j.1471-4159.1999.0722415.x. [DOI] [PubMed] [Google Scholar]
- BASTIANETTO S., RAMASSAMY C., POIRIER J., QUIRION R. Dehydroepiandrosterone (DHEA) protects hippocampal cells from oxidative stress-induced damage. Mol. Brain Res. 1999;66:35–41. doi: 10.1016/s0169-328x(99)00002-9. [DOI] [PubMed] [Google Scholar]
- BECKMAN J.S., CHEN J., CROW J.P., YE Y.Z. Reactions of nitric oxide, superoxide and peroxynitrite with superoxide dismutase in neurodegenration. Prog. Brain Res. 1994b;103:371–380. doi: 10.1016/s0079-6123(08)61151-6. [DOI] [PubMed] [Google Scholar]
- BECKMAN J.S., CHEN J., ISCHIROPOULOS H., CROW J.P. Oxidative chemistry of peroxynitrite. Methods Enzymol. 1994a;233:229–240. doi: 10.1016/s0076-6879(94)33026-3. [DOI] [PubMed] [Google Scholar]
- BONAIUTO C., MCDONALD P.P., ROSSI F., CASSATELLA M.A. Activation of nuclear factor-kappa B by beta-amyloid peptides and interferon-gamma in murine microglia. J. Neuroimmunol. 1997;77:51–56. doi: 10.1016/s0165-5728(97)00054-4. [DOI] [PubMed] [Google Scholar]
- BREDT D.S., HWANG P.M., SNYDER S.H. Localization of nitric oxide synthase indicating a neural role for nitric oxide. Nature. 1990;347:768–770. doi: 10.1038/347768a0. [DOI] [PubMed] [Google Scholar]
- BUTTERFIELD D.A., KOPPAL T., SUBRAMANIAM R., YATIN S. Vitamin E as an antioxidant/free radical scavenger against amyloid beta- peptide-induced oxidative stress in neocortical synaptosomal membranes and hippocampal neurons in culture: insights into Alzheimer's disease. 1999;10:141–149. doi: 10.1515/revneuro.1999.10.2.141. [DOI] [PubMed] [Google Scholar]
- CALLAHAN M.J., LIPINSKI W.J., BIAN F., DURHAM R.A., PACK A., WALKER L.C. Augmented senile plaque load in aged female beta-amyloid precursor protein-transgenic mice. Am. J. Pathol. 2001;158:1173–1177. doi: 10.1016/s0002-9440(10)64064-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CECCATELLI S., LUNDBERG J.M., ZHANG X., AMAN K., HOKFELT T. Immunohistochemical demonstration of nitric oxide synthase in the peripheral autonomic nervous system. Brain Res. 1994;656:381–395. doi: 10.1016/0006-8993(94)91483-4. [DOI] [PubMed] [Google Scholar]
- COTMAN C.W., ANDERSON A.J. A potential role for apoptosis in neurodegeneration and Alzheimer's disease. Mol. Neurobiol. 1995;10:19–45. doi: 10.1007/BF02740836. [DOI] [PubMed] [Google Scholar]
- DAWSON D.A. Nitric oxide and focal cerebral ischemia: multiplicity of actions and diverse outcome. Cerebrovac. Brain Meta. Rev. 1994;6:299–324. [PubMed] [Google Scholar]
- DE MAN J.G., PELCKMANS P.A., BOECKXSTAENS G.E., BULT H., OOSTERBOSCH L., HERMAN A.G., VAN MAERCKE Y.M. The role of nitric oxide in inhibitory non-adrenergic non-cholinergic neurotransmission in the canine lower oesophageal sphincter. Br. J. Pharmacol. 1991;103:1092–1096. doi: 10.1111/j.1476-5381.1991.tb12305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DIMMELER S., ZEIHER A.M. Nitric oxide and apoptosis: another paradigm for the double-edged role of nitric oxide. Nitric Oxide. 1997;1:275–281. doi: 10.1006/niox.1997.0133. [DOI] [PubMed] [Google Scholar]
