

Abstract
Alterations of the vessel structure, which is mainly determined by smooth muscle cells through cell growth and/or cell death mechanisms, are characteristic of diabetes complications. We analysed the influence of high glucose (22 mM) on cultured human aortic smooth muscle cell growth and death, as hyperglycaemia is considered one of the main factors involved in diabetic vasculopathy.
Growth curves were performed over 96 h in medium containing 0.5% foetal calf serum. Cell number increased by 2–4 fold over the culture period in the presence of 5.5 mM (low) glucose, while a 20% reduction in final cell number was observed with high glucose. Under serum-free conditions, cell number remained constant in low glucose cultures, but a 40% decrease was observed in high glucose cultures, suggesting that high glucose may induce increased cell death rather than reduced proliferation. Reduced final cell number induced by high glucose was also observed after stimulation with 5 or 10% foetal calf serum.
The possible participation of oxidative stress was investigated by co-incubating high glucose with different reactive oxygen species scavengers. Only catalase reversed the effect of high glucose. Intracellular H2O2 content, visualized with 2′,7′-dichlorofluorescein and quantified by flow cytometry, was increased after high glucose treatment.
To investigate the cell death mechanism induced by high glucose, apoptosis and necrosis were quantified. No differences were observed regarding the apoptotic index between low and high glucose cultures, but lactate dehydrogenase activity was increased in high glucose cultures.
In conclusion, high glucose promotes necrotic cell death through H2O2 formation, which may participate in the development of diabetic vasculopathy.
Keywords: Hyperglycaemia, vascular smooth muscle, reactive oxygen species, cell death
Introduction
Hyperglycaemia has been identified as the main factor contributing to the development of vascular diseases associated to diabetes mellitus (The Diabetes Control and Complications Trial Research Group, 1993; Stratton et al., 2000), although the mechanisms linking high glucose levels and diabetic vasculopathy are not well defined. In the last years, however, an increasing number of reports point at enhanced oxidative stress as a key factor in the development of vascular abnormalities in diabetes (Baynes, 1991; Giugliano et al., 1996). In this way, high glucose either by itself or through mechanisms of nonenzymatic glycosylation may favour an enhanced production of reactive oxygen species (ROS), such as superoxide anions, hydrogen peroxide or hydroxyl radicals (Giugliano et al., 1996).
In addition to exhibiting impaired vasoactive function (de vries et al., 2000), diabetic vessels may undergo changes in their structure due to remodelling processes. Such remodelling processes are mainly determined by alterations in the rates of vascular smooth muscle growth (proliferation or hypertrophy) and vascular smooth muscle death (necrosis or apoptosis), together with extracellular matrix expansion (Rasmussen & Heickendorff, 1989; Rumble et al., 1997; Fukumoto et al., 1998). Once established, these structural alterations, which are related to vascular diseases like atherosclerosis or vascular hypertrophy (Soulis et al., 1999), accrue cardiovascular morbidity and mortality in diabetic patients.
In this way, there are previous reports indicating that high glucose may influence vascular remodelling as it can modify the rate of cell growth and cell death of different cell types of the vasculature, like pericytes, endothelial cells or smooth muscle cells (Chibber et al., 1994; Graier et al., 1995; Yasunari et al., 1996). Additionally, a growing number of reports suggest a role for ROS as modulators of vascular smooth muscle growth by promoting either cell proliferation both in vivo and in vitro (Konneh et al., 1995; Li et al., 1997) or cell hypertrophy (Peiró et al., 1998). In addition, ROS can also induce cell death in vascular smooth muscle through different mechanisms (Li et al., 1997).
To date most of the studies designed to analyse the effect of high glucose on vascular smooth muscle cell growth and cell death parameters have been performed using cells derived from different experimental animal models. In the present work, we aimed to analyse the influence of high glucose levels on cell growth and death using vascular smooth muscle cultures derived from human vessels, with special focus on the possible role of ROS.
Methods
Cell culture
Human aortic smooth muscle cell (HASMC) cultures were obtained by enzymatic dissociation of the aortas obtained from three organ donors (age, 37±3 years) at Hospital Universitario de Getafe, according to Spanish legal dispositions. The aortic fragments were cleaned free of the adventitia layer and the endothelium was removed by gently rubbing the lumen of the vessel. The fragments were then cut into small pieces and digested by incubation for 90 min in Dulbecco's Modified Eagle's Medium (DMEM; Biological Industries, Beit Hamek, Israel) containing 0.1% BSA (Sigma Chemical Co., St. Louis, Missouri, U.S.A.) and 4 mg ml−1 collagenase (type II, Sigma Chemical Co.). After washing twice with fresh DMEM, the resulting cell suspension was plated onto 25-cm2 culture flasks (Costar-Corning, New York, U.S.A.) in DMEM containing 10% foetal calf serum (FCS; Biological Industries), 100 μg ml−1 streptomycin, 100 u ml−1 penicillin, and 2.5 μg ml−1 Amphotericin B (Sigma Chemical Co.). Cell characterization was performed based on both cell morphology and indirect immunofluorescence staining of α-smooth muscle actin, as previously described by others (Dubey et al., 1992). At confluence, HASMC were passaged using a 0.02% EDTA-0.05% trypsin solution and split in a 1 : 2 ratio. In the present study, cultures between passages 2 and 10 were used.
