

Abstract
Inhibition of Cav1.2 by antagonist 1,4 dihydropyridines (DHPs) is associated with a drug-induced acceleration of the calcium (Ca2+) channel current decay. This feature is contradictorily interpreted as open channel block or as drug-induced inactivation. To elucidate the underlying molecular mechanism we investigated the effects of (+)- and (−)-isradipine on Cav1.2 inactivation gating at different membrane potentials.
α11.2 Constructs were expressed together with α2-δ- and β1a- subunits in Xenopus oocytes and drug-induced changes in barium current (IBa) kinetics analysed with the two microelectrode voltage clamp technique. To study isradipine effects on IBa decay without contamination by intrinsic inactivation we expressed a mutant (V1504A) lacking fast voltage-dependent inactivation.
At a subthreshold potential of −30 mV a 200-times higher concentration of (−)-isradipine was required to induce a comparable amount of inactivation as by (+)-isradipine. At +20 mV the two enantiomers were equally efficient in accelerating the IBa decay.
Faster recovery from (−)- than from (+)-isradipine-induced inactivation at −80 mV in a Cav1.2 construct (τ(−)-isr.(Cav1.2)=0.74 s<τ(+)-isr.(Cav1.2)=2.85 s) and even more rapid recovery of V1504A (τ(−)-isr.(V1504A)=0.39 s<τ(+)-isr.(V1504A)=1.98 s) indicated that drug-induced determinants and determinants of intrinsic inactivation (V1504) stabilize the DHP-induced channel conformation in an additive manner.
In the voltage range between −25 and 20 mV where the channels inactivate predominantly from the open state the (+)- and (−)-isradipine-induced acceleration of the IBa decay in V1504A displayed similar voltage-dependence as intrinsic fast inactivation of Cav1.2.
Our data suggest that the isradipine-induced acceleration of the Cav1.2 current decay reflects enhanced fast voltage-dependent inactivation and not open channel block.
Keywords: Calcium channels, voltage-dependent inactivation, isradipine, receptor models
Introduction
The calcium (Ca2+) channel blocker (or Ca2+ antagonist) isradipine is widely used for the treatment of hypertension and angina (Frishman & Michaelson, 1997). Isradipine belongs to the chemical class of the 1,4 dihydropyridines (DHPs) and exerts its therapeutic effect primarily by blocking Cav1.2 (L-type Ca2+ channels, see Ertel et al., 2000 for nomenclature) in the vascular system (Lund-Johansen, 1993).
Cav1.2 are hetero-oligomeric protein complexes consisting of a pore forming α1-subunit, auxiliary β- (β1-β4) and α2-δ-subunits modulating voltage-dependence, expression density and channel kinetics (Catterall, 1994; Walker & De Waard, 1998; Hering et al., 2000). The receptor determinants for DHPs are localized on the pore forming α1-subunit (α11.2) of Cav1.2 (Striessnig et al., 1998). Mutational analysis revealed nine amino acid residues in segments IIIS5, IIIS6 and IVS6 of α12.1 that confer high affinity and stereoselective interaction with isradipine (Tang et al., 1993; Grabner et al., 1996; Peterson et al., 1996; 1997; Sinnegger et al., 1997; Ito et al., 1997).
Enantioselectivity is a hallmark of the interaction of isradipine with the Cav1.m superfamily (Glossmann & Ferry, 1985). High concentrations of isradipine also inhibit low threshold calcium channels. This low affinity interaction of isradipine with non-Cav1.m (non-L-type channels) occurs, however, in a non-stereoselective manner (Berjukow et al., 1996).
Cav1.2 inhibition by DHPs is commonly described in terms of a modulated receptor mechanism (Hille, 1978). A popular version of the modulated receptor hypothesis suggests high affinity DHP-binding to the inactivated Ca2+ channel conformation. This hypothesis is based on the well-documented DHP-induced leftward shift of the of the Cav1.2 availability curve (Bean, 1984; Sanguinetti & Kass, 1984).
However, Ca2+ channels inactivate by at least two (‘fast' and ‘slow') voltage-dependent mechanisms and in some channel types by an additional Ca2+-dependent inactivation (Brehm & Eckert, 1978; Lee et al., 1999; Hering et al., 2000 for review). Most previous studies did not differentiate between DHP interactions with different inactivated channel conformations. We have recently shown that the high affinity (+)-enantiomer of isradipine promotes a channel state resembling intrinsic fast inactivation (Berjukow et al., 2000).
The mechanism of the DHP-induced changes in Ca2+ channel gating remains, however, unclear. In particular, the DHP-induced acceleration of the IBa decay observed in many previous studies is controversially interpreted as either reflecting open channel block or, alternatively, drug-induced inactivation.
In the present study we have, therefore, analysed the effects of the two isradipine enantiomers on channel inactivation at different membrane potentials. We report here that (+)- and (−)-isradipine enhance channel transitions to a (drug-induced) inactivated channel conformation at subthreshold voltages where Cav1.2 inactivate predominantly from the resting state. At more depolarized voltages where Cav1.2 inactivate predominantly via the open channel conformation drug-effects on the current decay display a similar voltage-dependency as intrinsic fast inactivation. Our data suggest that the DHP-induced acceleration of the IBa decay at depolarized voltages reflects enhanced inactivation and not open channel block. An analysis of the recovery from drug-induced inactivation revealed that intrinsic determinants of fast inactivation (V1504) and drug-induced inactivation determinants contribute in an additive manner to the stability of the DHP-induced channel state.