- DINERMAN J.L., DAWSON T.M., SCHELL M.J., SNOWMAN A., SNYDER S.H. Endothelial nitric oxide synthase localized to hippocampal pyramidal cells: implications for synaptic plasticity. Proc. Natl. Acad. Sci. U.S.A. 1994;91:4214–4218. doi: 10.1073/pnas.91.10.4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DORÉ S., KAR S., QUIRION R. Insulin-like growth factor I protects and rescues hippocampal neurons against beta-amyloid and human amylin induced toxicity. Proc. Natl. Acad. Sci. U.S.A. 1997;94:4772–4777. doi: 10.1073/pnas.94.9.4772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EL AGNAF O.M., MAHIL D.S., PATEL B.P., AUSTEN B.M. Oligomerization and toxicity of beta-amyloid-42 implicated in Alzheimer's disease. Biochem. Biophys. Res. Commun. 2000;273:1003–1007. doi: 10.1006/bbrc.2000.3051. [DOI] [PubMed] [Google Scholar]
- ELIASSON M.J.L., HUANG Z., FERRANTE R.J., SASAMATA M.E., MOLLIVER M.E., SNYDER S.H., MOSKOWITZ M.A. Neuronal nitric oxide snthase activation and peroxynitrite formation in ischemic stroke linked to neural damage. J. Neurosci. 1999;19:5910–5918. doi: 10.1523/JNEUROSCI.19-14-05910.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FORLONI G., MANGIAROTTI F., ANGERETTI N., LUCCA E., DE SIMONI M.G. Beta-amyloid fragment potentiates IL-6 and TNF-alpha secretion by LPS in astrocytes but not in microglia. Cytokine. 1997;9:759–762. doi: 10.1006/cyto.1997.0232. [DOI] [PubMed] [Google Scholar]
- FURFINE E.S., HARMON M.F., PAITH J.E., GARVEY E.P. Selective inhibition of constitutive nitric oxide synthase by L-NG- nitroarginine. Biochemistry. 1993;32:8512–8517. doi: 10.1021/bi00084a017. [DOI] [PubMed] [Google Scholar]
- FURFINE E.S., HARMON M.F., PAITH J.E., KNOWLES R.G., SALTER M., KIFF R.J., DUFFY C., HAZELWOOD R., OPLINGER J.A., GARVEY E.P. Potent and selective inhibition of human nitric oxide synthases. Selective inhibition of neuronal nitric oxide synthase by S-methyl-L- thiocitrulline and S-ethyl-L- thiocitrulline. J. Biol. Chem. 1994;269:26677–26683. [PubMed] [Google Scholar]
- GAHTAN E., OVERMIER J.B. Inflammatory pathogenesis in Alzheimer's disease: biological mechanisms and cognitive sequeli. Neurosci. Biobehav. Rev. 1999;23:615–633. doi: 10.1016/s0149-7634(98)00058-x. [DOI] [PubMed] [Google Scholar]
- GARVEY E.P., OPLINGER J.A., FURFINE E.S., KIFF R.J., LASZLO F., WHITTLE B.J., KNOWLES R.G. 1400W is a slow, tight binding, and highly selective inhibitor of inducible nitric-oxide synthase in vitro and in vivo. J. Biol. Chem. 1997;272:4959–4963. doi: 10.1074/jbc.272.8.4959. [DOI] [PubMed] [Google Scholar]
- GIOVANNONI G., HEALES S.J., LAND J.M., THOMPSON E.J. The potential role of nitric oxide in multiple sclerosis. Mult. Scler. 1998;4:212–216. doi: 10.1177/135245859800400323. [DOI] [PubMed] [Google Scholar]
- GOOD P.F., WERNER P., HSU A., OLANOW C.W., PERL D.P. Evidence of neuronal oxidative damage in Alzheimer's disease. Am. J. Pathol. 1996;149:21–28. [PMC free article] [PubMed] [Google Scholar]
- GOODWIN J.L., KEHRLI M.E., , Jr, UEMURA E. Integrin Mac-1 and beta-amyloid in microglial release of nitric oxide. Brain Res. 1997;768:279–286. doi: 10.1016/s0006-8993(97)00653-7. [DOI] [PubMed] [Google Scholar]