Cell counting
HASMC were cultured into 24-well culture plates for 96 h in DMEM containing 0.5% FCS and either 5.5 mM or 22 mM glucose. For each experiment, the culture medium, either alone or containing different drugs, was renewed every 24 h. At different time points, cell number per well was determined by fixing cultures with 1% glutaraldehyde followed by cell nuclei staining with a 1% crystal violet solution (Fluka, Buchs, Switzerland), as previously described (Peiró et al., 1995). After extensive washing with deionized water, crystal violet was extracted from cells with 10% acetic acid and the resulting colour was quantified by measuring absorbance at 595 nm with an EL-340 automated microplate reader (Bio-Tek Instruments, Winooski, Vermont, U.S.A.). A standard curve was carried out to establish a relationship between absorbance and cell number per well (r=0.98).
Intracellular hydrogen peroxide (H2O2) detection
HASMC plated into glass coverslips inserted into 24-well plates were cultured as described above. When confluent, HASMC were loaded with 30 μM of the H2O2-sensitive fluorescent probe 2′,7′-dichlorofluorescein diacetate (DCF-DA; Molecular Probes, Leiden, The Netherlands) for 4 h at 37°C. Once inside the cell, nonfluorescent DCF-DA is deacetylated enzymatically by cellular esterases to 2′,7′-dichlorofluorescein (DCF), which remains trapped in the cell. DCF fluoresces in the presence of intracellular peroxides. After loading, HASMC were washed with phosphate-buffered saline and treated with 5.5 mM glucose or 22 mM glucose for 24 h. As a positive control, H2O2 (1 mM) was added to some wells incubated with 5.5 mM glucose 10 min before the end of the treatment. Intracellular fluorescence was then quantified using a FACScan (Becton Dickinson, Franklin Lakes, New Jersey, U.S.A.) and visualized with an Eclipse TE300 epifluorescence microscope (Nikon, Tokyo, Japan), using an excitation wavelength of 485 nm and an emission wavelength of 530 nm.
Determination of apoptosis
Apoptotic HASMC were identified based on chromatin morphology, as previously described (Peiró et al., 2000). Briefly, at the indicated time periods, the chromatin binding dye Hoescht 33342 (Molecular Probes) was added to cell cultures at a concentration of 5 μM for 30 min at 37°C. Afterwards, HASMC were collected together with the cell supernatants, centrifuged, and the resulting cell pellet was resuspended in FCS-containing medium. Cell nuclei were observed using an epifluorescence microscope at 40×magnification. Nuclei showing chromatin margination or fragmentation were considered apoptotic. For every treatment, 500 nuclei from random microscopic fields were counted by a blinded observer.
As another method to identify apoptotic cells, the terminal deoxyribonucleotidyl transferase-mediated dUTP-digoxigenin nick-end labelling (TUNEL) assay was performed using a commercial kit (Oncor, Gaithersburg, MD, U.S.A.), as previously described (Peiró et al., 2000). Nuclei were counterstained using mounting medium containing 1 μM 4′,6-diamidino-2-phenylindole (DAPI; Molecular Probes). For every treatment, 500 nuclei from random microscopic fields were observed under fluorescence microscopy by a blinded observer.
Lactate dehydrogenase release
As a marker of cytotoxicity, lactate dehydrogenase (LDH) was measured in the supernatant of HASMC submitted to the different experimental conditions. LDH was quantified as absorbance at 490 nm using a commercial kit (Boehringer Mannheim, Mannheim, Germany). A standard curve was performed to relate absorbance with LDH activity, using known concentrations of commercially available LDH. Maximum LDH release was calculated in cultures lysed with 1% Triton X-100.
Drugs
Superoxide dismutase (EC 1.15.1.1) from bovine erythrocytes, catalase (EC 1.11.1.6) from bovine liver, L-lactic dehydrogenase from porcine heart (EC 1.1.1.37), D-mannitol, N,N′-dimethylthiourea, deferoxamine mesylate and, unless otherwise stated, all other reagents were purchased from Sigma Chemical Co.
Statistical analysis
Results are expressed as mean±s.e.mean. The statistical analysis was evaluated by unpaired Student's t-test for single data points or by analysis of variance (ANOVA) for curves, with the level of significance chosen at P<0.05. n denotes the number of independent experiments.
Results
Effect of glucose concentration on cell counting
HASMC were cultured for 96 h in DMEM containing 5.5 mM (low) or 22 mM glucose (high) glucose and cell number per well was determined every 48 h. As shown in Figure 1A, in this cell type there was a significant increase in cell number per well over the culture period both in low and high glucose cultures when stimulated with 0.5% FCS (P<0.05, ANOVA). However, in high glucose cultures, the mean final cell number per well was approximately 20% lower compared with low glucose cultures. When an intermediate concentration of glucose (11 mM) was used, the final cell number per well remained unmodified compared with HASMC cultured in low glucose conditions (98.90±5.39% of cell counting obtained with low glucose, n=3).
Figure 1.
