

Abstract
The aim of the present study was to investigate the effects of 2,5-di-t-butyl-1,4-benzohydroquinone (BHQ), an inhibitor of the sarco-endoplasmic reticulum Ca2+-ATPase (SERCA), on the whole-cell voltage-dependent L-type Ca2+ current (ICa(L)) of freshly isolated smooth muscle cells from the rat tail artery using the patch-clamp technique.
BHQ, added to the perfusion solution, reduced ICa(L) in a concentration- (IC50=66.7 μM) and voltage-dependent manner. This inhibition was only partially reversible.
BHQ shifted the voltage dependence of the steady-state inactivation curve to more negative potentials by 7 mV in the mid-potential of the curve, without affecting the activation curve as well as the time course of ICa(L) inactivation.
Preincubation of the cells either with 10 μM cyclopiazonic acid, a SERCA inhibitor, or with 3 mM diethyldithiocarbamate, an inhibitor of intracellular superoxide dismutase (SOD), did not modify BHQ inhibition of ICa(L). On the contrary, this effect was no longer evident when SOD (250 u ml−1) was added to the perfusion medium.
Either in the presence or in the absence of cells, BHQ gave rise to superoxide anion formation, which was markedly inhibited by the addition of SOD.
These results indicate that, at micromolar concentrations, BHQ inhibits vascular ICa(L) by giving rise to the formation of superoxide anion which in turn impairs the channel function.
Keywords: BHQ, superoxide anion, Ca2+ channel blocker, rat tail artery smooth muscle, whole-cell L-type Ca2+ current
Introduction
2,5-Di-t-butyl-1,4-benzohydroquinone (BHQ) has been described as a rather selective sarco-endoplasmic reticulum Ca2+-ATPase (SERCA) inhibitor (Moore et al., 1987) and has been extensively used as a pharmacological tool to study the role of intracellular Ca2+ stores in response to various agonists of – inter alias – smooth muscle contraction. However, BHQ is not very specific, as it hits other sites of action besides SERCA. BHQ was shown, in fact, to reduce passive Ca2+ leakage from internal stores of permeabilized A7r5 vascular smooth muscle cells (Missiaen et al., 1992) and to inhibit plasma membrane Ca2+ influx in parotid acinar cells (Foskett & Wong, 1992), rat thymic lymphocytes (Mason et al., 1991) and GH3 pituitary cells (Nelson et al., 1994). Previous studies performed in this laboratory have shown that BHQ affects both endothelial and smooth muscle functions in rat aorta rings in vitro. BHQ induced endothelium-dependent relaxation by stimulating the NO synthase pathway, via activation of Ca2+ influx through Ni2+-sensitive Ca2+ channels in the endothelial cells (Fusi et al., 1999). Moreover, it exhibited pleiotypic effects on rings deprived of endothelium (Fusi et al., 1998; 2000) by inducing both a myotonic response, dependent on the activation of Ca2+ influx via a Ni2+-sensitive pathway, and a myolytic effect, possibly dependent on either the depletion of intracellular Ca2+ stores or antagonism of Ca2+ entry via L-type Ca2+ channels (Fusi et al., 1998). The latter effect was similar to that of the dihydropyridine Ca2+ antagonists since BHQ inhibition of Ca2+-induced contraction, at high K+ concentration, was fully antagonized by Bay K 8644 as well as by increasing the extracellular Ca2+ concentration, while it was augmented by increasing membrane depolarization.
Reactive oxygen species (ROS) are considered as multifunctional molecules, from one side being important for the signal transduction in cardiovascular cells (Wolin, 2000), from the other exerting deleterious effects on ion transport proteins that operate transmembrane signal transduction (Kourie, 1998), such as Ca2+ ion channels (Guerra et al., 1996). Superoxide anion is a negatively charged free radical that undergoes rather selective chemical reactions with the components of biological systems. Since BHQ, in aqueous solution, very easily undergoes spontaneous oxidation giving rise to the formation of superoxide anion (Fusi et al., 1999) we thought that the purported inhibition of L-type Ca2+ channels was a consequence of the damaging of L-type Ca2+ channels operated by this radical. The aim of the present work was to investigate the effects of BHQ on whole cell L-type Ca2+ currents (ICa(L)) in rat tail artery isolated myocytes. The present data show that BHQ inhibits ICa(L). This effect is ascribable to the generation of superoxide anion rather than to a direct action of BHQ on the channel components.
Methods
Cell isolation procedure
Smooth muscle cells were freshly isolated by collagenase treatment of tail artery obtained from male rats (350–450 g), previously anaesthetized with a mixture of Ketavet® (Gellini, Italy) and Rompum® (Bayer, Germany), decapitated and exsanguinated. Tail was immediately removed, cleaned of skin and placed in low Ca2+ (0.1 mM) physiological salt solution (PSS) (see below for composition) at 4°C. The main tail artery was dissected free, removing the connective tissue. A small piece (1 cm) was cut out at about 2–3 cm from the base of the tail and placed in low Ca2+ PSS at 4°C, for 30–40 min. The vessel was then transferred into 1 ml of 0.1 mM Ca2+ PSS containing 1 mg ml−1 collagenase (type XI), 1 mg ml−1 soybean trypsin inhibitor and 1 mg ml−1 BSA, gently bubbled with a 95% O2–5% CO2 gas mixture. After 40–50 min of incubation at 37°C, tissue was carefully washed with Ca2+-free PSS. Individual smooth muscle cells were then obtained by gentle agitation of tissue with a Pasteur pipette in 1 ml Ca2+-free PSS, until the solution became cloudy. The cells were used for experiments within 10 h after isolation. During this time, they were stored at 4°C in 0.1 mM PSS containing BSA. An aliquot of the cell suspension (5–10 μl) was transferred to a small recording chamber (250 μl) mounted on the stage of an inverted phase-contrast microscope (TE300, Nikon, Japan) and monitored with a video camera (JVC TK-1280E). Cells freshly isolated exhibited an ellipsoid form and were able to respond to phenylephrine that normally causes contraction of the vessel in situ or in vitro. The morphological changes observed in contracting cells (10 to 20% shortening with formation of membranous evaginations) appeared similar to those described by Ives et al. (1978). After adhesion of the cells to the glass bottom of the chamber (10 min), they were continuously superfused by means of a peristaltic pump (LKB 2132), at a flow rate of 800 μl min−1, with 5 mM Ca2+ PSS. Electrophysiological responses were tested at room temperature (22–24°C) only in cells that were phase dense.