- GUIDARELLI A., PALOMBA L., CANTONI O. Peroxynitrite-mediated release of arachidonic acid from PC12 cells. Br. J. Pharmacol. 2000;129:1539–1541. doi: 10.1038/sj.bjp.0703275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HA H.C., SNYDER S.H. Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc. Natl. Acad. Sci. U.S.A. 1999;96:13978–13982. doi: 10.1073/pnas.96.24.13978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAASS C., SELKOE D.J. Cellular processing of beta-amyloid precursor protein and the genesis of amyloid beta-peptide. Cell. 1993;75:1039–1042. doi: 10.1016/0092-8674(93)90312-e. [DOI] [PubMed] [Google Scholar]
- HALEY J.E., WILCOX G.L., CHAPMAN P.F. The role of nitric oxide in hippocampal long-term potentiation. Neuron. 1992;8:211–216. doi: 10.1016/0896-6273(92)90288-o. [DOI] [PubMed] [Google Scholar]
- HARA H., HUANG P.L., PANAHIAN N., FISHMAN M.C., MOSKOWITZ M.A. Reduced brain edema and infarction volume in mice lacking the neuronal isoform of nitric oxide synthase after reversible MCA occlusion. J. Cereb. Blood Flow Metab. 1996;16:605–611. doi: 10.1097/00004647-199607000-00010. [DOI] [PubMed] [Google Scholar]
- HESS D.T., PATTERSON S.I., SMITH D.S., SKENE J.H.P. Neuronal growth cone collapse and inhibition of protein fatty acylation by nitric oxide. Nature. 1993;366:562–565. doi: 10.1038/366562a0. [DOI] [PubMed] [Google Scholar]
- HOGG N., SINGH R.J., JOSEPH J., NEESE F., KALYANARAMAN B. Reactions of nitric oxide with nitronyl nitroxides and oxygen: prediction of nitrite and nitrate formation by kinetic simulation. Free Radic. Res. 1995;22:47–56. doi: 10.3109/10715769509147527. [DOI] [PubMed] [Google Scholar]
- HONDA S., MIGITA K., HIRAI Y., UEKI Y., YAMASAKI S., URAYAMA S., KAWABE Y., FUKUDA T., KAWAKAMI A., KAMACHI M., KITA M., IDA H., AOYAGI T., EGUCHI K. Induction of COX-2 expression by nitric oxide in rheumatoid synovial cells. Biochem. Biophys. Res. Commun. 2000;268:928–931. doi: 10.1006/bbrc.2000.2228. [DOI] [PubMed] [Google Scholar]
- HU J., AKAMA K.T., KRAFFT G.A., CHROMY B.A., VAN ELDIK L.J. Amyloid-beta peptide activates cultured astrocytes: morphological alterations, cytokine induction and nitric oxide release. Brain Res. 1998;785:195–206. doi: 10.1016/s0006-8993(97)01318-8. [DOI] [PubMed] [Google Scholar]
- HUANG H.M., OU H.C., HSIEH S.J. Amyloid beta peptide impaired carbachol but not glutamate-mediated phosphoinositide pathways in cultured rat cortical neurons. Neurochem. Res. 2000;25:303–312. doi: 10.1023/a:1007592007956. [DOI] [PubMed] [Google Scholar]
- HUANG Z., HUANG P.L., MA J., MENG W., AYATA C., FISHMAN M.C., MOSKOWITZ M.A. Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-L-arginine. J. Cereb. Blood Flow Metab. 1996;16:981–987. doi: 10.1097/00004647-199609000-00023. [DOI] [PubMed] [Google Scholar]
- HULSTAERT F., BLENNOW K., IVANOIU A., SCHOONDERWALDT H.C., RIEMENSCHNEIDER M., DE DEYN P.P., BANCHER C., CRAS P., WILTFANG J., MEHTA P.D., IQBAL K., POTTEL H., VANMECHELEN E., VANDERSTICHELE H. Improved discrimination of AD patients using beta-amyloid(1–42) and tau levels in CSF. Neurology. 1999;52:1555–1562. doi: 10.1212/wnl.52.8.1555. [DOI] [PubMed] [Google Scholar]
- IGNARRO L.J., BUGA G.M., WOOD K.S., BYRNS R.E., CHAUDHURI G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc. Natl. Acad. Sci. U.S.A. 1987;84:9265–9269. doi: 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- II M., SUNAMOTO M., OHNISHI K., ICHIMORI Y. beta-Amyloid protein-dependent nitric oxide production from microglial cells and neurotoxicity. Brain Res. 1996;720:93–100. doi: 10.1016/0006-8993(96)00156-4. [DOI] [PubMed] [Google Scholar]