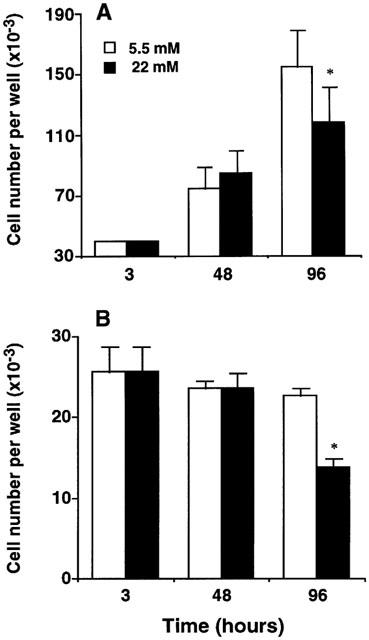
Effect of 5.5 and 22 mM glucose on human aortic smooth muscle (HASMC) cell counting. (A) HASMC were cultured up to 96 h in culture medium containing 0.5% FCS. Under both glucose concentrations, there was an increase in cell number per well over the culture period (P<0.05, ANOVA). (B) Similar experiments were performed using serum-free culture medium. Cell number per well was unmodified in low glucose cultures over the culture period, whereas decreased final cell number was observed in high glucose cultures (P<0.05, ANOVA). Data are expressed as mean±s.e.mean of three independent experiments. *P<0.05 versus low glucose, t-test.
When the growth curves were reproduced using FCS-free DMEM supplemented with 0.1% BSA, cell number per well was not significantly modified over the culture period in low glucose cultures (P>0.05, ANOVA; Figure 1B). On the contrary, a significant decrease in cell number, particularly within the 48–96 h period, was observed in HASMC cultured with high glucose (P<0.05, ANOVA; Figure 1B).
To assess whether high glucose exerted a similar inhibitory effect on final cell number in the presence of more potent proliferative stimuli, growth curves were performed using growing concentrations of FCS (0.5, 5 and 10%). As shown in Figure 2, there was a FCS concentration-dependent increase in final cell density (P<0.05, ANOVA). However, for every FCS concentration, treatment with high glucose resulted in a significant reduction of final cell number (P<0.05, t-test).
Figure 2.

Effect of 5.5 and 22 mM glucose on human aortic smooth muscle (HASMC) cell counting after 96 h of stimulation with 0.5, 5 or 10% FCS. Under both glucose concentrations there was a concentration-dependent increase in final cell number per well (P<0.05, ANOVA). For every FCS concentration, final cell number per well was diminished in the presence of high glucose. *P<0.05 versus low glucose, t-test. Data are expressed as mean±s.e.mean of three independent experiments.
Role of hyperosmolarity in the decrease in cell number induced by high glucose
To clarify whether the effect of high glucose on cell counting was a consequence of hyperosmolarity, the growth curves in response to 0.5% FCS were reproduced using DMEM containing 5.5 mM glucose plus 16.5 mM sucrose. After 96 h of culture, sucrose did not significantly modify the cell number per well obtained in the presence of low glucose, whereas, again, high glucose diminished cell number by around 20% (Figure 3).
Figure 3.

Role of hyperosmolarity on the effect of high glucose on cell counting. Human aortic smooth muscle cells were cultured up to 96 h in medium containing 0.5% FCS. Data are expressed as mean±s.e.mean of three independent experiments. *P<0.05 versus low glucose, ttest.
Role of ROS in the decrease in cell counting induced by high glucose
To study whether ROS were participating in the decrease in cell counting induced by high glucose, cell counting was performed after 96 h of culture in medium containing 0.5% FCS and high glucose supplemented with different ROS scavengers. Figure 4A shows that superoxide dismutase (200 u ml−1) did not significantly alter the effect of high glucose on cell counting. Similarly, neither deferoxamine (10 μM), mannitol (1 mM) or dimethylthiourea (1 mM) did modify the effect of high glucose (Figure 4A). On the contrary, catalase (200 u ml−1) completely reversed the effect of high glucose, yielding a cell number per well similar to that obtained in low glucose cultures (Figure 4A). Indeed, as shown in Figure 4B, the addition of H2O2 (10 μM) to low glucose medium mimicked the effects of high glucose (87.99±1.92% and 81.73±3.79% of cell number obtained with low glucose alone, respectively; P<0.05, n=4). The effect of H2O2 was abolished by co-incubation with 200 u ml−1 catalase (Figure 4B).
Figure 4.
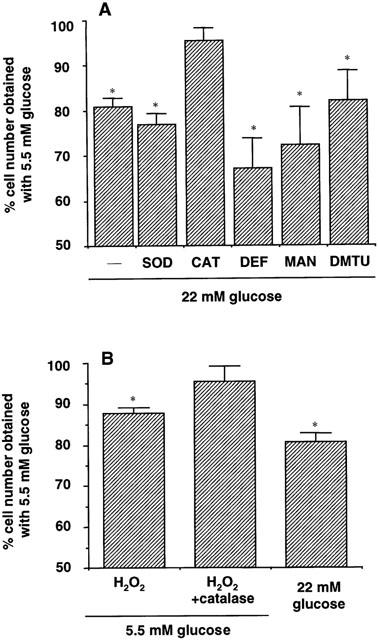
(A) Effect of different reactive oxygen species scavengers on the decrease in cell counting induced by high glucose. Human aortic smooth muscle cells were cultured for 96 h in medium containing 0.5% FCS and 22 mM glucose either alone or supplemented with 200 u ml−1 superoxide dismutase (SOD), 200 u ml−1 catalase (CAT), 10 μM deferoxamine (DEF), 1 mM mannitol (MAN) or 1 mM dimethylthiourea (DMTU). (B) Effect of H2O2 (10 μM) either alone or in the presence of 200 u ml−1 catalase, on cell counting after 96 h of culture in medium containing 0.5% FCS and 5.5 mM glucose. The effect of 22 mM glucose on cell counting is also shown. Data are expressed as mean±s.e.mean of three independent experiments. *P<0.05 versus low glucose, t-test.