Whole-cell patch clamp recording
The whole-cell configuration of the patch-clamp technique (Hamill et al., 1981) was employed to voltage-clamp the smooth muscle cells. Recording electrodes were pulled from borosilicate glass capillaries (WPI, Berlin, Germany) and fire polished to give a pipette resistance of 2–5 MΩ when filled with the internal solution. A low-noise, high-performance Axopatch 200B (Axon Instruments, U.S.A.) patch-clamp amplifier, driven by an IBM computer in conjunction with an A/D, D/A board (DigiData 1200 A/B series interface, Axon Instruments, U.S.A.) was used to generate and apply voltage pulses to the clamped cells and to record the corresponding membrane currents. Current signals were low-pass filtered at 1 kHz and digitized at 3 kHz before being stored on the computer hard disk. Long-lasting nifedipine-blockable inward currents through L-type Ca2+ channels, in 5 mM Ca2+-containing PSS, were measured over a range of test potentials (250 ms) from −55 to 45 mV from a holding potential (Vh) of −80 or −50 mV. Data were obtained after the current amplitude had been stabilized (usually 8–10 min after the whole-cell configuration was obtained). The ICa(L) did not run down over the next 50 to 60 min, under these conditions.
Steady-state inactivation curves were obtained using the double-pulse protocol. After various levels of the conditioning potential were applied for 5 s, followed by a short (5 ms) return to the holding potential, a test pulse (250 ms) to 5 mV was delivered to evoke the current.
Activation curves were derived from the current-voltage relationships (see Figure 1). Conductance (G) was calculated from the equation G=ICa(Em−ECa), where ICa is the peak current elicited by depolarizing test pulses to the various potentials and ECa is the reversal potential (obtained from the extrapolated current–voltage curves in Figure 1). Gmax is the maximal Ca2+ conductance (calculated at potentials above 10 mV). The points for G/Gmax were plotted against the membrane potential as a relative amplitude.
Figure 1.
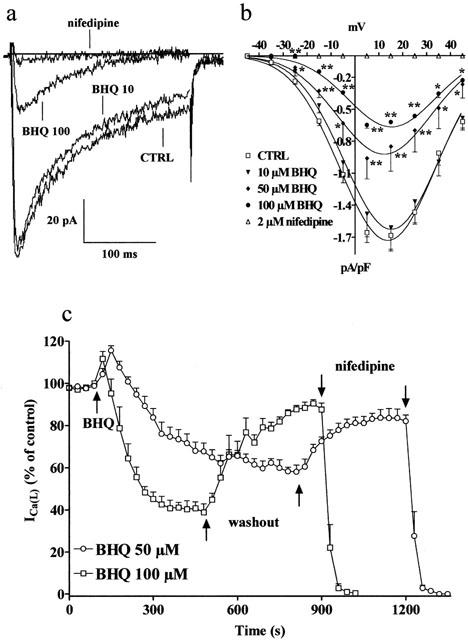
Inhibitory action of BHQ on ICa(L). (a) Original recordings of whole-cell ICa(L) in rat tail artery myocytes elicited with 250 ms depolarizing voltage-clamp pulses from a Vh of −50 mV to test potentials of 5 mV measured in the absence (CTRL) or presence of BHQ (10 and 100 μM). ICa(L) suppression by 2 μM nifedipine is also shown. (b) Current-voltage relationships constructed before the addition of BHQ (CTRL) and in the presence of 10, 50 and 100 μM BHQ or 2 μM nifedipine. Data points are means±s.e.mean of three to 11 cells (n=3–11). *P<0.05, **P<0.01, Dunnett's post test. (c) Time course of ICa(L) inhibition induced by BHQ. BHQ (50 or 100 μM) was applied, at the time indicated by the arrow, by superfusion and peak currents were recorded during a typical depolarization from −50 to 5 mV applied every 30 s and subsequently normalized according to the current recorded just before BHQ application. Wash-out of drug allows for partial recovery of inhibition. Data points are means±s.e.mean of six cells (n=5).
Potassium current was blocked with 30 mM tetraethylammonium (TEA) in the PSS and Cs+ in the internal solution. Values were corrected for the leak using 2 μM nifedipine, which was assumed to block completely ICa(L).
Superoxide anion detection
Superoxide anion was detected spectrophotometrically by a modification of the method described by McCord & Fridovich (1969). In brief, 30 μM cytochrome c and 50 μM BHQ, in 2 ml of 5 mM Ca2+ PSS, were mixed to polystyrene cuvettes of a spectrophotometer (Shimadzu UV-160) at room temperature (22–24°C). Superoxide anion generation was assessed by measuring the increase in absorbance at 550 nm associated with the reduction of cytochrome c. The increase in absorbance was recorded continuously for 40 min.
Solutions and chemicals
Ca2+-free PSS contained (in mM): NaCl 110; KCl 5.6; HEPES 10; taurine 20; glucose 20; MgCl2 1.2; Na-pyruvate 5 (pH 7.4). ICa was always recorded in 5 mM Ca2+-containing PSS: this concentration was shown to cause maximal peak current in arterial smooth muscle cells (Bolton et al., 1988).
The internal solution (pCa 8.4) consisted of (in mM): CsCl 100; HEPES 10; EGTA 11; MgCl2 2; CaCl2 1; Na-pyruvate 5; succinic acid 5; oxalacetic acid 5; Na2ATP 3 and phosphocreatine 5; pH was adjusted to 7.4 with CsOH.