- KIBBE M.R., LI J., NIE S., WATKINS S.C., LIZONOVA A., KOVESDI I., SIMMONS R.L., BILLIAR T.R., TZENG E. Inducible nitric oxide synthase (iNOS) expression upregulates p21 and inhibits vascular smooth muscle cell proliferation through p42/44 mitogen-activated protein kinase activation and independent of p53 and cyclic guanosine monophosphate. J. Vasc. Surg. 2000;31:1214–1228. doi: 10.1067/mva.2000.105006. [DOI] [PubMed] [Google Scholar]
- KOPPAL T., DRAKE J., YATIN S., JORDAN B., VARADARAJAN S., BETTENHAUSEN L., BUTTERFIELD D.A. Peroxynitrite-induced alterations in synaptosomal membrane proteins: insight into oxidative stress in Alzheimer's disease. J. Neurochem. 1999;72:310–317. doi: 10.1046/j.1471-4159.1999.0720310.x. [DOI] [PubMed] [Google Scholar]
- KRÖNCKE K.-D., FEHSEL K., KOLB-BACHOFEN V. Inducible nitric oxide synthase and its product nitric oxide, a small molecule with complex biological activities. Biol. Chem. Hoppe-Seyler. 1995;376:327–343. doi: 10.1515/bchm3.1995.376.6.327. [DOI] [PubMed] [Google Scholar]
- LINDEN D.J., CONNOR J.A. Cellular mechanisms of long-term depression in the cerebellum. Curr. Opin. Neurol. 1993;3:401–406. doi: 10.1016/0959-4388(93)90133-j. [DOI] [PubMed] [Google Scholar]
- LORENZO A., YANKNER B.A. Beta-amyloid neurotoxicity requires fibril formation and is inhibited by congo red. Proc. Natl. Acad. Sci. U.S.A. 1994;91:12243–12247. doi: 10.1073/pnas.91.25.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MATTSON M.P., CHENG B., DAVIS D., BRYANT K., LIEBERBURG I., RYDEL R.E. beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J. Neurosci. 1992;12:376–389. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MEDA L., CASSATELLA M.A., SZENDREI G.I., OTVOS L., JR, BARON P., VILLALBA M., FERRARI D., ROSSI F. Activation of microglial cells by beta-amyloid protein and interferon- gamma. Nature. 1995;374:647–650. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- MELDRUM B., GARTHWAITE J. Excitatory amino acid neurotoxcity and neurodegenerative disease. Trends Pharmacol. 1990;11:379–387. doi: 10.1016/0165-6147(90)90184-a. [DOI] [PubMed] [Google Scholar]
- MOORE W.M., WEBBER R.K., JEROME G.M., TJOENG F.S., MISKO T.P., CURRIE M.G. L-N6-(1-iminoethyl)lysine: a selective inhibitor of inducible nitric oxide synthase. J. Med. Chem. 1994;37:3886–3888. doi: 10.1021/jm00049a007. [DOI] [PubMed] [Google Scholar]
- NIMS R.W., COOK J.C., KRISHNA M.C., CHRISTODOULOU D., POORE C.M., MILES A.M., GRISHAM M.B., WINK D.A. Colorimetric assays for nitric oxide and nitrogen oxide species formed from nitric oxide stock solutions and donor compounds. Methods Enzymol. 1996;268:93–105. doi: 10.1016/s0076-6879(96)68012-4. [DOI] [PubMed] [Google Scholar]
- NITTA A., MURASE K., FURUKAWA Y., HAYASHI K., HASEGAWA T., NABESHIMA T. Memory impairment and neural dysfunction after continuous infusion of anti-nerve growth factor antibody into the septum in adult rats. Neuroscience. 1993;57:495–499. doi: 10.1016/0306-4522(93)90001-v. [DOI] [PubMed] [Google Scholar]
- SALGO M.G., PRYOR W.A. Trolox inhibits peroxynitrite-mediated oxidative stress and apoptosis in rat thymocytes. Arch. Biochem. Biophys. 1996;333:482–488. doi: 10.1006/abbi.1996.0418. [DOI] [PubMed] [Google Scholar]