Methods
α1 cDNAs
The study was performed on the Cav1.2 channel construct Lh (named herein α1L, Grabner et al., 1996). α1L is a construct corresponding to rabbit cardiac α1C-a cDNA (Mikami et al., 1989) with part of the amino terminus replaced by carp α11.1 sequence as described (Grabner et al., 1996; Berjukow et al., 1999). Mutation V1504A was previously described in Berjukow et al., 1999. Amino acid numbering of V1504A is according to α1C-a cDNA sequence.
Electrophysiology
Inward barium currents (IBa) were studied with two microelectrode voltage-clamp of Xenopus oocytes 2–7 days after microinjection of approximately equimolar cRNA mixtures of α1L or V1504A (Grabner et al., 1996) (0.3 ng–50 nl) together with α2-δ(0.2 ng–50 nl) and βIa(0.1 ng–50 nl) cRNA as described previously (Grabner et al., 1996). The corresponding constructs were named herein α1L/α2-δ/βIa or V1504A channels.
All experiments were carried out at room temperature in a bath solution with the following composition (mM): Ba(OH)2 40, NaOH 50, HEPES 5, CsOH 2 (pH adjusted to 7.4 with methanesulphonic acid). Voltage-recording and current-injecting microelectrodes were filled with 2.8 M CsCl, 0.2 M CsOH, 10 mM EGTA, 10 mM HEPES (pH 7.4) and had resistances of 0.4–2.2 MΩ. Tonic (resting-state-dependent) Ca2+ channel block was estimated as steady-state peak IBa inhibition during short (100 ms) test pulses from −80 to 20 mV at a frequency of 0.033 Hz. Drug effects on IBa inactivation were analysed as isradipine-induced current decay during 3 s test pulses from −80 to 20 mV. The dose-response curves of IBa inhibition were fitted using the Hill equation: IBa,drug/IBa,control (in %)=(100-A)/(1+(C/IC50)nH)+A, where IC50 is the concentration at which IBa inhibition is half maximal, C is the applied drug concentration, A is the fraction of IBa that is not blocked and nH is the Hill coefficient.
Recovery from inactivation was studied with a conventional double-pulse protocol after depolarizing Ca2+ channels during a 3 s prepulse (to 20 mV) at a holding potential of −80 mV. 30 ms-test pulses (to 20 mV) were applied at various time intervals after the conditioning prepulse. Peak IBa values were normalized to the peak current measured during the prepulse and the time course of IBa recovery from inactivation was fitted to a mono- or biexponential function (IBa,recovery=Afast×exp(-t/τfast)+Aslow×exp(−t/τslow)+C). The time constants of IBa inactivation were estimated by fitting the current decay to a mono or biexponential function. Data are given as mean±s.e.mean statistical significance was calculated according to Student's unpaired t-test.
Results
(+)- and (−)-isradipine effects on peak and late current of Cav1.2
Isradipine binding to Cav1.m is highly stereoselective with the (+)-enantiomer being more potent than (−)-isradipine (Glossmann & Ferry, 1985). To elucidate the functional consequences of this stereoselective drug-channel interaction we compared the inhibition of barium currents through Cav1.2 constructs by both enantiomers. First we expressed the α1L-subunit (Berjukow et al., 1999) together with the auxiliary α2-δ- and β1a-subunits in Xenopus oocytes and studied (+)- and (−)-isradipine effects on IBa under two microelectrode voltage clamp (see Methods). Peak IBa through α1L/α2-δ/β1a channels were much less efficiently inhibited by (−)-isradipine (IC50=34±8 μM, Figure 1A–C) than by the (+)-enantiomer (IC50=327±41 nM, Berjukow et al., 2000) which is in line with our previous observations (Berjukow et al., 1996) and data of Handrock et al. (1999). Ca2+ channel inhibition by both enantiomers was accompanied by a drug-induced acceleration of the current decay (Figure 1A,B). In order to quantify this effect we plotted the IBa decay during a 3 s test pulse versus the applied drug concentrations. Surprisingly, as shown in Figure 1D, (+)- and (−)-isradipine displayed no enantioselectivity with respect to their effects on IBa decay (IC50,(+)-isr.=78±18 nM, IC50,(−)-isr.=95±22 nM, P>0.05).
Figure 1.
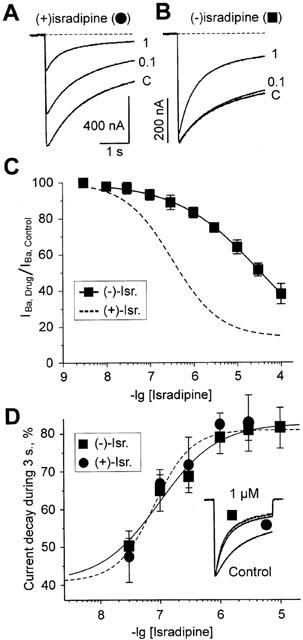
Peak current inhibition and modulation of inactivation kinetics in α1L/α2-δ/β1a channels by (+)- and (−)-isradipine. (A,B) Barium currents through α1L/α2-δ/β1a channels during membrane depolarizations from −80 mV to 20 mV in control and in the presence of different concentrations of (+)- and (−)-isradipine (in μM). (C) Concentration-response relationships of peak IBa inhibition of α1L/α2-δ/β1a channels by (+)- (dashed line, from Berjukow et al., 2000) and (−)-isradipine. Channel block was estimated as the ratio of peak current in the presence of the respective enantiomer compared to peak IBa in control. Data points represent the mean values from 4–11 experiments. The IC50 and the Hill coefficient (nH) for peak current block by (−)-isradipine were obtained by best fit of the data points to the general dose-response equation (see Methods) yielding: IC50=34±8 μM, nH=0.45±0.03, n=4. (D) Acceleration of IBa decay by (+)- and (−)-isradipine estimated as late current inhibition during a 3 s depolarizing test pulse from −80 to 20 mV. The inset shows three superimposed normalized IBa through α1L/α2-δ/β1a in control and the presence of 1 μM (+)- and (−)-isradipine.