The osmolarity of PSS was adjusted to 330 mosmol, that of internal solution to 300 mosmol (Stansfeld & Mathie, 1993) with use of an osmometer (Osmostat OM 6020, Menarini Diagnostics, Italy).
The chemicals used were: collagenase (type XI), TEA, diethyldithiocarbamate (DETCA), superoxide dismutase (SOD), cyclopiazonic acid (CPA), BHQ, and nifedipine (Sigma Chimica, Italy). BHQ and CPA, dissolved directly in dimethylsulphoxide (DMSO), and nifedipine, dissolved in ethanol, were diluted at least 1000 times in the PSS, before use. The resulting concentrations of DMSO and ethanol (below 0.1%) did not alter the currents (data not shown). Final drug concentrations are stated in the text.
After control measurements, each cell was exposed to drugs by perfusing the experimental chamber with a drug-containing PSS.
Statistical analysis
Acquisition and analysis of data were accomplished using pClamp 8.0.1.01 software (Axon Instruments, U.S.A.) and GraphPad Prism version 3.02 (GraphPad Software, U.S.A.). Data are reported as means±s.e.mean; n is the number of animals (indicated in parentheses). Statistical analysis and determinations of significance with ANOVA (followed by Dunnett's post tests) or Student's t-test for unpaired and paired samples, as appropriate, were performed using GraphPad InStat version 3.02 (GraphPad Software, U.S.A.). In all comparisons, P<0.05 was considered significant.
The current–voltage relationships were constructed using the peak values (leakage corrected) from the original traces of currents.
Results
Effects of BHQ on ICa(L)
In rat tail artery myocytes, starting from Vh of either −80 or −50 mV, a nifedipine-sensitive ICa(L) was the predominant calcium current. Its amplitude was maximal around 15 mV, with threshold activation occurring at about −35 mV. BHQ significantly inhibited, in a concentration-dependent manner, the peak inward current at all potentials tested (Figure 1a,b); this inhibition, however, was not accompanied by a significant shift of the current–voltage curve along the voltage axis. The concentration-response curve for BHQ (data not shown) gave an IC50 of 66.7±2.1 μM. Figure 1c shows the time course of the effects of BHQ on the current. After ICa(L) had reached steady values, BHQ (50 or 100 μM) was added to the bath solution. This produced a transient enhancement followed by a gradual decrease of the current (over 4–10 min); the rate of inhibition increased by increasing the concentration of BHQ. This decrease was only partially reversible (on the time scale of the recordings) upon wash-out (up to 84% of control current). Furthermore, the transient stimulation of ICa(L) observed after BHQ addition was no more evident in the presence of 1 μM nifedipine (data not shown).
ECa calculated from the extrapolated current–voltage curves (Figure 1b) obtained under control conditions (58.9±2.3 mV, n=5) was not modified by 50 μM BHQ (55.1±3.5 mV, n=5). BHQ inhibited ICa(L) to a degree that depended upon the holding potential applied (Figure 2). In fact, the inhibition of the current generated by a depolarizing voltage-clamp pulse from Vh of −80 mV to test potential of 5 mV was 36.5±5.7% (n=6) whereas that from a Vh of −50 mV was 51.8±6.9% (n=5).
Figure 2.
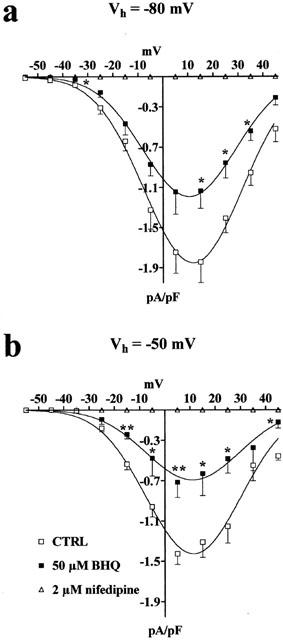
Effect of changes in Vh on BHQ-induced inhibition of ICa(L). (a,b) Current–voltage relationships obtained in the absence (CTRL) or presence of 50 μM BHQ. ICa(L) suppression by 2 μM nifedipine is also shown. Data points are means±s.e.mean of five to seven cells (n=5–7). *P<0.05, **P<0.01, Student's t-test for unpaired samples.
Effect of CPA on BHQ inhibition of ICa(L)
To assess whether sarcoplasmic reticulum was involved in BHQ inhibition of ICa(L), cells were treated with the SERCA inhibitor CPA (Seidler et al., 1989), before BHQ addition. As shown in Figure 3, 10 μM CPA significantly reduced ICa(L) and the extent of this inhibition was directly correlated to the holding potential employed. In fact, the inhibition of the current generated by a depolarizing voltage-clamp pulse from Vh of −80 mV to test potential of 5 mV was 24.3% whereas that from Vh of −50 mV was 41.0%. When BHQ (up to 100 μM) was added, the transient stimulation of ICa(L) (see above) was no more evident (data not shown). Furthermore, 50 μM BHQ reduced ICa(L) at both Vh of −80 and −50 mV by 21.5±7.5% (n=4) and 61.8±7.6% (n=3), respectively (as compared to CPA alone, taken at 100%), which were not significantly different from the inhibition measured in the presence of BHQ alone (see above).
Figure 3.
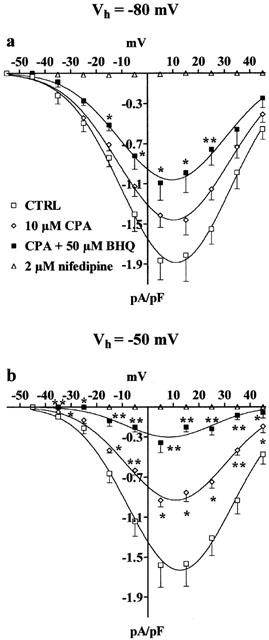
Effect of CPA on BHQ-induced inhibition of ICa(L). Current–voltage relationships obtained from Vh of −80 (a) or −50 mV (b) under control conditions (CTRL), in the presence of 10 μM CPA and in the presence of CPA plus 50 μM BHQ. ICa(L) suppression by 2 μM nifedipine is also shown. Data points are means±s.e.mean of three to five cells (n=3–5). *P<0.05, **P<0.01, Dunnett's post test.