Intracellular content of H2O2
The fluorescent probe 2′,7′-dichlorofluorescein was used to detect intracellular levels of H2O2. As shown in Figure 5A, after 24 h of incubation, intracellular fluorescence was around 1.5 fold higher in HASMC cultured with high glucose medium compared with HASMC cultured with low glucose (20.30±4.97 versus 13.05±2.2 arbitrary fluorescence units; P<0.05, n=4). Co-incubation with catalase (200 u ml−1) reduced high glucose-induced fluorescence levels to 1.43±0.05 arbitrary units (P<0.05, n=4). When H2O2 (1 mM) was added to low glucose cultures as a positive control, fluorescence levels were markedly increased (135.11±7.72 arbitrary units; P<0.05, n=4). Figure 5B shows the changes in intracellular fluorescence as viewed under a fluorescence microscope.
Figure 5.
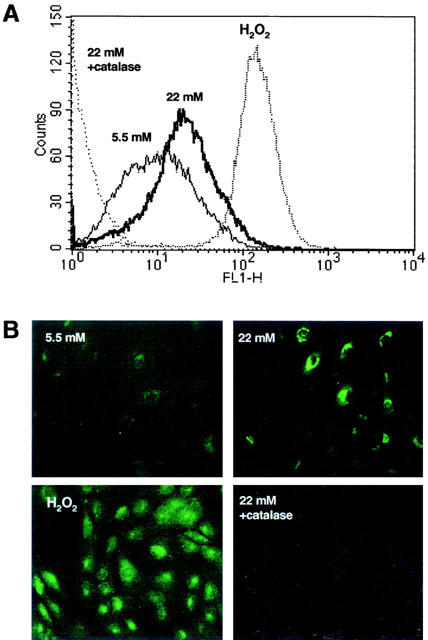
(A) Representative diagram of intracellular H2O2 quantified by flow cytometry in human aortic smooth muscle cells loaded with the fluorescent probe 2′,7′-dichlorofluorescein and cultured with medium containing either 5.5 mM glucose, 22 mM glucose, 1 mM H2O2 or 22 mM glucose plus 200 u ml−1 catalase. (B) Intracellular H2O2 content as visualized by epifluorescence microscopy in the aboved described culture conditions.
Proapoptotic and cytotoxic effects of high glucose on HASMC
To assess whether high glucose induced HASMC cell death through an apoptotic mechanism, apoptotic cells were quantified by chromatin staining and TUNEL in both low and high glucose cultures. As shown in Table 1, high glucose did not modify the number of apoptotic nuclei, as assessed by chromatin morphology. Used as a positive control, thapsigargin, an endoplasmic reticulum calcium ATPase inhibitor, used at a concentration of 1 μM markedly enhanced the number of apoptotic nuclei. In the same way, the number of labelled nuclei by the TUNEL assay after 72 h of culture was significantly enhanced in thapsigargin-treated HASMC but not in high glucose cultures compared to low glucose cultures (Table 1).
Table 1.
Quantification of apoptosis in human aortic smooth muscle cell cultures submitted to low or high glucose concentrations

However, when necrotic cell death was evaluated by the release of LDH in the culture supernatant over the 96 h culture period, high glucose supernatants were found to contain significantly higher levels of LDH than low glucose supernatants (P<0.05, ANOVA; Figure 6). This higher release of LDH could be observed at every time point analysed (P<0.05 between 5.5 and 22 mM for every time point, t-test).
Figure 6.
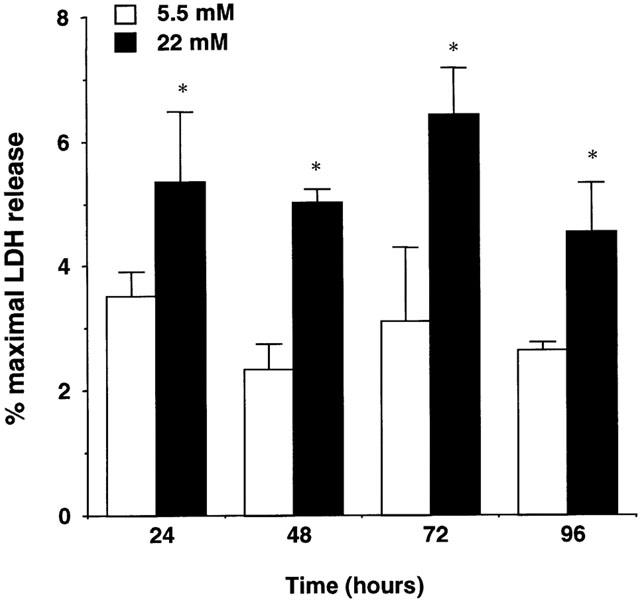
Quantification of lactate dehydrogenase (LDH), as a marker of necrosis, in the supernatant of human aortic smooth muscle cells cultured for 96 h in medium containing 0.5% FCS and 5.5 or 22 mM glucose. The release of LDH over the culture period was significantly enhanced in cultures containing high glucose levels (P<0.05, ANOVA). Results are expressed as percentage of maximal LDH release obtained after cell lysis with 0.1% Triton X-100. Mean basal LDH release in the presence of 5.5 mM glucose was 10.09±0.31 mu ml−1. Data represent mean±s.e.mean of three independent experiments. *P<0.05 versus low glucose, t-test.