Next we investigate if the different potencies of (+)- and (−)-isradipine to inhibit peak IBa (Figure 1A–C) would correlate with their abilities to induce inactivation at subthreshold potentials. (+)- and (−)-isradipine induced an additional component in the onset of resting state inactivation of α1L/α2-δ/β1a (Figure 2A, see also Berjukow et al., 2000). The mean time constant of the (−)-isradipine-induced inactivation at −30 mV ranged between 0.5 and 0.7 s (100 nM–100 μM) which was quite similar to the rates observed for the (+)-enantiomer (between 0.5 and 0.73 s in 10 nM–10 μM, see Figure 2C). The corresponding amplitude coefficients (Afast) of this drug-induced component in channel inactivation increased with increasing drug concentrations (Figure 2D). Plotting Afast versus the applied (+)- and (−)-isradipine concentrations revealed a significantly higher potency of the (+)-enantiomer. As shown in Figure 2B, an about 200-times higher concentration of (−)-isradipine was required to induce a comparable inactivation of resting channels as by (+)-enantiomer (IC50 72±5 nM for (+)-isradipine (n=3) and 13±4 μM for (−)-isradipine (n=4)).
Figure 2.
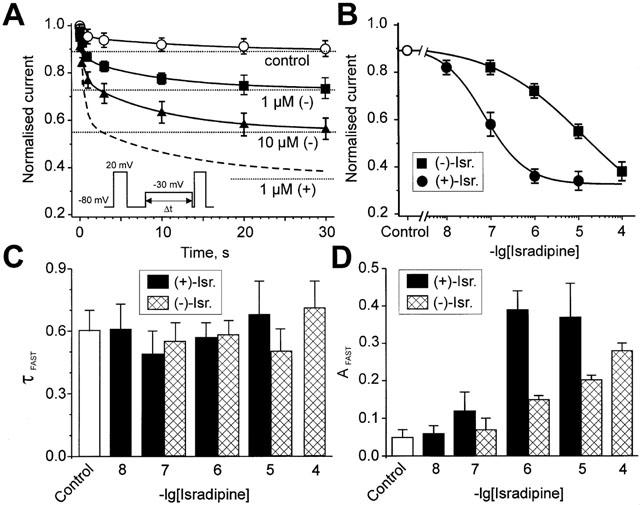
Drug-induced closed-state inactivation of α1L/α2-δ/β1a channels at the subthreshold potential of −30 mV. (A) Effect of prepulses of variable duration from −80 mV to −30 mV on the peak current evoked by a subsequent test pulse to 20 mV (see inset). The smooth curves are biexponential functions fitted to the time course of mean IBa inactivation of α1L/α2-δ/β1a channels (IBa/normalised=Afast×exp(−τ/τfast)+Aslow×exp(−τ/τslow)+C). Asymptotic values are represented by the dotted lines. The parameters of the fit in control: Afast=0.05, τfast=0.62 s, Aslow=0.06, τslow=13.0 s, C=0.89; in 1 μM (−)-isradipine: Afast=0.15, τfast=0.58 s, Aslow=0.1, τslow=14.0 s, C=0.73; in 10 μM (−)-isradipine: Afast=0.21, τfast=0.49 s, Aslow=0.22, τslow=11 s, C=0.56. The time course of the biexponential fit to 1 μM (+)-isradipine-induced inactivation (dashed line from Berjukow et al., 2000) is shown for comparison. (B) Drug-induced steady-state inactivation (dotted lines in A) induced by different concentrations of (+)- and (−)-isradipine is plotted as a function of the applied drug-concentration. Fitting of the data points to the dose-response equation yielded for (−)-isradipine: IC50=13±5 μM, nH=0.41±0.03 (n=4) and for (+)-isradipine: IC50=72±5 nM, nH=0.92±14 (n=3). (C) Kinetics of the fast component of drug-induced inactivation by (+)- and (−)-isradipine. The corresponding mean time constants (τfast) of the biexponential fits (shown in A) are indicated for different drug-concentrations. (D) The amplitude coefficient of the fast component (Afast, see A) is illustrated for different drug concentration. (+)- (black bars) and (−)-isradipine- (hatched bars) induced fast inactivation at −30 mV are illustrated at different drug concentrations.
(+)- and (−)-isradipine-induced acceleration of IBa decay is voltage-dependent
To gain further insights into the molecular mechanism underlying the isradipine effects on IBa decay we made use of mutant V1504A lacking fast voltage-dependent inactivation (Berjukow et al., 1999; 2000). This enabled us to investigate drug effects on IBa decay without contamination by intrinsic voltage-dependent inactivation.