Effect of SOD and DETCA on BHQ inhibition of ICa(L)
To clarify the possible role of superoxide anion possibly generated by BHQ, under the present experimental conditions, in BHQ inhibition of ICa(L), in some experiments exogenous SOD was added. The addition of SOD (250 u ml−1) produced a modest increase of ICa(L) (Figure 4b, d). The further addition of 50 μM BHQ did not significantly affect the current–voltage relationship (Figure 4c, d). The enhancement of the current promoted by BHQ addition, however, was still transiently evident after SOD addition (data not shown).
Figure 4.
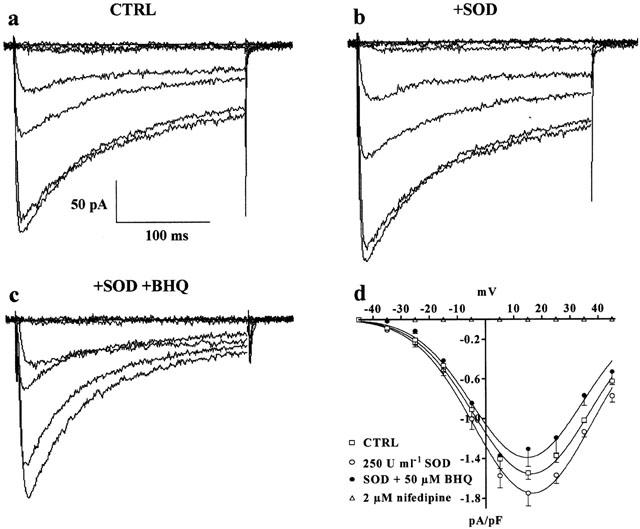
Effect of SOD on BHQ-induced inhibition of ICa(L). (a–c) Representative recordings illustrating whole-cell ICa(L). The current traces, elicited with 250 ms depolarizing voltage-clamp pulses from Vh of −50 mV to test potentials of −75 to 45 mV in increments of 20 mV, were recorded in the absence (CTRL) (a) or presence of 250 u ml−1 SOD (b) and SOD plus 50 μM BHQ (c). (d) Current–voltage relationships obtained in the absence (CTRL) or presence of SOD and SOD plus BHQ. ICa(L) suppression by 2 μM nifedipine is also shown. Data points are means±s.e.mean of four cells (n=3).
To test whether endogenous SOD could prevent, at least in part, BHQ effect on ICa(L), DETCA, an inhibitor of intracellular superoxide dismutase, was used. Incubation of the cells with 3 mM DETCA for up to 25 min reduced ICa(L), elicited by depolarization pulse to 5 mV from Vh −50 mV, to 75.0±2.3% of control values (data not shown). The subsequent addition of 50 μM BHQ caused a further reduction of ICa(L) by 58.5±2.2% (n=4) of that measured in the presence of DETCA (taken as 100%). This inhibition was comparable to that observed with BHQ alone (see above).
Superoxide anion formation from BHQ
When added to the perfusion medium, either in the absence or in the presence of cells, BHQ gave rise to the formation of superoxide anion, as shown by the reduction of cytochrome c (see Methods section). At the maximum concentration of BHQ tested (50 μM), this was almost linear with time and averaged 519.1±28.9 pmols min−1. In the presence of 500 u ml−1 SOD, the amount of superoxide anion generated by BHQ was undetectable with the method used.
Effect of BHQ on steady-state inactivation and activation curves for ICa(L)
The voltage dependence of BHQ inhibition was analysed by determining the steady-state inactivation curve of ICa(L). BHQ significantly shifted the steady-state inactivation curve to more negative potentials (Figure 5a,b). However, when SOD (250 u ml−1) was present in the perfusion PSS, this shift was no more evident. The 50% inactivation potentials evaluated by Boltzmann fitting were −24.31±1.27 mV (control, n=7), −23.39±1.03 mV (SOD, n=6), −32.27±1.68 mV (BHQ, n=4, P<0.01 vs control, Dunnett's post test) and −28.15±1.24 (BHQ plus SOD, n=6). On the contrary, the slope factor (−7.32±0.37 mV, control) was not affected by BHQ (−9.27±1.88 mV) as well as by SOD (−7.14±0.25 mV) and SOD plus BHQ (−7.30±0.24 mV). Noticeably, the blockade of ICa(L) by BHQ was still evident even at negative conditioning potentials. In fact, after conditioning pulses to −80 mV for 5 s, ICa(L) was still reduced to 64.4±11.0% by 50 μM BHQ (Figure 5b).
Figure 5.
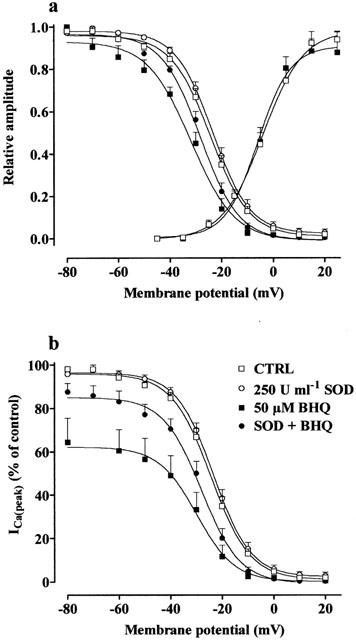
Effect of BHQ on activation and inactivation curves. Steady-state inactivation curves, obtained in the absence (CTRL) and presence of 50 μM BHQ, 250 u ml−1 SOD or BHQ plus SOD, were fitted to the Boltzmann equation. Peak current values were used. The steady-state inactivation curve was obtained using the double-pulse protocol (see Methods section). The current measured during the test pulse is plotted against membrane potential and expressed as relative amplitude (a) and in per cent of control (b). Activation curves were obtained from the current–voltage relationships of Figure 1 and fitted to the Boltzmann equation (see Methods section). Relative amplitude is plotted against membrane potential. Each point represents the means±s.e.mean of four to seven cells (n=3).