Discussion
Hyperglycaemia has been identified as the main factor contributing, either through direct or indirect mechanisms, to the pathogenesis of vascular disease in diabetes by producing biochemical and metabolic alterations that lead to both functional and structural alterations in diabetic vessels (The Diabetes Control and Complication Trial Research Group, 1993; Stratton et al., 2000). Vascular remodelling in diabetes, as the result of alterations in the rates of vascular smooth muscle growth and/or death, together with extracellular matrix expansion, contributes to cardiovascular disease, one of the main complications of long-term diabetes (Nathan, 1993).
In the present work, experimental evidence is presented, indicating that upon incubation of HASMC in a hyperglycaemic medium (22 mM), cell counting after 96 h of stimulation with 0.5% FCS was significantly lower than that observed in normoglycaemic conditions (5.5 mM). Such a difference was due to a reduced up-shift in cell number, particularly during the 48–96 h period. The effect of high glucose could not be attributable to hyperosmolarity, as HASMC cultured with 5.5 mM glucose plus 16.5 mM sucrose (osmolarity control) did not mimic the effect of high glucose.
This inhibitory effect of high glucose on final cell density was also observed in the presence of higher proliferative stimuli (5 and 10% FCS). However, we chose the 0.5% FCS concentration to study the mechanisms underlying the effect of high glucose, as in HASMC cultures this FCS concentration yielded a 2 fold increase in cell number all over the experimental period, compared to a 15–20 fold increase observed in the presence of 5 or 10% FCS. As the replication rate of adult vascular smooth muscle cells in the vessel wall in vivo is quite low (Jackson & Schwartz, 1992), the 0.5% FCS concentration could be more appropriate to investigate the effects of high glucose on HASMC.
A first mechanism that could account for the reduced cell counting observed under hyperglycaemic conditions could be a retarded growth rate of HASMC. Indeed, in other vascular and non- vascular cell types, like endothelial cells (Umeda et al., 1991; Graier et al., 1995; La Selva et al., 1996), retinal pericytes (Chibber et al., 1994) or mesangial cells (Trachtman et al., 1994), a delayed replication rate has been described when cultured in the presence of high glucose concentrations. Concerning VSMC, however, some previous reports have shown an opposite effect for high glucose, which has been mainly identified as a growth promoter for vascular smooth muscle obtained from rat, pig or rabbit (Graier et al., 1995, Yasunari et al., 1996; 1997; Sharpe et al., 1998). In the present study, however, high glucose did not behave as a growth promoter for HASMC. A possible explanation for such different effects may reside in the experimental protocol. Thus, Yasunari et al. (1996) pre-incubated rat VSMC in high glucose up to two passages prior to the experiments to mimic chronic hyperglycaemia, while the aim of the present work is to study a more acute model of hyperglycaemia. However, it is also possible that the different effect of high glucose may be due to a different behaviour of VSMC of human origin. Indeed, most of the previous work performed without high glucose pre-incubation has shown a proliferative effect of high glucose for vascular smooth muscle cultures but in all cases VSMC were derived from non-human species (Graier et al., 1995; Yasunari et al., 1997; Sharpe et al., 1998).
A second mechanism that could explain the reduced cell counting induced by hyperglycaemia is that high glucose may not decrease the proliferation rate of HASMC, but rather promote HASMC death, therefore impairing the balance between both mechanisms. The results obtained in our experimental conditions suggest that this is likely to be the case. Indeed, cell counting performed in serum-free non-proliferating conditions showed that HASMC number remained constant in low glucose cultures, while in high glucose cultures there was a significant decrease in cell number, especially during the last 48 h period, which could be only attributable to cell death, as there was no proliferation of HASMC. Concerning this point, there are several reports demonstrating that glucose at high concentrations can act as an inducer of cell death in the vasculature, although most of the studies have been performed in endothelial cells (Baumgartner-Parzer et al., 1995; Du et al., 1999; Ho et al., 1999).
In endothelial cultures, the main death pattern followed after exposure to high glucose concentrations seems to be apoptosis (Baumgartner-Parzer et al., 1995; Du et al., 1998; Ho et al., 1999). On the contrary, high glucose has recently been reported to prevent cultured VSMC apoptosis induced by serum starvation or Fas ligand through a protein kinase C-dependent mechanism (Hall et al., 2000). In the present work, we failed to see any evidence for an apoptotic death pattern after exposure of HASMC to high glucose. Indeed, neither chromatin morphology nor the TUNEL assay revealed any significant difference between the apoptotic index of low and high glucose cultures, which was very low in both cases (around 0.5–1% of total). In order to validate these results, HASMC were treated with the endoplasmic reticulum calcium ATPase inhibitor thapsigargin, which has been previously reported to induce apoptosis in this cell type (Peiró et al., 2000), resulting in a marked increase of the apoptotic index. Contrasting with the lack of a proapoptotic effect of high glucose, the supernatants of HASMC cultured with a hyperglycaemic medium contained significantly higher amounts of LDH all over the experimental period, indicating a loss of integrity of the plasma membrane. The enhancement of LDH release, a widely accepted marker of cytotoxicity, in the absence of any evidence for apoptosis, indicates that HASMC death in a hyperglycaemic environment may occur through a mechanism of cell necrosis.