Peak current inhibition of V1504A by (−)-isradipine was more pronounced (IC50=2.2±0.2 μM, n=4) than in α1L/α2-δ/β1a channels (IC50=34±8 μM, n=4, Figure 3A,B, see also Berjukow et al., 2000). Both enantiomers accelerated the IBa decay to comparable extents (Figure 3B, see also Figure 1D for similar result in the Cav1.2 construct).
Figure 3.
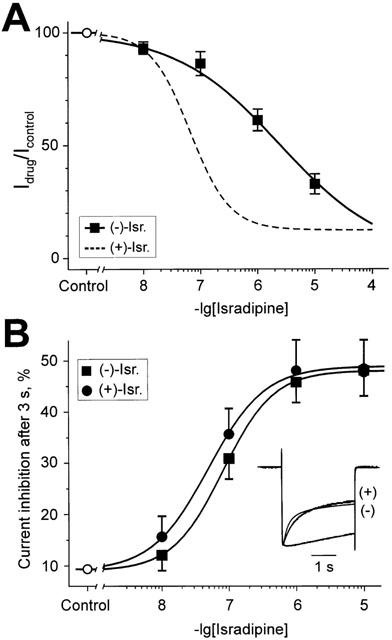
Inhibition of peak IBa and (+)- and (−)-isradipine-induced inactivation in mutant V1504A. (A) Concentration-response relationships of peak IBa inhibition of V1504A channels by (−)-isradipine. Data points represent the mean values from 3–5 experiments. The IC50 and the Hill coefficient (nH) for peak current block by (−)-isradipine were obtained by best fit of the data points to the dose-response equation yielding: IC50=2.2±0.2 μM, nH=0.53±0.03; The dashed line represents the fitted concentration response curve for channel block by (+)-isradipine (from Berjukow et al., 2000). (B) (+)- and (−)-isradipine accelerate IBa inactivation of V1504A. (+)- and (−)-induced effects on IBa decay were estimated as current inhibition during a 3 s depolarizing test pulse from −80 to 20 mV in per cent. Inset: Superimposed normalized IBa illustrating (+)- and (−)-isradipine-induced acceleration of the current decay compared to control.
Plotting the time constants of (+)- and (−)-isradipine (1 μM) induced IBa decay versus the applied test potentials revealed that both enantiomers accelerate the current decay in a voltage-dependent manner. Moreover, the drug-induced component of IBa decay in mutant V1504A and the time constants of intrinsic fast voltage-dependent inactivation of α1L/α2-δ/β1a displayed almost identical voltage-dependence. These data suggest that both enantiomers restore fast voltage-dependent inactivation in mutant V1504A rather than blocking open channels (Figure 4).
Figure 4.
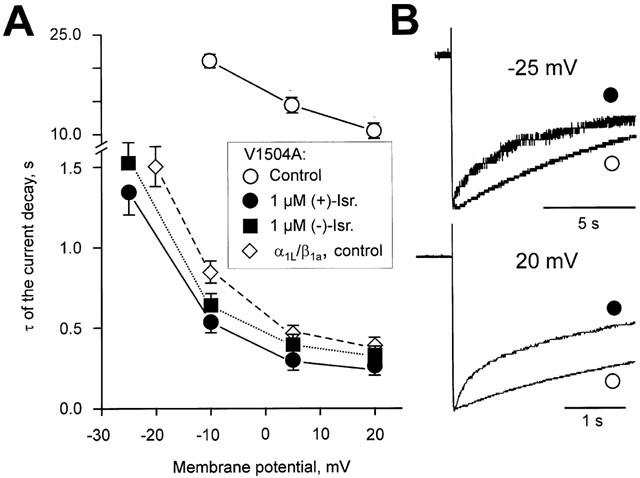
Voltage-dependent acceleration of the IBa decay by (+)- and (−)-isradipine. (A) The time constants of IBa decay of V1504A and α1L/α2-δ/β1a were estimated by fitting single (V1504A, control) or bi-exponential functions to the current decay. Currents were elicited by 30 s steps from a holding potential −80 mV to the indicated voltages. Open circles represent the time constant of V1504A inactivation in control, filled circles and squares represent the (+)- and (−)-isradipine induced current decay time constants in V1504A. Open diamonds show the fast voltage-dependent inactivation time constant of α1L/α2-δ/β1a in control. (B) Representative IBa of the mutant V1504A elicited by 10 s depolarizing steps to −25 mV and 3 s steps to 20 mV from −80 mV in control (open circles) or the presence of 1 μM (+)-isradipine (filled circles).
Different stabilities of (+)- and (−)-isradipine-induced inactivated states
The stability of the DHP-induced channel conformation can be assessed by analysing the recovery from drug-induced inactivation (Berjukow et al., 2000). In the present study we compared the stabilities of the (−)- and (+)-isradipine-induced channel states. As shown in Figure 5, IBa of α1L/α2-δ/β1a recover at −80 mV in control with monoexponential kinetics (τrec.control≈0.65 s). Recovery of α1L/α2-δ/β1a channels from (+)-isradipine-induced inactivation at −80 mV (1 μM (+)-isradipine) was previously found to occur at an about four times slower rate (illustrated by the dashed line in Figure 5A, from Berjukow et al., 2000). The faster recovery of α1L/α2-δ/β1a channels from (−)-isradipine-induced inactivation (Figure 5A,C, τrec,(−)-israd. (1 μM)=0.74±0.07, n=4) suggested that the (−)-isradipine-induced channel conformation is less stable than the (+)-isradipine-induced state.
Figure 5.