Activation curves, obtained from the current–voltage relationships of Figure 1 and fitted to the Boltzmann equation, are shown in Figure 5a. The 50% activation potential as well as the slope factor obtained from individual experiments (−2.26±2.96 and 8.41±1.14 mV, control, n=5) were not affected by 50 μM BHQ (−4.98±1.75 and 6.47±0.98 mV, n=5).
Effect of BHQ on ICa(L) inactivation rate
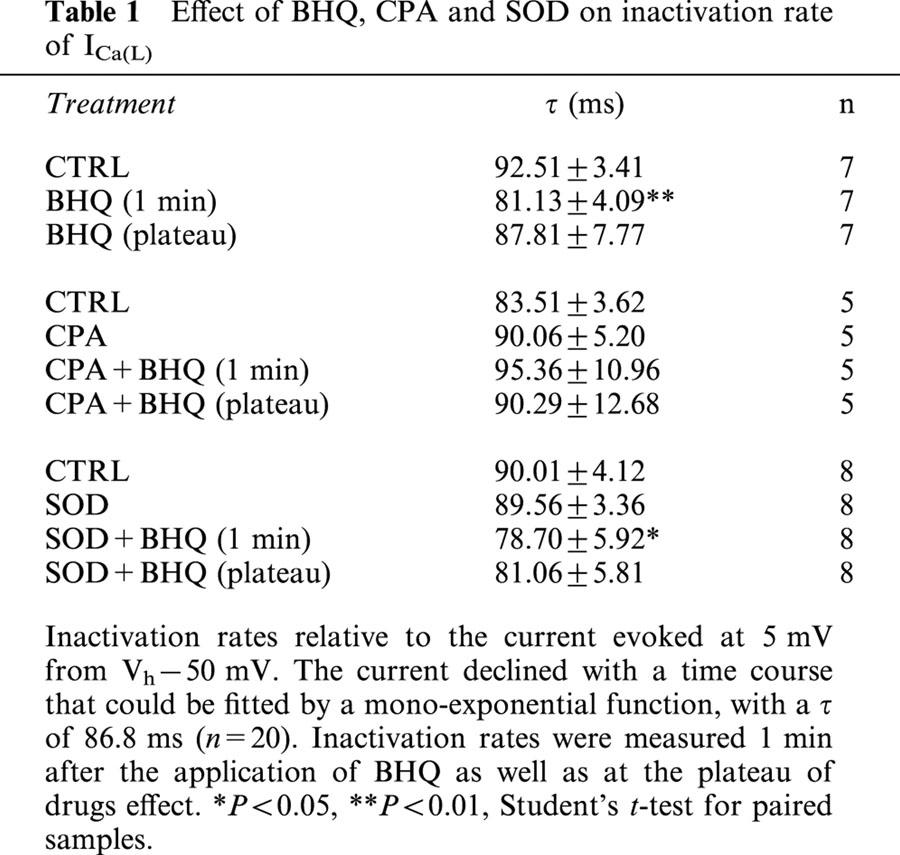
The effect of BHQ on the kinetic of ICa(L) inactivation was analysed under various experimental conditions, as summarized in Table 1. Inactivation rates were measured 1 min after the application of drug as well as at the plateau of BHQ inhibition. The current evoked at 5 mV from Vh −50 mV declined with a time course that could be fitted by a mono-exponential equation, with a τ of 86.8 ms. The addition to the perfusion medium of either CPA or SOD had no effects on the inactivation rate. Furthermore, BHQ significantly reduced τ immediately after application; this effect then slowly disappeared. This transient effect caused by BHQ was still observed in cells treated with SOD but not in cells treated with CPA.
Table 1.
Effect of BHQ, CPA and SOD on inactivation rate of ICa(L)
Discussion
BHQ and thapsigargin, but not CPA, three structurally unrelated inhibitors of SERCA (Nakamura et al., 1992; Seidler et al., 1989; Thastrup et al., 1990), have been shown to inhibit plasma membrane Ca2+ influx in various mammalian cell types (Nelson et al., 1994; Buryi et al., 1995; Foskett & Wong, 1992; Geiszt et al., 1995; Rossier et al., 1993). Among these, only the electrophysiological study supplied the direct evidence that BHQ impairs the function of L-type Ca2+ channels in GH3 pituitary cells (Nelson et al., 1994), acting either directly at the channel or exerting its effect without the requirement for diffusible cytoplasmic components. Furthermore, very recently, BHQ but not thapsigargin or CPA, was shown to block N-, P-, and Q- but not T-, L-, or R-type ICa in central and peripheral neurons (Scamps et al., 2000). The present investigation shows that BHQ inhibits ICa(L) in rat tail artery myocytes via generation of superoxide anion. This concept is supported by the evidence that BHQ-induced inhibition was reverted by the presence of SOD.
Hydroquinones undergo auto-oxidation (Marklund & Marklund, 1974) to form semi-quinone radicals and donate an electron to molecular oxygen, thereby giving rise to the superoxide anion. It has already been shown (Fusi et al., 1999) that BHQ, in aqueous solution, at physiological pH, gives rise to superoxide anion formation strongly inhibitable by the addition of SOD. This has been confirmed also in the present study.
Superoxide anion does not cross cell membrane, thus it can react only in the compartment where it is generated (Stamler, 1996). However, BHQ is a lipophilic, membrane permeable molecule, capable of generating superoxide anion at both sides of the plasmalemma. Therefore, superoxide anion might affect Ca2+ channel function from the inner and/or from the outer mouth of the channel. If this hypothesis is correct, one would expect BHQ antagonism either to be reversed by exogenous SOD or to be exacerbated by inhibition of endogenous (i.e. intracellular) SOD. Our results, however, confirmed only the first hypothesis, since inhibition of intracellular SOD activity with DETCA did not modify BHQ inhibition. On the other hand, exogenous SOD, which does not penetrate the cell, could inactivate BHQ-formed superoxide anion only extracellularly. Thus, superoxide anion formed extracellularly from BHQ seems to be responsible for BHQ effect on ICa(L).