Concerning the mechanisms mediating the cytotoxic effect of high glucose on HASMC, oxidative stress, a key factor in the development of diabetic complications, arises as the main candidate. Indeed, many studies have evidenced an increased oxidative status in vascular cells submitted to hyperglycaemic conditions, based either on direct quantification of ROS or determinations of the activity of antioxidant enzymes and peroxidation metabolites (Sharpe et al., 1998; Ho et al., 2000). Depending on the different cellular parameters studied, different ROS, like superoxide anions, hydrogen peroxide or hydroxyl radicals, have been identified as the effectors of high glucose (Graier et al., 1996; Ho et al., 2000). In the present study, the possible species mediating high glucose-induced cytotoxicity in HASMC cultures was investigated by co-incubating high glucose with different selective ROS scavengers. The observation that either superoxide dismutase or different hydroxyl radicals scavengers failed to prevent the effect of high glucose permitted the discarding of both superoxide anions and hydroxyl radicals as the principal mediators of the effects of high glucose. On the contrary, catalase totally prevented the effect of high glucose on HASMC counting, therefore pointing at hydrogen peroxide as the ROS responsible for HASMC necrosis. Supporting this, the addition of hydrogen peroxide to HASMC cultures in the presence of low glucose mimicked the effects of high glucose on cell counting.
Furthermore, a direct evidence of an increased intracellular hydrogen peroxide content in high glucose-treated cultures was provided using the fluorescent probe 2′,7′-dichlorofluorescein, which specifically detects this ROS (Hunt et al., 1990; Ho et al., 2000). After 24 h of culture, HASMC cultured in the presence of high glucose showed a 50% increase in hydrogen peroxide content compared to HASMC cultured with low glucose, this increase in fluorescence being prevented by the addition of catalase. Using cultured endothelial cells, other authors have reported a time-dependent increase in intracellular H2O2 content after several hours of culture with high glucose (Ho et al., 2000). Therefore, it seems reasonable to conclude that glucose-induced cell death in HASMC cultures is mediated by hydrogen peroxide. Interestingly, superoxide anions and hydrogen peroxide have recently been shown to exert differential actions on cultured VSMC (Griendling & Harrison, 1999). Indeed, superoxide seems to be more related with growth mechanisms, while hydrogen peroxide is associated with cell death processes (Li et al., 1997). In addition, using cultured VSMC we have previously reported that superoxide anions can promote cell growth through a hypertrophic mechanism (Peiró et al., 1998), while in the present work we present evidence for a cytotoxic effect of hydrogen peroxide in the same cell type.
In conclusion, in the present experimental conditions, short periods of hyperglycaemia do not enhance cell growth as it occurs in other animal species but rather induces cultured human vascular smooth muscle necrosis through the production of intracellular hydrogen peroxide. Although extrapolating to in vivo conditions must be handled with caution, it can be suggested that an acute enhancement of oxidative stress following hyperglycaemia, may induce the loss of medial myocytes, which, together with other related phenomena, like inflammation of the surrounding tissue, may represent an additional factor contributing to vascular lesions in diabetes.
Acknowledgments
We are grateful to Drs Antonio Cuadrado and Daniel Martín from Departamento de Bioquímica, Universidad Autónoma de Madrid, for their kind help with the flow cytometry analysis. Supported by grants from CICYT (SAF 98-0010), FISS (99/0246), FEDER (2FD97-0445-CO2), and Pharmamar.