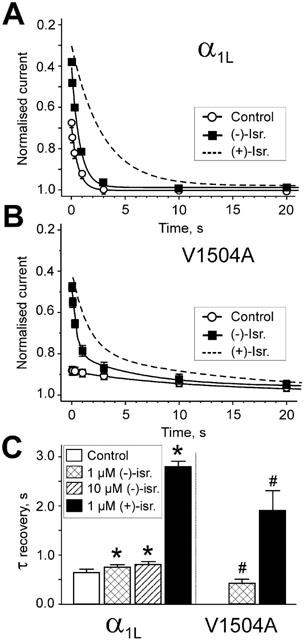
Recovery of α1L/α2-δ/β1a and V1504A channels from (+)- and (−)-isradipine-induced inactivation. Time course of IBa recovery from (−)-isradipine-induced inactivation after a 3 s conditioning prepulses to 20 mV (holding potential −80 mV). Test pulses to 20 mV were applied at various time intervals after the conditioning pulses. Peak IBa during the test pulses were normalized to peak IBa measured during the conditioning prepulse. Smooth curves in panels represent mono- or biexponential functions fitted to IBa recovery of α1L/α2-δ/β1a (A) and V1504A (B) channels in control and in 1 μM (−)-isradipine. The parameters of fit were in (A) for control recovery (○): A=0.33, τ=0.61 s, C=1; recovery in 1 μM (−)-isradipine: A=0.63, τ=0.74 s, C=0.97. Broken line illustrates the corresponding time course of IBa recovery in 1 μM (+)-isradipine (fitted curve form Berjukow et al., 2000). (B) Recovery of V1504A in control (○): Aslow=0.11, τslow=13.4 s, C=0.98 and the presence of 1 μM (−)-isradipine: Afast=0.42, τfast=0.39 s, Aslow=0.21, τslow=11.1 s, C=0.98. Dashed line illustrates recovery of IBa in 1 μM (+)-isradipine (data from Berjukow et al., 2000). (C) Time constant of recovery from fast inactivation in control (white column), 1 μM and 10 μM (−)-isradipine (hatched columns). Black columns illustrate the corresponding recovery from (+)-isradipine-induced inactivation (data from Berjukow et al., 2000). α1L/α2-δ/β1a and V1504A channels recover significantly faster from (−)-isradipine-induced inactivation compared to (+)-isradipine. Note that the inactivation deficient mutant V1504A recovered faster from (+)- and (−)-isradipine-induced inactivation than α1L/α2-δ/β1a channels (*P<0.01 compared to α1L/α2-δ/β1a in control. #P<0.01 compared to recovery of the respective enantiomers in α1L/α2-δ/β1a).
A similar trend was observed for construct V1504A displaying a significantly faster recovery from (−)-isradipine-induced inactivation (τrec,(−)-israd. (1 μM)=0.39±0.09 s, n=3) compared to (+)-enantiomer (τrec,(+)-israd. (1 μM)=1.98±0.48 s, n=5, Berjukow et al., 2000) (Figure 5B). Interestingly, recovery of V1504A from (+)- and (−)-isradipine-induced inactivation occurred at a significantly faster rate than corresponding recovery of α1L/α2-δ/β1a (Figure 5C). These data suggest that (+)- and (−)-isradipine stabilize the drug-induced channel conformation to different extents. Furthermore, residue V1504 has obviously an additional stabilizing effect on the DHP-induced channel state.
Discussion
Mutational analysis of voltage-gated Ca2+ channels enabled first insight into the localization of putative DHP-binding determinants (Striessnig et al., 1998 for review). The molecular mechanism of Ca2+ channel block by DHPs is, however, still controversial (Hering et al., 1998). Recent studies on Ca2+ channel mutants with impaired inactivation enable for the first time a profound analysis of the role of the inactivated channel state in block by DHPs. In a preceding study we have demonstrated that the high affinity (+)-enantiomer of isradipine stabilizes an inactivated channel conformation resembling fast voltage-dependent inactivation (Berjukow et al., 2000).
However, the molecular mechanisms of antagonist DHP-induced changes in Ca2+ channel kinetics are still controversial. Drug-induced changes in Ca2+ channel gating are either interpreted as open channel block (e.g. Lacinova & Hofmann, 1998; Handrock et al., 1999) or, alternatively, as drug-induced channel inactivation (Berjukow et al., 2000).
We made, therefore, an attempt to characterize the role of intrinsic fast voltage-dependent channel inactivation in DHP action by analysing the IBa inhibition by the two isradipine enantiomers (Figure 1A–C, see also previous results of Berjukow et al., 1996; Handrock et al., 1999). Particular attention was paid to the molecular mechanism underlying the drug-induced changes in Ca2+ channel current decay.
Ca2+ channel current inhibition by isradipine correlates with resting-state-dependent inactivation
A key observation of our study was the close correlation between the different potencies of the two enantiomers to inhibit peak IBa and their different effectiveness to promote inactivation of resting channels (Figures 1 and 2). Hence, at −30 mV where Cav1.2 enter the inactivated state predominantly from the closed resting state an almost 200 times higher concentration of the (−)-enantiomer was required to inactivate the channels by a similar extent as by (+)-isradipine (IC50,(+)-isr.=72±5 nM vs IC50,(−)-isr.=13±5 μM) (Figure 2B). This corresponds to the stereoselective inhibition of peak current by the two enantiomers (IC50,(+)-isr.=327±41 nM vs IC50,(−)-isr.=34±8 μM). A correlation between resting channel block (at −80 mV, Figure 1A) and isradipine-induced inactivation at −30 mV (Figure 2B) supports the hypothesis that the two block components are closely interrelated (Figure 5). It is tempting to speculate that the two isradipine enantiomers bind to resting channels with different affinities and enhance voltage-dependent inactivation during subsequent membrane depolarizations. Such a scenario is in line with the results of our preceding study demonstrating that (+)-isradipine promotes inactivation in Cav1.2 without necessarily binding with high affinity to an inactivated channel conformation (Berjukow et al., 2000).