The mechanism of ROS-induced modifications of ion transport pathways involves: (1) oxidation of sulfhydryl groups located in the channel protein; (2) peroxidation of membrane phospholipids; and (3) inhibition of membrane-bound regulatory enzymes and impairment of oxidative phosphorylation and ATP levels (Kourie, 1998). Several reports demonstrate that vascular ion channels are potentially controlled by multiple redox-linked mechanisms (Wolin, 2000) and raise the possibility of significant regional and/or tissue differences in redox-sensitive modulatory properties of L-type Ca2+ channels in the cardiovascular system (Campbell et al., 1996). In particular, ROS have been shown to decrease ICa as well as to reduce dihydropyridines binding sites in guinea-pig ventricular myocytes (Guerra et al., 1996). The latter phenomenon seems to involve both a direct effect on the channel (oxidation of sulfhydryl groups; Iesaki & Wolin, 2000) and lipoperoxidation. It has been shown that the incubation of liver microsomes with BHQ (up to 100 μM) for 1 h, did not result in a measurable loss of protein thiols (Moore et al., 1987). At present, it is not clear whether BHQ oxidises sulfhydryl groups of the channel in rat vascular myocytes or causes lipid peroxidation of its surrounding. We can only speculate that the production of superoxide anion may result in a modification of channel function (Wolin, 2000) which, however, can be recovered, almost completely, following washout. Lipid peroxidation is likely to be responsible for the irreversible component of BHQ-induced inhibition of ICa(L), although we can not rule out the possibility either that it could reflect channel rundown or that BHQ, owing to its high hydrophobicity, may not have been effectively removed by simple wash-out. Scamps et al. (2000) indicated the pore-forming α1-subunit as the site of action of BHQ. Interestingly, α1C subunit is also the target of oxidants which cause the inhibition of ICa(L) (Fearon et al., 1999). Therefore, it is conceivable that superoxide anion produced by BHQ might target the same subunit of L-type channels in vascular smooth muscle cells.
Scamps et al. (2000) observed that the onset of ICa inhibition by BHQ in neurological preparations was fast and complete within a few seconds. On the contrary, in smooth muscle cells, the rate of inhibition, which was concentration-dependent, reached its maximum effect only after 4–10-min incubation time (present study). Two hypotheses may be put forward to explain this difference in the kinetic of inhibition between neurones and smooth muscle cells. Firstly, this difference could be the consequence of the higher sensitivity of nerves towards oxidative stress as compared to muscles. In fact, the IC50 value obtained here was 2 fold that reported in nerves. Secondly, this effect may be related to the different pore-forming α1-subunit responsible for ICa(L) in smooth muscle (α1C; Lory et al., 1991) and in differentiated NG108-15 cells (α1D; Scamps et al., 2000).
BHQ inhibition of ICa(L) was voltage-dependent, as suggested both by the reduction of the inhibition following membrane potential change from −50 to −80 mV and by BHQ-induced shift of the steady-state inactivation curve to more negative potentials, leaving unaltered the sigmoid shape of the curve. Since this effect indicates the stabilization of Ca2+ channels in their inactivated state (see Bean, 1984), we can hypothesise that the main effect of superoxide anion on the channel is that of stabilizing it in the inactivated state. The voltage-dependence of BHQ blockade also indicates that superoxide anion might impair Ca2+ entry into cells that are depolarized (perhaps during anoxia or as a consequence of the hypoxic damage), while leaving unaffected the cells with normal, i.e. more negative resting potentials (Bean, 1989). Furthermore, the blockade of ICa(L) by BHQ was not abolished even at negative Vh (Wegener et al., 2000), indicating that blocked channels can not be restored to the available pool by hyperpolarization that normally permits drug release from the weakly binding closed-state (McDonald et al., 1994). Further experiments, however, are necessary in order to clarify this aspect.
Previous works (Nelson et al., 1994; Scamps et al., 2000) have suggested that the molecular mechanism of BHQ inhibition is not dependent on depletion of intracellular calcium store sites following SERCA inhibition (i.e. ICa(L) inhibition due to Ca2+-induced current inactivation arising from Ca2+ mobilization from internal stores). The present results further support this hypothesis, since preincubation of the cells with CPA did not antagonize BHQ inhibition of the current. Low et al. (1996) suggested that, in the rat tail artery, SR does not play a key role in the maintenance of vascular contractility, Ca2+ fluxes across the plasma membrane having a major role. However, the data presented here suggest that SR might modulate ICa(L) at least transiently. In fact, application of BHQ to the bath caused a transient enhancement of current amplitude as well as a transient decrease of current inactivation rate. Both effects are likely to be the consequence of SERCA inhibition by BHQ, because they were not observed in cells pre-treated with CPA. The increase in current amplitude, reported also for thapsigargin in GH3 pituitary cells (Nelson et al., 1994), reflected influx of extracellular Ca2+ possibly through voltage dependent L-type Ca2+ channels and not through non-selective cation channels, as observed with CPA in cultured bovine pulmonary arterial endothelial cells (Pasyk et al., 1995), since it was blocked by the presence of nifedipine. The decrease of τ might instead reflect Ca2+-induced inactivation of the channel following the elevation of cytosolic Ca2+ near the cell membrane as a consequence of SERCA inhibition by BHQ (vanBreemen et al., 1995). Furthermore, since the inhibition of ICa(L) due to both BHQ and CPA were additive, it is conceivable that these agents act through different mechanisms.
In conclusion, these results indicate that BHQ acts indirectly on voltage-dependent Ca2+ channels of rat tail artery myocytes, causing the inhibition of ICa(L). The mediator of such inhibition seems to be superoxide anion, which might impair the channel function from the extracellular space.