Abbreviations
- DCF-DA
2′,7′-dichlorofluorescein diacetate
- DMEM
Dulbecco's Modified Eagle's Medium
- FCS
foetal calf serum
- HASMC
human aortic smooth muscle cells
- H2O2
hydrogen peroxide
- LDH
lactate dehydrogenase
- ROS
reactive oxygen species
References
- BAUMGARTNER-PARZER S.M., WAGNER L., PETTERMANN M., GRILLARI J., GESSL A., WALDHAUS W. High-glucose triggered apoptosis in cultured endothelial cells. Diabetes. 1995;44:1323–1327. doi: 10.2337/diab.44.11.1323. [DOI] [PubMed] [Google Scholar]
- BAYNES J.W. Role of oxidative stress in development of complications in diabetes. Diabetes. 1991;40:405–412. doi: 10.2337/diab.40.4.405. [DOI] [PubMed] [Google Scholar]
- CHIBBER R., MOLINATTI P.A., WONG J.S.K., MIRLEES D., KOHNER E.M. The effect of aminoguanidine and tolrestat on glucose toxicity in bovine retinal capillary pericytes. Diabetes. 1994;43:758–763. doi: 10.2337/diab.43.6.758. [DOI] [PubMed] [Google Scholar]
- DE VRIES A.S., VERBEUREN T.J., VAN DE VOORDE J., LAMEIRE N.H., VANHOUTTE P.M. Endothelial dysfunction in diabetes. Br. J. Pharmacol. 2000;130:963–974. doi: 10.1038/sj.bjp.0703393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DU X.L., SUI G.Z., STOCKKLAUSER-FÄRBER K., WEISS J., ZINK S., SCHWIPPERT B., WU Q.X., TSCHÖPE D., RÖSEN P. Induction of apoptosis by high proinsulin and glucose in cultured human umbilical vein endothelial cells is mediated by reactive oxygen species. Diabetologia. 1998;41:249–256. doi: 10.1007/s001250050900. [DOI] [PubMed] [Google Scholar]
- DU X., STOCKKLAUSER-FÄRBER K., RÖSEN P. Generation of reactive oxygen intermediates, activation of NF-kB, and induction of apoptosis in human endothelial cells by glucose: role of nitric oxide synthase. Free Rad. Biol. Med. 1999;27:752–763. doi: 10.1016/s0891-5849(99)00079-9. [DOI] [PubMed] [Google Scholar]
- DUBEY R.K., ROY A., OVERBECK H.W. Culture of renal arteriolar smooth muscle cells. Mitogenic responses to angiotensin II. Circ. Res. 1992;71:1143–1152. doi: 10.1161/01.res.71.5.1143. [DOI] [PubMed] [Google Scholar]
- FUKUMOTO H., NAITO Z., ASANO G., ARAMAKI T. Immunohistochemical and morphometric evaluation of coronary atherosclerotic plaques associated with myocardial infarction and diabetes mellitus. J. Atheroscl. Thromb. 1998;5:29–35. doi: 10.5551/jat1994.5.29. [DOI] [PubMed] [Google Scholar]
- GIUGLIANO D., CERIELLO A., PAOLISSO G. Oxidative stress and diabetic vascular complications. Diabetes Care. 1996;19:257–267. doi: 10.2337/diacare.19.3.257. [DOI] [PubMed] [Google Scholar]
- GRAIER W.F., GRUBENTHAL I., DITTRICH P., WASCHER T.C., KOSTNER G.M. Intracellular mechanism of high D-glucose-induced modulation of vascular cell proliferation. Eur. J. Pharmacol. 1995;294:221–229. doi: 10.1016/0014-2999(95)00534-x. [DOI] [PubMed] [Google Scholar]
- GRAIER W.F., SIMECEK S., KUKOVETZ W.R., KOSTNER G.M. High D-glucose-induced changes in endothelial Ca2+/EDRF signaling are due to generation of superoxide anions. Diabetes. 1996;45:1386–1396. doi: 10.2337/diab.45.10.1386. [DOI] [PubMed] [Google Scholar]
- GRIENDLING K.K., HARRISON D.G. Dual role of reactive oxygen species in vascular growth. Circ. Res. 1999;85:562–563. doi: 10.1161/01.res.85.6.562. [DOI] [PubMed] [Google Scholar]
- HALL J.F., MATTER C.M., WANG X., GIBBONS G.H. Hyperglycemia inhibits vascular smooth muscle cell apoptosis through a protein linase C-dependent pathway. Circ. Res. 2000;87:574–580. doi: 10.1161/01.res.87.7.574. [DOI] [PubMed] [Google Scholar]
- HO F.M., LIU S.G., LIAU C.S., HUANG P.J., SHIAH S.G., LIN-SHIAU S.Y. Nitric oxide prevents apoptosis of human endothelial cells from high glucose exposure during early stage. J. Cell. Biochem. 1999;75:258–263. doi: 10.1002/(sici)1097-4644(19991101)75:2<258::aid-jcb8>3.3.co;2-v. [DOI] [PubMed] [Google Scholar]
- HO F.M., LIU S.H., LIAU C.S., HUANG P.J., LIN-SHIAU S.Y. High glucose-induced apoptosis in human endothelial cells is mediated by sequential activation of c-jun NH2-terminal kinase and caspase-3. Circulation. 2000;101:2618–2624. doi: 10.1161/01.cir.101.22.2618. [DOI] [PubMed] [Google Scholar]
- HUNT J.V., SMITH C.C.T., WOLFF S.P. Autoxidative glycosylation and possible involvement of peroxides and free radicals in LDL modification by glucose. Diabetes. 1990;39:1420–1424. doi: 10.2337/diab.39.11.1420. [DOI] [PubMed] [Google Scholar]
- JACKSON C.L., SCHWARTZ S.M. Pharmacology of smooth muscle cell replication. Hypertension. 1992;20:713–736. doi: 10.1161/01.hyp.20.6.713. [DOI] [PubMed] [Google Scholar]
- KONNEH M.K., RUTHERFORD C., LI S.R., ANGGARD E.E., FERNS G.A. Probucol inhibits the intimal response to balloon catheter injury in the carotid artery of the cholesterol-fed rat. Atherosclerosis. 1995;113:29–39. doi: 10.1016/0021-9150(94)05423-g. [DOI] [PubMed] [Google Scholar]