Open channel block or drug-induced inactivation?
At more positive voltages where Cav1.2 inactivate predominantly via the open state drug-induced changes in the current decay are more difficult to interpret. Some authors believe that DHP-effects on IBa decay reflect predominantly the rate of open channel block (Lacinova & Hofmann, 1998; Handrock et al., 1999) while others interpret the kinetics of drug-induced acceleration of the Ca2+ channel current decay as enhanced inactivation (Berjukow et al., 2000). We have, therefore, compared the concentration dependencies of (+)- and (−)-isradipine effects on the IBa decay and corresponding drug-induced inactivation at −30 mV where channel inactivation occurs predominantly from the resting state.
(+)-Isradipine-induced inactivation at −30 mV (IC50=72±2 nM, n=3) and the corresponding acceleration of the current decay at 20 mV (IC50=79±18 nM, n=3) displayed similar concentration dependencies (Figures 1D and 2B). At least for the high affinity (+)-enantiomer this finding supports the view that the drug effects at both potentials (−30 and 20 mV) reflect drug-induced transitions to an inactivated state (see Figures 1D, 2A and 3B) and not an open channel block.
This hypothesis is also indirectly supported by experiments of Lacinova & Hofmann (1998), indicating that open channel block by isradipine requires two orders of magnitude higher concentrations of isradipine than drug-induced inactivation.
However, a comparison of the effects of (+)- and (−)-isradipine on the IBa decay of α1L/α2-δ/β1a and V1504A at 20 mV revealed a loss in enantioselectivity. At this potential both enantiomers were almost equally efficient in accelerating the calcium channel current decay (Figures 1D and 3B).
The most interesting finding of our study was, however, the prominent voltage-dependency of (+)- and (−)-isradipine effects on the current decay. This result provides for the first time convincing evidence that the isradipine effects on the IBa decay reflect voltage-dependent state transitions to an inactivated channel conformation and not an open channel block (Figure 4). Making use of mutant V1504A we were able to analyse the kinetics of (+)- and (−)-isradipine-induced changes in IBa kinetics during 30 s depolarizing test pulses to different voltages. In the absence of drug IBa of V1504A decayed at a slow rate that could be approximated by fitting a single exponential function (τinact.=22 s at −10 mV and τinact.=13 s at 20 mV). (+)- and (−)-isradipine (1 μM) induced an additional voltage-dependent component in the IBa decay with a time constant τinact.,(−)-isr.=1.5±0.2 s and τinact.,(+)-isr.=1.4±0.1 s at −25 mV and τinact.,(−)-isr.=0.33±0.03 s and τinact.,(+)-isr.=0.29±0.04 s at 20 mV. A comparison with intrinsic fast inactivation of α1L/α2-δ/β1a revealed an almost identical voltage-dependence (Figure 4).
The (+)-isradipine-induced acceleration of the IBa decay observed in this and our previous study (Berjukow et al., 2000, Figures 1, 3 and 4B) is in contrast to results of Handrock et al. (1999) who observed similar effects only in the presence of high concentrations of the (−)-enantiomer. Handrock et al. (1999) considered the possibility that the lack of effect of (+)-isradipine in their study could be due to the strong block of Ipeak by the (+)-enantiomer, which might have masked the effect on the current decay. The lower DHP-sensitivity of Cav1.2 expressed in Xenopus oocytes (i.e. less pronounced peak current inhibition compared to the more highly sensitive mammalian expression system used by Handrock et al., 1999) apparently provided more optimal conditions to analyse drug effects on current kinetics.
Drug-induced and intrinsic determinants of Cav1.2 inactivation
Our finding that α1L/α2-δ/β1a recover faster from (−)- than from (+)-isradipine-induced inactivation and the observation that impaired fast voltage-dependent inactivation of mutant V1504A is associated with an even faster recovery from (+)- and (−)-isradipine-induced inactivation provided important new insights into the molecular mechanism of DHP action. These data suggest that the stability of the inactivated states induced by the two enantiomers is crucially dependent on determinants of intrinsic fast voltage-dependent inactivation (i.e. V1504). The faster IBa recovery from (−)-isradipine-induced inactivation (compared to (+)-isradipine, Figures 2 and 5) illustrates the different efficiency of the two enantiomers to stabilize the drug-induced channel conformation (Figure 5).
Taken together, our data support the hypothesis that the drug-induced acceleration in the current decay at depolarized voltages (−25–+20 mV) reflect the kinetics of (+)- and (−)-isradipine-induced inactivation rather than open channel block. The differences in repriming kinetics in the presence of the two enantiomers apparently characterize the different stabilities of the drug-induced inactivated channel conformations. Similarities between the (+)- and (−)-isradipine-induced conformational changes in Cav1.2 were highlighted by the almost identical onset kinetics (Figure 2A).