Acknowledgments
We wish to thank Dr Lara Vittori for the assistance in some experiments and Dr Luciana Volpi and Prof Alberto Auteri (Istituto di Semeiotica Medica, Facoltà di Medicina e Chirurgia, Università degli Studi di Siena) for help in the measurement of solutions osmolarity. This work was supported by MURST Cofin '98 and a grant from Ministero degli Affari Esteri (Rome, Italy) under Law 212 (26-2-1992).
Abbreviations
- BHQ
2,5-di-t-butyl-1,4-benzohydroquinone
- DETCA
diethyldithiocarbamate
- DMSO
dimethylsulphoxide
- CPA
cyclopiazonic acid
- G
conductance
- ICa(L)
L-type Ca2+ current
- PSS
physiological salt solution
- ROS
reactive oxygen species
- SERCA
sarco-endoplasmic reticulum Ca2+-ATPase
- SOD
superoxide dismutase
- TEA
tetraethylammonium
- Vh
holding potential
References
- BEAN B.P. Nitrendipine block of cardiac calcium channels: high-affinity binding to the inactivated stat. Proc. Natl. Acad. Sci. U.S.A. 1984;81:6388–6392. doi: 10.1073/pnas.81.20.6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BEAN B.P. Classes of calcium channels in vertebrate cells. Annu. Rev. Physiol. 1989;51:367–384. doi: 10.1146/annurev.ph.51.030189.002055. [DOI] [PubMed] [Google Scholar]
- BOLTON T.B., AARONSON P.I., MACKENZIE I. Voltage-dependent calcium channel in intestinal and vascular smooth muscle cells. Ann. N.Y. Acad. Sci. 1988;522:32–42. doi: 10.1111/j.1749-6632.1988.tb33340.x. [DOI] [PubMed] [Google Scholar]
- BURYI V., MOREL N., SALOMONE S., KERGER S., GODFRAIND T. Evidence for a direct interaction of thapsigargin with voltage-dependent Ca2+ channel. Naunyn-Schmiedeberg's Arch. Pharmacol. 1995;351:40–45. doi: 10.1007/BF00169062. [DOI] [PubMed] [Google Scholar]
- CAMPBELL D.L., STAMLER J.S., STRAUSS H.C. Redox modulation of L-type calcium channels in ferret ventricular myocytes. Dual mechanism regulation by nitric oxide and S-nitrosothiols. J. Gen. Physiol. 1996;108:277–293. doi: 10.1085/jgp.108.4.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FEARON I.M., PALMER A.C.V., BALMFORTH A.J., BALL S.G., VARADI G., PEERS C. Modulation of recombinant human cardiac L-type Ca2+ channel α1c subunits by redox agents and hypoxia. J. Physiol. 1999;514:629–637. doi: 10.1111/j.1469-7793.1999.629ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FOSKETT J.K., WONG D. Calcium oscillations in parotid acinar cells induced by microsomal Ca2+-ATPase inhibition. Am. J. Physiol. 1992;262:C656–C663. doi: 10.1152/ajpcell.1992.262.3.C656. [DOI] [PubMed] [Google Scholar]
- FUSI F., GORELLI B., VALOTI M., MARAZOVA K., SGARAGLI G.P. Effects of 2,5-di-t-butyl-1,4-benzohydroquinone (BHQ) on rat aorta smooth muscle. Eur. J. Pharmacol. 1998;346:237–243. doi: 10.1016/s0014-2999(98)00056-9. [DOI] [PubMed] [Google Scholar]
- FUSI F., MARAZOVA K., PESSINA F., GORELLI B., VALOTI M., FROSINI M., SGARAGLI G.P. On the mechanisms of the antispasmodic action of some hindered phenols in rat aorta rings. Eur. J. Pharmacol. 2000;394:109–115. doi: 10.1016/s0014-2999(00)00152-7. [DOI] [PubMed] [Google Scholar]
- FUSI F., VALOTI M., FROSINI F., SGARAGLI G.P. 2,5,Di-t-butyl-1,4-benzohydroquinone induces endothelium-dependent relaxation of rat thoracic aorta. Eur. J. Pharmacol. 1999;366:181–187. doi: 10.1016/s0014-2999(98)00932-7. [DOI] [PubMed] [Google Scholar]
- GEISZT M., KALDI K., SZEBERENYI J.B., LIGETI E. Thapsigargin inhibits Ca2+ entry into human neutrophil granulocytes. Biochem. J. 1995;305:525–528. doi: 10.1042/bj3050525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUERRA L., CERBAI E., GESSI S., BOREA P.A., MUGELLI A. The effect of oxygen free radicals on calcium current and dihydropyridine binding sites in guinea-pig ventricular myocytes. Br. J. Pharmacol. 1996;118:1278–1284. doi: 10.1111/j.1476-5381.1996.tb15534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAMILL O.P., MARTY A., NEHER E., SAKMANN B., SIGWORTH F.J. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- IESAKI T., WOLIN M.S. Thiol oxidation activates a novel redox-regulated coronary vasodilator mechanism involving inhibition of Ca(2+) influx. Arterioscler. Thromb. Vasc. Biol. 2000;20:2359–2365. doi: 10.1161/01.atv.20.11.2359. [DOI] [PubMed] [Google Scholar]
- IVES H.E., SCHULTZ G.S., GALARDY R.E., JAMIESON J.D. Preparation of functional smooth muscle cells from the rabbit aorta. J. Exp. Med. 1978;148:1400–1413. doi: 10.1084/jem.148.5.1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOURIE J.I. Interaction of reactive oxygen species with ion transport mechanisms. Am. J. Physiol. 1998;275:C1–C24. doi: 10.1152/ajpcell.1998.275.1.C1. [DOI] [PubMed] [Google Scholar]
- LORY P., VARADI G., SCHWARTZ A. Molecular insights into regulation of L-type Ca channel function. News Physiol. Sci. 1991;6:277–281. [Google Scholar]