- SASAKI M., GONZALEZ-ZULUETA M., HUANG H., HERRING W.J., AHN S., GINTY D.D., DAWSON V.L., DAWSON T.M. Dynamic regulation of neuronal NO synthase transcription by calcium influx through a CREB family transcription factor-dependent mechanism. Proc. Natl. Acad. Sci. U.S.A. 2000;97:8617–8622. doi: 10.1073/pnas.97.15.8617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SATO N., KAMINO K., TATEISHI K., SATOH T., NISHIWAKI Y., YOSHIIWA A., MIKI T., OGIHARA T. Elevated amyloid beta protein(1-40) level induces CREB phosphorylation at serine-133 via p44/42 MAP kinase (Erk1/2)-dependent pathway in rat pheochromocytoma PC12 cells. Biochem. Biophys. Res. Commun. 1997;232:637–642. doi: 10.1006/bbrc.1997.6341. [DOI] [PubMed] [Google Scholar]
- SCHMIDT H.H.H.W., WILKE P., EVERS B., BÖHME E. Enzymatic formation of nitrogen oxides from L-arginine in bovine brain cytosol. Biochem. Biophys. Res. Commun. 1989;165:284–291. doi: 10.1016/0006-291x(89)91067-x. [DOI] [PubMed] [Google Scholar]
- SELKOE D.J. Alzheimer's disease: a central role for amyloid [see comments] J. Neuropathol. Exp. Neurol. 1994;53:438–447. doi: 10.1097/00005072-199409000-00003. [DOI] [PubMed] [Google Scholar]
- SIMS N.R. Energy metabolism, oxidative stress and neuronal degeneration in Alzheimer's disease. Neurodegeneration. 1996;5:435–440. doi: 10.1006/neur.1996.0059. [DOI] [PubMed] [Google Scholar]
- SMITH M.A., RICHEY HARRIS P.L., SAYRE L.M., BECKMAN J.S., PERRY G. Widespread peroxynitrite-mediated damage in Alzheimer's disease. J. Neurosci. 1997;17:2653–2657. doi: 10.1523/JNEUROSCI.17-08-02653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STUEHR D.J., MARLETTA M.A. Mammalian nitrate biosynthesis: Mouse macrophages produce nitrite and nitrate in response to Escherichia cdi lipopolysaccharide. Proc. Natl. Acad. Sci. U.S.A. 1985;82:7738–7742. doi: 10.1073/pnas.82.22.7738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TANG Y., YAMADA K., KANOU Y., MIYAZAKI T., XIONG X., KAMBE F., MURATA Y., SEO H., NABESHIMA T. Spatiotemporal expression of BDNF in the hippocampus induced by the continuous intracerebroventricular infusion of beta-amyloid in rats. Mol. Brain Res. 2000;80:188–197. doi: 10.1016/s0169-328x(00)00158-3. [DOI] [PubMed] [Google Scholar]
- WALLACE M.N., GEDDES J.G., FARQUHAR D.A., MASSON M.R. Nitric oxide synthase in reactive astrocytes adjacent to beta-amyloid plaques. Exp. Neurol. 1997;144:266–272. doi: 10.1006/exnr.1996.6373. [DOI] [PubMed] [Google Scholar]
- WEI H., LEEDS P.R., QIAN Y., WEI W., CHEN R., CHUANG D. beta-amyloid peptide-induced death of PC 12 cells and cerebellar granule cell neurons is inhibited by long-term lithium treatment. Eur. J. Pharmacol. 2000;392:117–123. doi: 10.1016/s0014-2999(00)00127-8. [DOI] [PubMed] [Google Scholar]
- YANG S.N., HSIEH W.Y., LIU D.D., TSAI L.M., TUNG C.S., WU J.N. The involvement of nitric oxide in synergistic neuronal damage induced by beta-amyloid peptide and glutamate in primary rat cortical neurons. Chin. J. Physiol. 1998;41:175–179. [PubMed] [Google Scholar]
- YANKNER B.A., DAWES L.R., FISHER S., VILLA-KOMAROFF L., OSTER-GRANITE M.L. Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer's disease. Science. 1989;245:417–420. doi: 10.1126/science.2474201. [DOI] [PubMed] [Google Scholar]