A distinction between DHP-effects on fast and slow inactivation of Ca2+ channels in further studies appears to be essential for a deeper understanding of their interaction with the Cav1.2 molecule. Our present results support a model where (+)- and (−)-isradipine promote fast voltage-dependent inactivation, although with different efficiency (Figure 6).
Figure 6.
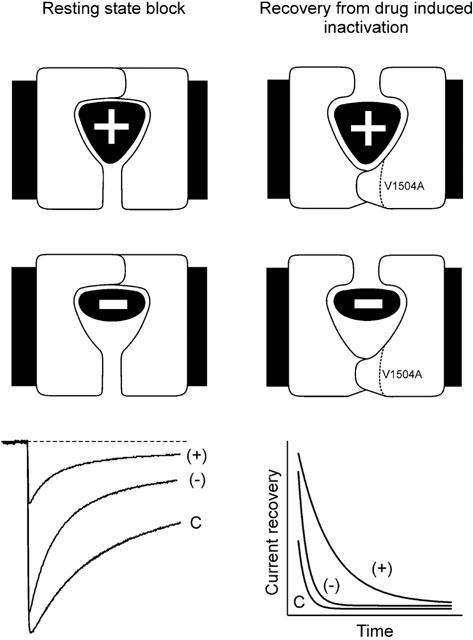
The two isradipine enantiomers differentially affect the stability of the drug-induced channel conformation. The scheme summarizes the principle findings of the present study. The left column illustrates the stronger tonic block of construct α1L/α2-δ/β1a by (+)- compared to the (−)-enantiomer (illustrated by different fit of the two components in a schematic binding pocket). Superimposed currents in control (c), 1 μM (+)- and 1 μM (−)-isradipine (lower panel) are from Figure 1. The scheme on the right illustrates the lower stability of the (−)-isradipine-induced inactivated state in constructs α1L/α2-δ/β1a and V1504A (Figure 5). The (−)-enantiomer is suggested to induce an inactivated state that is less stable than the channel conformation induced by (+)-isradipine. The experimental evidence for this hypothesis is illustrated in the lower panel summarizing the slower recovery of α1L/α2-δ/β1a from (+)- than from (−)-isradipine-induced inactivation (data from Figure 5).
Acknowledgments
We thank Stanislav Sokolov for valuable comments on the manuscript, Dr A. Schwartz for providing the α1C-a and α2-δ cDNA and E. Margreiter for expert technical assistance. This work was supported by Fonds zur Förderung der Wissenschaftlichen Forschung Grant 12649-MED (S. Hering), Grant 12828-MED (S. Hering), a grant of the Else-Kröner-Fresenius Stiftung and a grant from the Austrian National Bank (S. Hering).
Abbreviations
- DHP
1,4-dihydropyridines
- IBa
Ba2+ current
References
- BEAN B.P. Nitrendipine block of cardiac calcium channels: high-affinity binding to the inactivated state. Proc. Natl. Acad. Sci. U.S.A. 1984;81:6388–6392. doi: 10.1073/pnas.81.20.6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERJUKOW S., DORING F., FROSCHMAYR M., GRABNER M., GLOSSMANN H., HERING S. Endogenous calcium channels in human embryonic kidney (HEK293) cells. Br. J. Pharmacol. 1996;118:748–754. doi: 10.1111/j.1476-5381.1996.tb15463.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERJUKOW S., GAPP F., ACZEL S., SINNEGGER M.J., MITTERDORFER J., GLOSSMANN H., HERING S. Sequence differences between alpha1C and alpha1S Ca2+ channel subunits reveal structural determinants of a guarded and modulated benzothiazepine receptor. J. Biol. Chem. 1999;274:6154–6160. doi: 10.1074/jbc.274.10.6154. [DOI] [PubMed] [Google Scholar]
- BERJUKOW S., MARKSTEINER R., GAPP F., SINNEGGER M.J., HERING S. Molecular mechanism of calcium channel block by isradipine. Role of a drug induced inactivated channel conformation. J. Biol. Chem. 2000;275:22114–22120. doi: 10.1074/jbc.M908836199. [DOI] [PubMed] [Google Scholar]
- BREHM P., ECKERT R. Calcium entry leads to inactivation of calcium channel in Paramecium. Science. 1978;202:1203–1206. doi: 10.1126/science.103199. [DOI] [PubMed] [Google Scholar]
- CATTERALL W.A. Molecular properties of a superfamily of plasma-membrane cation channels. Curr. Opin. Cell. Biol. 1994;6:607–615. doi: 10.1016/0955-0674(94)90083-3. [DOI] [PubMed] [Google Scholar]
- ERTEL E.A., CAMPBELL K.P., HARPOLD M.M., HOFMANN F., MORI Y., PEREZ-REYES E., SCHWARTZ A., SNUTCH T.P., TANABE T., BIRNBAUMER L., TSIEN R.W., CATTERALL W.A. Nomenclature of voltage-gated calcium channels. Neuron. 2000;25:533–535. doi: 10.1016/s0896-6273(00)81057-0. [DOI] [PubMed] [Google Scholar]
- FRISHMAN W.H., MICHAELSON M.D. Use of calcium antagonists in patients with ischemic heart disease and systemic hypertension. Am. J. Cardiol. 1997;79:33–38. doi: 10.1016/s0002-9149(97)00270-1. [DOI] [PubMed] [Google Scholar]
- GLOSSMANN H., FERRY D.R. Assay for calcium channels. Methods Enzymol. 1985;109:513–550. doi: 10.1016/0076-6879(85)09112-1. [DOI] [PubMed] [Google Scholar]