- LOW A.M., KOTECHA N., NEILD T.O., KWAN C.Y., DANIEL E.E. Relative contributions of extracellular Ca2+ and Ca2+ stores to smooth muscle contraction in arteries and arterioles of rat, guinea-pig, dog and rabbit. Clin. Exp. Pharmacol. Physiol. 1996;23:310–316. doi: 10.1111/j.1440-1681.1996.tb02829.x. [DOI] [PubMed] [Google Scholar]
- MARKLUND S., MARKLUND G. Involvement of the superoxide anion radical in the autoxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur. J. Pharmacol. 1974;47:469–474. doi: 10.1111/j.1432-1033.1974.tb03714.x. [DOI] [PubMed] [Google Scholar]
- MASON M.J., GARCIA-RODRIGUEZ C., GRINSTEIN S. Coupling between intracellular Ca2+ stores and the Ca2+ permeability of the plasma membrane. Comparison of the effects of thapsigargin, 2,5-di-(tert-butyl)-1,4-hydroquinone, and cyclopiazonic acid in rat thymic lymphocytes. J. Biol. Chem. 1991;266:20856–20862. [PubMed] [Google Scholar]
- MCCORD J.M., FRIDOVICH I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein) J. Biol. Chem. 1969;244:6049–6055. [PubMed] [Google Scholar]
- MCDONALD T.F., PELZER S., TRAUTWEIN W., PELZER D.J. Regulation and modulation of calcium channels in cardiac, skeletal, and smooth muscle cells. Physiol. Rev. 1994;74:365–507. doi: 10.1152/physrev.1994.74.2.365. [DOI] [PubMed] [Google Scholar]
- MISSIAEN L., DE SMEDT H., DROOGMANS G., CASTEELS R. 2,5-Di(tert-butyl)-1,4-benzohydroquinone and cyclopiazonic acid decrease the Ca2+ permeability of endoplasmic reticulum. Eur. J. Pharmacol. 1992;227:391–394. doi: 10.1016/0922-4106(92)90156-p. [DOI] [PubMed] [Google Scholar]
- MOORE G.A., MCCONKEY D.J., KASS G.E.N., O'BRIEN P.J., ORRENIUS S. 2,5-Di(tert-butyl)-1,4-benzohydroquinone - a novel inhibitor of liver microsomal Ca2+ sequestration. FEBS Lett. 1987;224:331–336. doi: 10.1016/0014-5793(87)80479-9. [DOI] [PubMed] [Google Scholar]
- NAKAMURA H., NAKASAKI Y., MATSUDA N., SHIGEKAWA M. Inhibition of sarcoplasmic reticulum Ca2+-ATPase by 2,5-di(tert-butyl)-1,4-benzohydroquinone. J. Biochem. 1992;112:750–755. doi: 10.1093/oxfordjournals.jbchem.a123970. [DOI] [PubMed] [Google Scholar]
- NELSON E.J., LI C.C.-R., BANGALORE R., BENSON T., KASS R.S., HINKLE P.M. Inhibition of L-type calcium-channel activity by thapsigargin and 2,5-t-butylhydroquinone, but not by cyclopiazonic acid. Biochem. J. 1994;302:147–154. doi: 10.1042/bj3020147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PASYK E., INAZU M., DANIEL E.E. CPA enhances Ca2+ entry in cultured bovine pulmonary arterial endothelial cells in a IP3-independent manner. Am. J. Physiol. 1995;268:H138–H146. doi: 10.1152/ajpheart.1995.268.1.H138. [DOI] [PubMed] [Google Scholar]
- ROSSIER M.F., PYTHON C.P., BURNAY M.M., SCHLEGEL W., VALLOTTON M.B., CAPPONI A.M. Thapsigargin inhibits voltage-activated calcium channels in adrenal glomerulosa cells. Biochem. J. 1993;296:309–312. doi: 10.1042/bj2960309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCAMPS F., VIGUES S., RESTITUITO S., CAMPO B., ROIG A., CHARNET P., VALMIER J. Sarco-endoplasmic ATPase blocker 2,5-di(tert-butyl)-1,4-benzohydroquinone inhibits N-, P-, and Q- but not T-, L-, or R-type calcium currents in central and peripheral neurons. Mol. Pharmacol. 2000;58:18–26. doi: 10.1124/mol.58.1.18. [DOI] [PubMed] [Google Scholar]
- SEIDLER N.W., JONA I., VEGH M., MARTONOSI A. Cyclopiazonic acid is a specific inhibitor of Ca2+-ATPase of sarcoplasmic reticulum. J. Biol. Chem. 1989;264:17816–17823. [PubMed] [Google Scholar]
- STAMLER J.S. A radical vascular connection. Nature. 1996;380:108–111. [PubMed] [Google Scholar]
- STANSFELD C., MATHIE A.Recording membrane currents of peripheral neurones in short-term culture Electrophysiology. A practical approach 1993Oxford: IRL Press; 3–28.Ed. Wallis, D.I., pp [Google Scholar]
- THASTRUP O.P., CULLEN P.J., DROBAK B.K., HANLEY M.R., DAWSON A.P. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proc. Natl. Acad. Sci. U.S.A. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAN BREEMEN C., CHEN Q., LAHER I. Superficial buffer barrier function of smooth muscle sarcoplasmic reticulum. Trends Pharmacol. Sci. 1995;16:98–105. doi: 10.1016/s0165-6147(00)88990-7. [DOI] [PubMed] [Google Scholar]
- WEGENER J.W., MEYRER H., RUPP J., NAWRATH H. Barnidipine block of L-type Ca2+ channel currents in rat ventricular cardiomyocytes. Br. J. Pharmacol. 2000;130:2015–2023. doi: 10.1038/sj.bjp.0703514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WOLIN M.S. Interactions of oxidants with vascular signaling systems. Arterioscler. Thromb. Vasc. Biol. 2000;20:1430–1442. doi: 10.1161/01.atv.20.6.1430. [DOI] [PubMed] [Google Scholar]