- LA SELVA M., BELTRAMO E., PAGNOZZI F., BENA E., MOLINATTI G.M., PORTA M. Thiamine corrects delayed replication and decreases production of lactate and advanced glycation end-products in bovine retinal and human umbilical vein endothelial cells cultured under high glucose conditions. Diabetologia. 1996;39:1263–1268. doi: 10.1007/s001250050568. [DOI] [PubMed] [Google Scholar]
- LI P., DIETZ R., VON HARSDORF R. Differential effect of hydrogen peroxide and superoxide anion on apoptosis and proliferation of vascular smooth muscle cells. Circ. Res. 1997;96:3602–3609. doi: 10.1161/01.cir.96.10.3602. [DOI] [PubMed] [Google Scholar]
- NATHAN D.M. Long-term complications of diabetes mellitus. N. Engl. J. Med. 1993;328:1676–1685. doi: 10.1056/NEJM199306103282306. [DOI] [PubMed] [Google Scholar]
- PEIRÓ C., ANGULO J., RORIGUEZ-MAÑAS L., LLERGO J.L., VALLEJO S., CERCAS E., SANCHEZ-FERRER C.F. Vascular smooth muscle cell hypertrophy induced by glycosylated human oxyhaemoglobin. Br. J. Pharmacol. 1998;125:637–644. doi: 10.1038/sj.bjp.0702097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PEIRÓ C., REDONDO J., RODRIGUEZ-MARTINEZ M.A., ANGULO J., MARIN J., SANCHEZ-FERRER C.F. Influence of endothelium on cultured vascular smooth muscle cell proliferation. Hypertension. 1995;25:748–751. doi: 10.1161/01.hyp.25.4.748. [DOI] [PubMed] [Google Scholar]
- PEIRÓ C., VALLEJO S., CERCAS E., LLERGO J.L., LAFUENTE N., MATESANZ N., RODRIGUEZ-MAÑAS L., SANCHEZ-FERRER C.F. Thapsigargin induces apoptosis in cultured human vascular smooth muscle cells. J. Cardiovasc. Pharmacol. 2000;36:676–680. doi: 10.1097/00005344-200011000-00018. [DOI] [PubMed] [Google Scholar]
- RASMUSSEN L.M., HEICKENDORFF L. Accumulation of fibronectin in aortas from diabetic patients: a quantitative immunohistochemical and biochemical study. Lab. Invest. 1989;61:440–446. [PubMed] [Google Scholar]
- RUMBLE J.R., COOPER M.E., SOULIS T., COX A., WU L., YOUSSEF S., JASIK M., GERUMS G. Vascular hypertrophy in experimental diabetes. Role of advanced glycation end products. J. Clin. Invest. 1997;99:1016–1027. doi: 10.1172/JCI119229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHARPE P.C., YUE K.K.M., CATHERWOOD M.A., MCMASTER D., TRIMBLE E.R. The effects of glucose-induced oxidative stress on growth and extracellular matrix gene expression of vascular smooth muscle cells. Diabetologia. 1998;41:1210–1219. doi: 10.1007/s001250051054. [DOI] [PubMed] [Google Scholar]
- SOULIS T., SASTRA S., THALLAS V., MORTENSEN S.B., WILKEN M., CLAUSEN J.T., BJERRUM O.J., PETERSEN H., LAU J., JERUMS G., BOEL E., COOPER M.E. A novel inhibitor of advanced glycation end-product formation inhibits mesenteric vascular hypertrophy in experimental diabetes. Diabetologia. 1999;42:472–479. doi: 10.1007/s001250051181. [DOI] [PubMed] [Google Scholar]
- STRATTON I.M., ADLER A.I., NEIL H.A.W., MATTHEWS D.R., MANLEY S.E., CULL C.A., HADDEN D., TURNER R.C., HOLMAN R.R., on behalf of the UK PROSPECTIVE DIABETES STUDY GROUP Association of glycaemia with macrovacular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. Br. Med. J. 2000;321:405–419. doi: 10.1136/bmj.321.7258.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THE DIABETES CONTROL AND COMPLICATIONS TRIAL RESEARCH GROUP The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med. 1993;329:977–986. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- TRACHTMAN H., FUTTERWEIT S., PRENNER J., HANON S. Antioxidants reverse the antiproliferative effect of high glucose and advanced glycosylation end products in cultured rat mesangial cells. Biochem. Biophys. Res. Commun. 1994;199:346–352. doi: 10.1006/bbrc.1994.1235. [DOI] [PubMed] [Google Scholar]
- UMEDA F., YAMAUCHI T., NAKASHIMA N., ONO H., NAWATA H., MASUKO M., NAKAYAMA K., TATEMATSU A. Glucose reduces PDGF production and cell proliferation of cultured vascular endothelial cells. Horm. Metab. Res. 1991;23:274–277. doi: 10.1055/s-2007-1003672. [DOI] [PubMed] [Google Scholar]
- YASUNARI K., KOHNO M., KANO H., YOKOKAWA K., HORIO T., YOSHIKAWA J. Possible involvement of phospholipase D and protein kinase C in vascular growth induced by elevated glucose concentration. Hypertension. 1996;28:159–168. doi: 10.1161/01.hyp.28.2.159. [DOI] [PubMed] [Google Scholar]
- YASUNARI K., KOHNO M., KANO H., YOKOKAWA K., MINAMI M., YOSHIKAWA J. Mechanisms of action of troglitazone in the prevention of high glucose-induced migration and proliferation of cultured coronary smooth muscle cells. Hypertension. 1997;81:953–962. doi: 10.1161/01.res.81.6.953. [DOI] [PubMed] [Google Scholar]