- GRABNER M., WANG Z., HERING S., STRIESSNIG J., GLOSSMANN H. Transfer of 1,4-dihydropyridine sensitivity from L-type to class A (BI) calcium channels. Neuron. 1996;16:207–218. doi: 10.1016/s0896-6273(00)80037-9. [DOI] [PubMed] [Google Scholar]
- HANDROCK R., RAO-SCHYMANSKI R., KLUGBAUER N., HOFMANN F., HERZIG S. Dihydropyridine enantiomers block recombinant L-type Ca2+ channels by two different mechanisms. J. Physiol. 1999;521:31–42. doi: 10.1111/j.1469-7793.1999.00031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HERING S., BERJUKOW S., ACZÉL S., TIMIN E.N. Ca2+ channel block and inactivation: common molecular determinants. Trends Pharmacol. Sci. 1998;19:439–443. doi: 10.1016/s0165-6147(98)01258-9. [DOI] [PubMed] [Google Scholar]
- HERING S., BERJUKOW S., SOKOLOV S., MARKSTEINER R., WEIß R.G., KRAUS R., TIMIN E.N. Molecular determinants of inactivation in voltage-gated Ca2+ channels. J. Physiol. 2000;528.2:237–249. doi: 10.1111/j.1469-7793.2000.t01-1-00237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HILLE B. Ionic channels in excitable membranes. Current problems and biophysical approaches. Biophys. J. 1978;22:283–294. doi: 10.1016/S0006-3495(78)85489-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ITO H., KLUGBAUER N., HOFMANN F. Transfer of the high affinity dihydropyridine sensitivity from L-type to non-L-type calcium channel. Mol. Pharm. 1997;52:735–740. doi: 10.1124/mol.52.4.735. [DOI] [PubMed] [Google Scholar]
- LACINOVA L., HOFMANN F. Isradipine interacts with the open state of the L-type calcium channel at high concentrations. Receptors Channels. 1998;6:153–164. [PubMed] [Google Scholar]
- LEE A., WONG S.T., GALLAGHER D., LI B., STORM D.R., SCHEUER T., CATTERALL W.A. Ca2+/calmodulin binds to and modulates P/Q-type calcium channels. Nature. 1999;399:155–159. doi: 10.1038/20194. [DOI] [PubMed] [Google Scholar]
- LUND-JOHANSEN P. Cardiac effects of isradipine in patients with hypertension. Am. J. Hypertens. 1993;6:294S–299S. doi: 10.1093/ajh/6.7.294s. [DOI] [PubMed] [Google Scholar]
- MIKAMI A., IMOTO K., TANABE T., NIIDOME T., MORI Y., TAKESHIMA H., NARUMIYA S., NUMA S. Primary structure and functional expression of the cardiac dihydropyridine-sensitive calcium channel. Nature. 1989;340:230–233. doi: 10.1038/340230a0. [DOI] [PubMed] [Google Scholar]
- PETERSON B.Z., JOHNSON B.D., HOCKERMAN G.H., ACHESON M., SCHEUER T., CATTERALL W.A. Analysis of the dihydropyridine receptor site of L-type calcium channels by alanine-scanning mutagenesis. J. Biol. Chem. 1997;272:18752–18758. doi: 10.1074/jbc.272.30.18752. [DOI] [PubMed] [Google Scholar]
- PETERSON B.Z., TANADA T.N., CATTERALL W.A. Molecular determinants of high affinity dihydropyridine binding in L-type calcium channels. J. Biol. Chem. 1996;271:5293–5296. doi: 10.1074/jbc.271.10.5293. [DOI] [PubMed] [Google Scholar]
- SANGUINETTI M.C., KASS R.S. Voltage-dependent block of calcium channel current in the calf cardiac Purkinje fiber by dihydropyridine calcium channel antagonists. Circ. Res. 1984;55:336–348. doi: 10.1161/01.res.55.3.336. [DOI] [PubMed] [Google Scholar]
- SINNEGGER M.J., WANG Z., GRABNER M., HERING S., STRIESSNIG J., GLOSSMANN H., MITTERDORFER J. Nine L-type amino acid residues confer full 1,4-dihydropyridine sensitivity to the neuronal calcium channel alpha1A subunit. Role of L-type Met1188. J. Biol. Chem. 1997;272:27686–27693. doi: 10.1074/jbc.272.44.27686. [DOI] [PubMed] [Google Scholar]
- STRIESSNIG J., GRABNER M., MITTERDORFER J., HERING S., SINNEGGER M.J., GLOSSMANN H. Structural basis of drug binding to L Ca2+ channels. Trends Pharmacol. Sci. 1998;19:108–115. doi: 10.1016/s0165-6147(98)01171-7. [DOI] [PubMed] [Google Scholar]
- TANG S., YATANI A., BAHINSKI A., MORI Y., SCHWARTZ A. Molecular localization of regions in the L-type calcium channel critical for dihydropyridine action. Neuron. 1993;11:1013–1021. doi: 10.1016/0896-6273(93)90215-d. [DOI] [PubMed] [Google Scholar]
- WALKER D., DE WAARD M. Subunit interaction sites in voltage-dependent Ca2+ channels: role in channel function. Trends Neurosci. 1998;21:148–154. doi: 10.1016/s0166-2236(97)01200-9. [DOI] [PubMed] [Google Scholar]